Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Inflammatory Pathophysiology and New Potential Biomarkers in Alzheimer’s Disease
Time:2021-01-17
Number:6782
Author Affiliations

Conditioning Medicine 2020. 3(6): 285-295.
Abstract
Alzheimer’s disease (AD) is a type of progressive neurodegenerative disorder that primarily affects a subject’s cognitive function and memory. It is often also described as a fatal degenerative dementing disease whose course varies considerably from a few months to more than 20 years in certain cases. Current understanding of the diagnosis and prognosis of the condition informs us of three major pathophysiological hallmarks, namely - amyloid beta (Aβ) plaque formation, neuroinflammation/neuronal degeneration or loss, and neurofibrillary tangles accumulation. Research so far has also determined the major cellular mediators - microglia, astrocytes, and neurons - and their respective pathways of inflammation have been observed. Although traditionally used biomarkers are effective, new studies of potential markers exhibit greater diagnostic accuracy. Cerebrospinal fluid (CSF) biomarker studies of Aβ variations as well as neurogranin pathways for diagnosis of AD prove that these existing traditional methods can be refined and deployed more effectively. The major advantage that peripheral blood mononuclear cell (PBMC) studies offer as potential biomarkers over CSF is the ability to examine events that occur at a molecular level at different stages of the disorder, and thus reflect the underlying mechanisms behind the foundation, onset, and progression of the disease. Although the claims made so far are fairly accurate and pave a new path for diagnoses, it is essential to adopt a multi-modal biomarker system for the most reliable prediction. This review will therefore provide a new take and a novel perspective on biochemical markers in AD.
Keywords: Alzheimer's disease, inflammation, biomarkers, cytokines, PBMCs, CSF
Abstract
Alzheimer’s disease (AD) is a type of progressive neurodegenerative disorder that primarily affects a subject’s cognitive function and memory. It is often also described as a fatal degenerative dementing disease whose course varies considerably from a few months to more than 20 years in certain cases. Current understanding of the diagnosis and prognosis of the condition informs us of three major pathophysiological hallmarks, namely - amyloid beta (Aβ) plaque formation, neuroinflammation/neuronal degeneration or loss, and neurofibrillary tangles accumulation. Research so far has also determined the major cellular mediators - microglia, astrocytes, and neurons - and their respective pathways of inflammation have been observed. Although traditionally used biomarkers are effective, new studies of potential markers exhibit greater diagnostic accuracy. Cerebrospinal fluid (CSF) biomarker studies of Aβ variations as well as neurogranin pathways for diagnosis of AD prove that these existing traditional methods can be refined and deployed more effectively. The major advantage that peripheral blood mononuclear cell (PBMC) studies offer as potential biomarkers over CSF is the ability to examine events that occur at a molecular level at different stages of the disorder, and thus reflect the underlying mechanisms behind the foundation, onset, and progression of the disease. Although the claims made so far are fairly accurate and pave a new path for diagnoses, it is essential to adopt a multi-modal biomarker system for the most reliable prediction. This review will therefore provide a new take and a novel perspective on biochemical markers in AD.
Keywords: Alzheimer's disease, inflammation, biomarkers, cytokines, PBMCs, CSF
Introduction
Alzheimer's disease (AD) is the leading cause of dementia worldwide (Reitz et al., 2011) and is said to account for up to 70% of cases of cognitive neural impairment in the elderly population (Cummings et al., 2002). Fundamentally, there are three pathophysiological alterations that take place in the AD brain - neurodegeneration and neuronal loss (Niikura et al., 2006), neurofibrillary tangles (NFT) aggregation in the intracellular space due to hyperphosphorylation of the protein tau (Metaxas et al., 2016), and amyloid beta (Aβ) plaque formation due to excessive deposition (Murphy et al., 2010). In addition to these pathological changes, synaptic degeneration is also observed.
Macrophage cells play a vital role in inflammation modulation in the AD brain in the presence of the aforementioned hallmarks, and they serve as key structures in neural development, as well as regulating synaptic health. These cells, namely microglia and astrocytes, are responsible for the production of various mediators of inflammation throughout the central nervous system (CNS) – radicals, monokines, chemokines, complements, and most importantly inflammatory cytokines (Griffin et al., 1998; Mrak et al., 1995; Town et al., 2005). A comprehensive cross-sectional combined analysis of a total of 175 different studies with a total of 51 analytes in 13,344 AD and 12,912 healthy control patients determined that elevated peripheral interleukins (IL) IL-1, IL-6, IL-18, and tumor necrosis factor (TNF-α) were found in patients with AD compared with healthy controls (Lai et al., 2017). Recently, transforming growth factor beta (TGF-β) has also been shown to majorly contribute to this process. This served as the basis upon which the major cytokines were picked for discussion.
Biomarkers are a broad category of ‘medical signs’ that can be measured accurately and reproducibly (Strimbu et al., 2010). In AD, cerebrospinal fluid (CSF) biomarkers have been serving as the primary pathological markers for several years given their accuracy and predictability. However, in recent advances, peripheral blood mononuclear cells (PBMCs), which are essentially blood cells with a single round nucleus such as lymphocytes, monocytes or macrophages (Pourahmad et al., 2015), have displayed selective responses to immunological and pathophysiological changes in the AD brain. The striking fact is that these cells offer differential diagnoses, as well as diagnoses pre-onset of severe symptoms, thus allowing earlier therapeutic actions and hypothetically a better morbidity rate.
In this review we will focus on microglia and astrocytes, and will additionally discuss the potential of neurons to act as mediator cells. We will discuss the main inflammatory cytokines that affect AD physiopathology and analyze their role in AD pathogenesis. We will also determine the potential of PBMCs as biomarkers, as well as debate the recent advances in traditionally used CSF biomarkers. The biological pathways that are involved serve as prime pathological hallmarks of AD.
Mediators of inflammation in AD pathology
The two major glial cell types – microglia and astrocytes – do act complementary in most instances as their recruitment is interchangeable and similar (Cohen et al., 2019; Harry 2013). These glial cells act as transmission bridges and efficient neural modulators that maintain synaptic homeostasis and have balanced physiological properties. Of the categories of macrophages present in the CNS, the microglia constitute the majority and they express a myriad of receptors that detect pathological insults as well as preserve cerebral homeostasis (Yin et al., 2017). Microglial activation serves as a pathophysiological feature of AD. Several reports suggest that the microglia inflammatory response is starkly increased, whereas the mediated clearance is compromised (Domingues et al., 2017; Sarlus et al., 2017; Subhramanyam et al., 2019). It is also clear that microglial dysfunction is a primary contributor to the onset and the prognosis of AD (Wang et al., 2019). Neuroinflammation in itself is considered an innate immunological response in the AD brain. Activated microglia arise from their naturally occurring resting state in response to varied stimuli and are expressed in M1 or M2 microglial states. The classic M1 state facilitates the production and release of pro-inflammatory cytokines (including the ones discussed in this review) as an immunological response to combat incoming pathogens. On the other hand, the M2 state cells play an opposing role by suppressing and regulating the impact of this inflammation by secreting anti-inflammatory cytokines to maintain cerebral homeostasis. Aβ plaques trigger microglial cell accumulation whose eventual activation leads to an inflammatory response, followed by complement activation, and finally amyloid clearance takes place through phagocytosis (Hickman et al., 2008; Magalingam et al., 2018). This is further supported by experimental models that prove neuronal loss in AD results due to disruption of synaptic plasticity and affected clearance of Aβ via chemotactic mechanisms (Edwards 2019; Streit et al., 2014). Recent studies have demonstrated that microglial cells can modulate the pathogenesis process and ameliorate cognitive deficiency (Wang et al., 2019). Mounting evidence also suggests that in addition to Aβ clearance, microglia also plays a role in preventing such deposits from converting into potentially toxic states by encasing them in a protective shield, thus preventing the accumulation of new amyloid (Condello et al., 2015). These studies therefore provide strong evidence of the multifaceted functions of microglia and have laid the foundation for further research of microglial- targeted therapeutics in AD.
Astrocyte cells in the CNS promote and regulate synaptogenesis and play varied roles in the maintenance of neurological function and structure. Additionally, these glial cells dynamically modulate neuronal and synaptic plasticity in order to control cognitive behavior and activity (Jensen et al., 2013; Medeiros et al., 2013). Complimentary to microglial activation, astrocytes also participate in inflammation propagation in AD by reacting to insults, especially after exposure to Aβ and expression of pro-inflammatory cytokines and chemokines (Farina et al., 2007; Wyss-Coray et al., 2003; Yamaguchi et al., 1998). They represent an array of receptors that are involved in the foundation of innate immunity. Following activation in response to tissue injury, inflammation, or extra-environmental insult (Sekar et al., 2015), astrocyte cells secrete soluble mediators such as chemokine (C-C motif) ligand 2 (CCL2) and B-cell activating factor (BAFF), which play a role in immune response in the CNS. These mediators have been observed to produce various synaptic disturbances as well as neural dystrophies in the AD brain as demonstrated through multiple studies of mouse models (Furman et al., 2012; Garwood et al., 2011). The secreted inflammatory cytokines consequently increase the activity of tau kinases and lead to the induction of tau hyperphosphorylation (Griffin et al., 2006; Kitazawa et al., 2011). Impaired astrocytic functioning and cell death occur in AD. On one hand astrocytes are known to amplify and expand the neuroinflammatory response, while on the other hand their remodeling is proven to repair injured tissue as well as promote neuroprotection (Sofroniew 2009; Sofroniew et al., 2010).
Although neurons themselves were originally suspected to act as passive cells during inflammation progression in neurodegenerative diseases such as AD, contemporary modern-day studies consistently identify them as competent enough to secrete their own inflammatory mediators including complements and cytokines (Davis et al., 2003; Orzyłowska et al., 1999; Tchelingerian et al., 1994; Terai et al., 1997). It was noted earlier that all the above-described mediators are known to exacerbate neuroinflammation and are present in significantly high numbers in AD pathophysiology. Therefore, it is safe to postulate that neuronal cells themselves contribute to insults and reactions in their immediate environment, and thus participate in their own degeneration in AD.
Inflammatory cytokines in the AD brain
Originally coined as solely monokines, chemokines, and lymphokines – cytokines are a broad group of non-structural peptides, proteins, and glycoproteins, usually in the range of 5000 to 20000 Da, which cater special interactions and communication between cells, including but not limited to immunity, inflammation etc. (Zhang et al., 2007) They fall under the category of secretory cells and do not exhibit any motif of amino acid sequences nor any 3D structure linkage, thus their grouping into classes takes place on the basis of their biochemical activities (Rubio-Perez et al., 2012).
Cytokine association with AD is mainly based on displaying heterogeneity and multiformity, reflecting the correlated phenotype and genotype features, and finally having history of altered quantities in AD subjects – over 20 different polymorphisms and their 13 different types of cytokines have since been identified (Zheng et al., 2016). It is known that cytokines undergo and facilitate identical intra and intercellular signaling mechanisms in cellular mediators such as astrocytes and microglia, like they display in the periphery. However, cytokine processes distinct to the CNS have since been suggested and research examining their role in AD is underway (Akiyama et al., 2000). It is to be noted that there is an interplay between pro and anti-inflammatory signaling pathways in the case of AD. Certain cytokines such as IL-6 and TGF- β exhibit multifunctional properties and can act as both pro-inflammatory and anti-inflammatory cellular mediators depending on the environment and conditions present. While pro-inflammatory pathways are assumed to exacerbate neuroinflammation, anti-inflammatory signaling is believed to provide a more neuroprotective effect and exhibit neurotrophic behavior such as improving synaptic plasticity and homeostasis.
Interleukin-1
IL-1 was one of the first cytokines to be analyzed for its impact on the brain and its tissues (Dinarello et al., 1993), and as a pro- inflammatory cytokine it evokes a myriad of intrinsic biological effects (Besedovsky et al., 1986). Encoded by the genes IL-1A and IL-1B respectively (Nicklin et al., 1994), IL-1 occurs primarily in two isoforms, namely IL-1α and IL-1β (Shaftel et al., 2008). According to recent studies, IL-1 elevation has not only been reported in acute detrimental conditions but has also been implicated in chronic neurodegeneration (Allan et al., 2005; Patel et al., 2003). This serves as a key component in the brain’s systematic response to neuroinflammation and additionally the recruitment of leukocytes to the CNS.
IL-1 levels have been proven to dramatically increase in the AD brain, especially in microglia, and this has been interlinked to the major hallmarks of AD pathophysiology, including Aβ plaques, NFTs, and tau phosphorylation (Griffin et al., 1989). More specifically, it was elevated IL-1β that was more prominently detected in the blood analysis of subjects with AD (Alvarez et al., 1996; Swardfager et al., 2010). In recent studies, it was observed that there were no IL-1 changes in the serum of control patients but the samples from AD subjects, especially in the earlier stages of the disease, saw escalated levels of IL-1α and IL-1β (Forlenza et al., 2009; Licastro et al., 2000). Thus, it can be conclusively stated that AD pathology is characterized by constantly fluctuating circulatory levels of the IL-1 cytokine and its family of receptors (Italiani et al., 2018).
Studies also show that IL-1 is known to interact with proteins, including but not limited α1-antichymotrypsin, β-amyloid precursor protein, α2-macroglobulin, and apolipoprotein E, which are considered prime risk factors in the field of neurological diseases, particularly in AD (Mrak et al., 2000). However, there is not adequate evidence to support the fact that increased IL-1 in AD solely stems from peripheral immune activation during disease pathogenesis. IL-1 signal expression hinderances are able to amend the inflammatory response in the AD brain and have also resulted in the reduction of tau pathology, as well as a partial curtail of specific oligomeric Aβ. For a cytokine that is consistently affiliated with Aβ plaque progression (Wang et al., 2015), advancements in the field have revealed a possible therapeutic benefit to AD physiopathology through IL-1β (Kitazawa et al., 2012). A preliminary study on a mouse model at the University of Rochester also revealed a significant reduction in plaque deposition rather that the predictive exacerbation, and further research on this gradual shift is underway (Rivera-Escalera et al., 2019).
Interleukin-6
Best known for its dual role as both an anti-inflammatory myokine and a pro-inflammatory cytokine (Elahi et al., 2020), IL-6 is vital in the regeneration and degeneration of neurons in the CNS and peripheral nervous system. Neurons and glial cells form the major sites of IL-6 synthesis (Gadient et al., 1997). In the case of AD, the IL-6 cytokine is primarily responsible for the stimulation of glia and similar neuronal modulators, and are involved in promotion of the synthesis of acute phase proteins (Querfurth et al., 2010), as well as the phosphorylation of tau through the deregulation of the cdk5/p35 pathway (Quintanilla et al., 2004). Neurogenesis is suppressed through a Janus kinase (JAK)2/signal transducer and activator of transcription (STAT)3 signaling in neural stem cells through IL-6 (Kong et al., 2019). It was also discovered that the activation of JAK2/STAT3 pathway and the mitogen activated protein kinase (MAPK) pathway results in the hyperphosphorylation of regular tau, and thus ultimately leading to NFT formation (Quintanilla et al., 2004). The protein complex nuclear factor kappa-light-chain- enhancer of activated B cells (NF-кB) is also stimulated through the contribution of the JAK signaling via the phosphoinositide 3-kinase (PI3K)/Akt pathway. The binding of NF-кB along with the amyloid precursor protein promoter leads to the synthesis of Aβ, eventually resulting in the formation of plaques (Ait- Ghezala et al., 2007).
Multiple studies have reached a general consensus that IL-6 levels trend toward an increase in the AD brain (Angelopoulos et al., 2008; Bauer et al., 1991; Singh et al., 1997; Wu et al., 2015), and in some cases of mild AD and cognitive impairment as well (Wu et al., 2015). This observation is key in the early stages of plaque formation due to its link to diffuse plaque accumulation (Huell et al., 1995) and is represented by Aβ induced expression in neuronal culture systems (Lee et al., 2009). Recent studies have revealed that subjects with severe AD tend to display increased plasma volume of IL-6 than controls or less severe patients (Kálmán et al., 1997). Thus, it can be inferred that IL-6 levels in the periphery tend to exhibit an increasing trend over time during AD. A comprehensive literature study by Brosseron et al. (2014) revealed that a large number of articles produced inter-individual variances in the levels of IL-6, greatly swaying from reported mean values – concluding that it is highly probable that small subject cohorts resulted in misleading numbers. These contrasting results ultimately dispute the role of IL-6 in AD pathophysiology, but it is certainly promising that the suggested intra-individual data research will paint a clearer picture.
Interleukin-18
IL-18 is a pro-inflammatory cytokine that is considered to be a relative of the IL-1 cytokine family as it shares many physical as well as cellular characteristics with them. The cellular mediators discussed in this paper – microglia, reactive astrocytes, and neurons are all sources of IL-18 in the CNS (Ojala et al., 2009). IL-18 is observed to increase in a variety of conditions paired with inflammation, AD-risk increasing diseases, and the same applies to AD pathophysiology itself. IL-18 elevation in the AD brain seems particularly high due to the fact that it has a longer half-life than a majority of the other cytokines. IL-18 expression enhances oxidative stress due to its pro-inflammatory behavior and thus it can also exacerbate Aβ production. Recent data suggest a direct influence of IL-18 on the survival of neuronal cells as well as upholding synaptic integrity and plasticity. IL-18 along with its receptor result in the activation of MAPK p38 as well as the c-Jun N-terminal kinase signaling pro-apoptotic pathways. This also suggests that IL-18 might possibly act as one of the apoptotic factors in AD progression as pathway activation is primarily mediated through the expression of p53 and Fas ligand (Sutinen et al., 2012). On the contrary, it was observed that decreased gene expression of IL-18 improved physiological function in the elderly male population, thus suggesting that the cytokine may indeed have a neuroprotective role (Frayling et al., 2007). Furthermore, it has also been shown that increased expression of the cytokine in the AD brain, as well as in peripheral blood, has been associated with cognitive impairment (Bossù et al., 2010; Scott et al., 2020). The accurate role of IL-18 in AD therefore remains inconclusive and requires further research.
TGF-β
TGF-β are peptide units that have a vital role in tissue synthesis, regeneration and internal environment balance (Border et al., 1997). There are mainly three identified isoforms of TGF-β, namely 1, 2, and 3, expressed in the CNS within microglia, astrocytes, and neurons. TGF- β plays a role in the response to brain damage and astrocytosis, glial cell activation and adjoining inflammatory response, regulation of AD risk factors like apolipoprotein E, accumulation of amyloid, and lastly cell death inhibition (da Cunha et al., 1993; Krieglstein et al., 1995; Ongali et al., 2018; van der Wal et al., 1993). They are therefore known to have a multifunctional role as both pro and anti- inflammatory cytokines in the pathogenesis of AD.
The most commonly studied isoform in AD, TGF-β1, exhibits increased levels in serum (Chao et al., 1994) and CSF (Chao et al., 1994), and is also present within senile plaques when compared to healthy controls (van der Wal et al., 1993). Although contrastingly, studies have shown decreased levels in the plasma of AD subjects (Masuda et al., 2017), thus explaining the cytokine’s role in neurodegeneration as well as increased neuroinflammation. Neuronal loss is a critical element in the early prognosis of AD thus CSF levels of TGF-β1, which are assumed to be more closely representative of levels in the AD brain itself, are considered vital and most studies indicate it is dramatically increased (Blasko et al., 2004; Border et al., 1997; Tarkowski et al., 2002; Zetterberg et al., 2004). Moreover, immunoreactivities were reported elevated in reactive glial cells in subjects with AD compared to controls for TGF-β receptors type 1 and 2 (Lippa et al., 1998).
There is increasing evidence that aging weakens the ability to regenerate neuronal cells both after an injury or under normal circumstances. It was discovered that TGF-β1 excessive overproduction from astrocytes leads to an almost complete inhibition of neurogenesis in old transgenic mice (Buckwalter et al., 2006). This role of TGF-β1 as a potent inhibitor at high levels should be noted as the cytokine’s behavior has been quoted to be neurotrophic under a variety of conditions in AD. A new novel perspective was also offered by Hu et al. (2019) where TGF-β1 displays therapeutic effects in AD by promoting PI3K/Akt/ and Wnt/β-catenin signaling, thereby restoring memory and synaptic plasticity in mice AD models (Hu et al., 2019).
TNF-α
TNF-α is a pro-inflammatory cytokine that plays a prominent role in the cytokine cascade during a response, and the level of its expression is correlated with neuronal damage and toxicity, cognitive impairment, and apoptosis (Rubio-Perez et al., 2012). Hippocampal long-term potentiation suppression by the cytokine eventually results in impaired neurological function and thus has been implicated in AD (Contreras et al., 2020; Tancredi et al., 1992). TNF-α is present in low quantities in a healthy subject, thereby its exact role remains elusive under normal conditions. It is however known that TNF-α is first predominantly produced in activated microglia under inflammatory states in response to pathological stimuli (Breder et al., 1993), as a precursor molecule that is membrane bound. It is then converted in an autocrine manner into a smaller active cytokine by the TNF-α converting enzyme (Perry et al., 2001). A similar process occurs in AD and glial activation is instigated by the Aβ precursor protein (Lue et al., 2001), and TNF-α contributes to neuronal degeneration and death (Janelsins et al., 2008). In the case of AD, TNF-α levels in the brain potentially alter peripheral levels of the cytokine and there are a number of studies that examine the variations in serum, plasma, and CSF from AD subjects, where they revealed elevated levels of TNF-α and its association with cognitive impairment and decline (Brosseron et al., 2014; Fillit et al., 1991; Holmes et al., 2009; McAlpine et al., 2008; Swardfager et al., 2010; Tarkowski et al., 1999). In contrast to the mentioned results, there have been reports of neuroprotective properties displayed by TNF-α in AD patients (Akiyama et al., 2000). Moreover, there is mounting evidence of vast individual fluctuation in TNF-α, which may mimic a state or disease dependent variation (Brosseron et al., 2014). Certain subjects do not explicitly cite an increase in the serum. In constrast, either a decrease (De Luigi et al., 2002) or no significant changes have been observed (Tarkowski et al., 2003; Yasutake et al., 2006). A more intensive study into the chronic inflammatory state is required to establish the precise effect of TNF-α on neuronal health in AD (Contreras et al., 2020).
New CSF biochemical markers
A myriad of studies have focused on the collection of CSF through a lumbar puncture (LP) as one of the prime and most promising diagnostic tools to understand the metabolism of the brain (Albert et al., 2011; Dubois et al., 2014; Duits et al., 2016; Engelborghs et al., 2017; Janelidze et al., 2018; McKhann et al., 2011; Sperling et al., 2011). This is often supported as LP is generally considered a safe, rational, and widely available procedure. Studies have also provided mounting evidence of varying concentrations of biomarkers in the early stages of AD, including pre-symptomatic scenarios, thereby facilitating earlier diagnoses (De Meyer et al., 2010; Thies et al., 1999). In addition to their use in the pre-clinical phase of AD progression, CSF biochemical markers are primarily used to reflect the pathophysiology of the brain in AD as CSF is sequestered from the immediate footprint of the periphery through minimal transportation of matter across the blood brain barrier, as well as the fact that interstitial fluid of the brain is in uninterrupted contact with CSF due to the bi-directional flow of molecules (Olsson et al., 2016). Thus, the examination of CSF components and their concentrations serves as a fruitful asset of biomarkers in the in-vivo detection of neurological disorders. We will examine traditionally measured CSF biochemical markers, along with new developments in diagnostic accuracy, differential diagnosis, and progression through the discussion of Aβ variations and neurogranin.
Traditionally used CSF biomarkers
Trans-membrane amyloid precursor protein cleaving by β- and γ-secretase results in the formation of Aβ1-42, the amyloid-β composed of 42 amino acids. In terms of AD pathology, the Aβ1-42 is identified to have decreased concentrations in the CSF, thus referring to a high prodromal AD and AD dementia diagnostic accuracy (Kuhlmann et al., 2017; Mattsson et al., 2009; Visser et al., 2009). The initial stages of Aβ plaques can be detected in the CSF much earlier than the final onset of symptoms, in some cases over 20 years, thus aiding in early diagnoses (Jansen et al., 2015). Proteins called tau are liberally found in the cytosol of neural cells where they are primarily involved in microtubule stabilization in axons. The excessive or abnormal phosphorylation of tau leads to the breakage of normal adult tau from microtubules and transforms it into NFT. In addition, the extracellular space experiences a surge in phosphorylated forms of tau and total tau, thus leading to increased concentrations of tau in the CSF during AD. The combined effects of NFT as well as plaque accumulation in AD results in a slow yet constant neuronal degeneration. In comparison to Aβ1-42 in the CSF, the tau protein biochemical markers undergo delayed changes in the pathological process of AD (Buchhave et al., 2012; Jack et al., 2013), and is usually correlated more vigorously with cognitive degeneration. Studies in the last two decades provide increased evidence of high protein tau levels, along with lower Aβ1-42 biomarker levels in the CSF catering to the high prediction accuracy of AD pathophysiological changes, thus indicating conclusively the presence of AD (Anoop et al., 2010; Buerger et al., 2006; Cedazo-Minguez et al., 2010; Hansson et al., 2006; Savva et al., 2009; Tapiola et al., 2009).
Aβ Variations in differential diagnosis
Although the presence of Aβ1-42 is a pathological hallmark in AD, several studies such as the case cohort study by van Oijen et al. (2006) hypothesize that a significant decrease in the marker occurs in the early stages of dementia, thus making the differentiation of AD and non-AD dementia challenging. To facilitate an improved accuracy, isoforms of amyloid-β in the CSF are being examined. The most abundant of those, Aβ1-40 or amyloid-β consisting of 40 amino acids, has demonstrated an increase in plasma concentration, and along with combined decrease in Aβ1-42, they indicate a higher risk of AD dementia. However, research has also shown a decrease of Aβ1-40 in dementia patients without AD, thereby elucidating the disease-specific variability in Aβ metabolism – increased or decreased presence and clearance (Lewczuk et al., 2015). Therefore, a recent focus effectively diminishing the inter-patient variability in the field involves the combination of the two prime isoforms into a Aβ1-42/Aβ1-40 ratio to act as a control for increased or decreased Aβ1-42 production regardless of AD pathology (Pannee et al., 2016; Struyfs et al., 2015). Differential dementia diagnosis has also been facilitated by Aβ1-40 in individuals with intermediate levels of phosphorylated tau-181 protein (P-tau181) (Slaets et al., 2013). Improved accord amongst amyloid markers was also observed in two other studies where the Aβ1-42/Aβ1- 40 ratio was utilized in contrast to an individual Aβ1-42 CSF concentration measure (Leuzy et al., 2016; Niemantsverdriet et al., 2017). There have also been recent advancements with the usage of carboxy-terminally truncated Aβ peptides Aβ1- 37 and Aβ1-38 to increase detection accuracy of AD from frontotemporal dementia or dementia with Lewy bodies (Bibl et al., 2006; Struyfs et al., 2015).
AD progression through Neurogranin
Since many of the discussed proteins serve purpose only until the mild cognitive impairment (MCI) stage of the AD, neurogranin, a post-synaptic protein, has emerged as a new candidate biomarker for the progression of AD, as it has been shown to have high potential as an indicator for synaptic degeneration or dysfunction, thus aiding in detection of disease progression (Thorsell et al., 2010). The primary function of neurogranin as a CSF marker is to detect cognitive decline and predict future impact in normal patients, therefore complementing the already diverse list of AD biochemical markers (Tarawneh et al., 2016). CSF levels of the markers Aβ1-42, P-tau181 and T-tau illustrate distinctive alterations between the controls and AD groups but did not explicitly demonstrate any differences between the controls and MCI groups according to the cross-sectional study quoted in the above literature. However, analysis of the CSF from MCI and AD patients against controls also indicated an elevating trend in the levels of neurogranin. The increased levels of neurogranin must therefore conclusively reflect synaptic breakdown and loss (Thorsell et al., 2010). It is critical to note that the CSF neurogranin was detected to have increased in AD patients rather than its plasma counterpart. A further negative correlation was detected between CSF neurogranin and the Aβ1-42/Aβ1-40 ratio (De Vos et al., 2015). A study by De Vos et al. (2016) also discovered the neurogranin trunc P75/BACE1 ratio accurately reflects synaptic integrity (a measure of cognitive decline), therefore reemphasizing the use of neurogranin as a biomarker for AD (De Vos et al., 2016).
Peripheral blood mononuclear cells as potential biomarkers
PBMCs play a key direct role in the neurodegeneration process and its pathophysiology along with immune response and communication. They closely replicate the environment of neural cells and thus can be standardized on the basis of their biological responses by the matching catalog of enzymes as other organ tissues. The examination of PBMC biochemical markers could change the diagnostic accuracy and stage analysis of AD, thus aiding in a better response to treatment as well as matters such as subgrouping of patients (Govoni et al., 1996). Although several plasma and blood cell markers have been tested before, this review will focus solely on two new peripheral mononuclear markers – monocytes, particularly cluster of differentiation (CD)14+ and CD16+, and T cells.
CD14+ and CD16+ Monocytes
Monocytes have been found to critically affect the pathology of AD (Gu et al., 2016). A recent case study that measured the changes in the blood profile of AD patients against control revealed no significant disparity in monocyte concentration. Nevertheless, it is vital to note that the function of monocytes and its behavior affects the pathogenesis of the disease. Studies in the past have proven enhanced functional levels of monocytes resulting in a considerable suppression of the progression of AD in a number of animal models (Chen et al., 2017; Darlington et al., 2015; Fiala et al., 2005).
It is assumed that the monocytes CD14+ and CD16+ increase in quantity in conditions that entail cytokine presentation since cytokines themselves are proficient at inducing these monocytes in-vivo (Ziegler-Heitbrock 1996). The CD16+ monocytes themselves contribute to around 5 to 10% of the total monocyte population in normal patients and are drastically modified in multiple pathophysiological conditions, exhibiting a unique phenotype (Ancuta et al., 2003). The CD14+ and CD16+ monocytes are prone to an increased potency in antigen presentations and results convey drastically increased levels of pro-inflammatory TNF and lower levels of anti-inflammatory IL-10. These cells have been shown to exhibit distinct phenotype and function, and the CD14+ CD16+ phenotypes were labeled proinflammatory based on higher expression of proinflammatory cytokines and higher potency in antigen presentation. These monocytes also respond to chemokines such as C-X-C motif chemokine ligand 12 (CXCL12) and C-X3-C motif chemokine ligand 1 (CX3CL1) and migrate in terms of their expression (Ziegler-Heitbrock 2007). Comparing AD patients in preliminary stages with normal controls revealed a higher CX3CL1 expression in the abluminal and endothelial compartments with mononuclear cells. This particular observation will assist in the differentiation of pre-symptom and early onset stages of AD (Vérité et al., 2018).
Although the ultimate role of increased CD14+ and CD16+ might seem unclear at this stage, the above evidence clearly proves that these cells lead to a much more effective and striking inflammation than normal monocytes. They also facilitate a high adherence of cells to the endothelium and migrate into other tissues due to the increased expression of adhesive molecules. To completely understand and discern the role of CD14+ and CD16+ monocytes, it is essential to investigate these cells after they have moved into tissues. Overall, there is an expanding library of literature that prove these monocytes increase in population in AD, and we are speculating that further research will analyze their prognostic role as well as their impact on other similar inflammatory conditions (Ziegler-Heitbrock 1996).
T Cells
Lymphocyte populations such as the CD4+ T cells can act as enticing biomarker candidates in AD as they are easily extracted in a minimally invasive fashion and are readily examined by flow cytometry (Rezai-Zadeh et al., 2009). Possible diagnostic applications can be implemented through the linkage of regulatory T cell (Tregs) profiles to the grading of AD due to the presence of indications of autoimmune mechanisms in the pathology of AD. Treg cells play an active role in the peripheral tolerance operations in relation to the development of the autoimmune disease, according to new studies. It is also widely known that regulatory T cells can possibly malfunction in peripheral immune surveillance mechanisms, such as through an impaired T cell suppressive function, increased reactivity, or enhanced resistance to the effector T cell machinery (Bettini et al., 2009). A study that measured the immune phenotype of regulatory T cells in AD revealed a stark decrease in the percentage of Tregs in AD patients in contrast to a similar cohort of healthy subjects. These findings also suggest an impaired immunosuppression in AD pathology and as discussed above, this decrease of Tregs therefore could contribute to the failure of Treg immune surveillance in AD (Ciccocioppo et al., 2019).
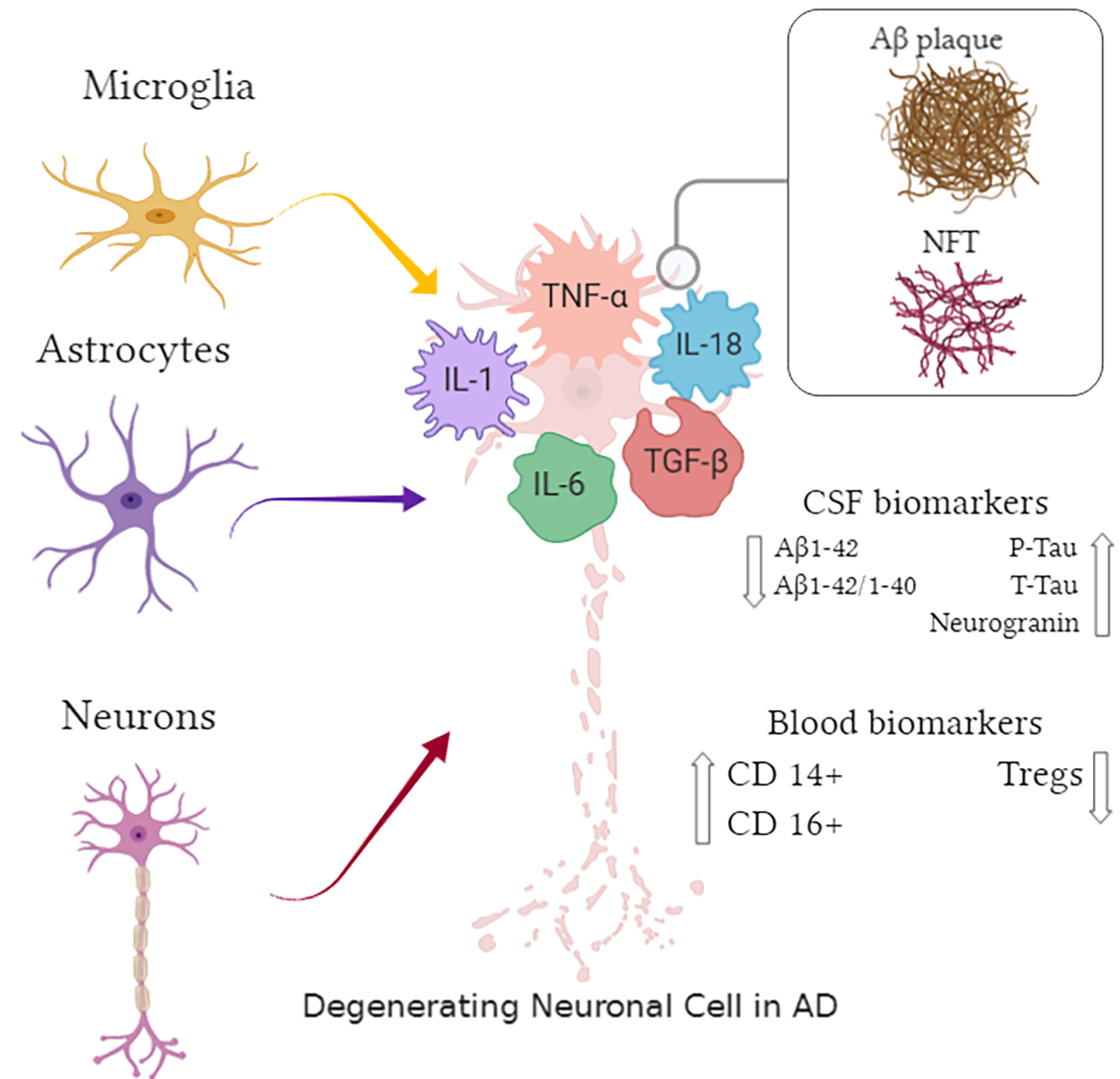
In a new window | Download PPT
Figure 1. Pathological hallmarks and biomarkers in Alzheimer's disease (AD): an overview. Arrows represent activation of cellular mediators; IL-1: interleukin -1; IL-6: interleukin-6; IL-18: interleukin-18; TNF-α: tumor necrosis factor alpha; TGF-β: transforming growth factor beta; Aβ: amyloid beta; P-tau: phosphorylated tau; T-tau: total tau; NFT: neurofibrillary tangles: Tregs: T-regulatory cells.
Conclusion
This review serves as a comprehensive breakdown of mediators, inflammatory modulators, and biomarkers in both the diagnosis and prognosis of AD. A variety of prominent cytokines and their pathways were discussed, along with a deeper dive into PBMCs and CSF components as potential biomarkers. A common trend observed throughout the majority of the studies indicate that the results obtained tend to show inter-individual variations and therefore inconsistencies. Overall, the effect as well as the behavior of each individual inflammatory modulator was summarized, anomalies were debunked, and novel therapeutic suggestions were made.
The current state of diagnosis for AD rests only well after a notable amount of neurodegeneration and cognitive impairment has taken place. Therefore, the outlook of this review is to primarily welcome potential biomarkers into the arena, which are able to accurately predict the pathophysiology of the condition at the earliest onset of symptoms, and thereby lead to a more balanced and well-rounded early treatment intervention. The PBMC biomarkers serve that very purpose and show promise in the early as well as in the differential diagnosis of AD and similar neurological conditions. Multiple research literatures have already analyzed CSF markers in great detail, although quite a bit of variation does indeed exist due to certain parameter accuracies in diagnosis. The Aβ variations have introduced disease-specific variability in the diagnosis of AD and thereby can be used to differentiate AD from other dementia syndromes. Similarly, recent advancements in neurogranin research in cognitive decline and synaptic integrity establishes them as strong candidates for progression rather than just diagnosis of the disease. The PBMC profile changes in the physiopathology of AD is still at its preliminary stage and are yet to be thoroughly analyzed. Nevertheless, there are a number of studies that report altered blood profiles in AD diagnosis and progression. Although the specificity and sensitivity of monocyte population in AD is quite scarcely established, there is sufficient evidence to support these cells as one of the most promising biomarkers for AD (Richartz-Salzburger et al., 2007). On the other hand, there is limited research on determining a definite leukocyte marker for AD. Whilst most of the described markers are associated with either the immune or the inflammatory response to the presence of AD, there is no conclusive evidence of a biomarker that solely changes with the progression of the disease itself. A non-biased approach to conduct conclusive research with large sample sizes in terms of peripheral leukocyte marker changes in AD will ultimately lead to greater comprehension of their utility. Such approaches will also assess the efficacy of therapeutics and unequivocally determine a standardized sensitivity and specificity (Frank et al., 2003; Rezai-Zadeh et al., 2009). It is essential to understand that a sole diagnostic tool nor a standalone biomarker is enough to forecast the progression or the onset of AD definitively. A multimodal combination of all these diagnostic determinants should be deployed for establishing an accurate and effective therapeutic strategy.
References
Girishkumar Sivakumar1
1University College London Division of Medicine, Undergraduate Medicine.
Sivakumar Viswanathan2
2Cardiovascular and Metabolic Disorder Programme, Duke-NUS Medical School , Singapore
Corresponding author:
Girishkumar Sivakumar
Email: girish2439@gmail.com
In a new window | Download PPT
Figure 1. Pathological hallmarks and biomarkers in Alzheimer's disease (AD): an overview. Arrows represent activation of cellular mediators; IL-1: interleukin -1; IL-6: interleukin-6; IL-18: interleukin-18; TNF-α: tumor necrosis factor alpha; TGF-β: transforming growth factor beta; Aβ: amyloid beta; P-tau: phosphorylated tau; T-tau: total tau; NFT: neurofibrillary tangles: Tregs: T-regulatory cells.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 6782 | 25 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA