Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Autophagy as a target for cardioprotection in acute myocardial infarction and heart failure
Time:2021-01-18
Number:11296
Author Affiliations

Conditioning Medicine 2020. 3(6): 264-273.
Abstract
Acute myocardial infarction (AMI) and heart failure (HF) that often follows are the leading causes of death and disability worldwide, exerting huge burdens on healthcare and economic resources. Therefore, new therapeutic approaches are required to reduce myocardial injury, improve cardiac repair, and prevent adverse myocardial remodeling, and the consequent HF. The death of cardiomyocytes (CMs) has been detected in the heart at all stages of AMI. In this context, autophagy, a catabolic process for the engulfment, degradation, and recycling of dysfunctional or damaged cellular components, plays a crucial role in CMs homeostasis, making it a key process in AMI and the development of HF. Autophagy induced by ischemia confers cardioprotection via the activation of adenosine monophosphate-activated protein kinase and inhibition of mTOR pathway, whereas further amplification of autophagy during reperfusion is maladaptive and exacerbates myocardial injury. As such, strategies that can modulate and normalize the autophagic response could prevent irreversible loss of cardiomyocytes in AMI and HF, thereby conferring cardioprotection. Here, we summarize the role of autophagy in AMI and HF as a potential target for cardioprotection, highlighting studies that focus on the development of new therapies that take advantage of autophagy modulation to prevent or delay the pathogenesis of AMI and progression to HF.
Keywords: autophagy, cardioprotections, acute myocardial infarction, heart failure, cell death
Abstract
Acute myocardial infarction (AMI) and heart failure (HF) that often follows are the leading causes of death and disability worldwide, exerting huge burdens on healthcare and economic resources. Therefore, new therapeutic approaches are required to reduce myocardial injury, improve cardiac repair, and prevent adverse myocardial remodeling, and the consequent HF. The death of cardiomyocytes (CMs) has been detected in the heart at all stages of AMI. In this context, autophagy, a catabolic process for the engulfment, degradation, and recycling of dysfunctional or damaged cellular components, plays a crucial role in CMs homeostasis, making it a key process in AMI and the development of HF. Autophagy induced by ischemia confers cardioprotection via the activation of adenosine monophosphate-activated protein kinase and inhibition of mTOR pathway, whereas further amplification of autophagy during reperfusion is maladaptive and exacerbates myocardial injury. As such, strategies that can modulate and normalize the autophagic response could prevent irreversible loss of cardiomyocytes in AMI and HF, thereby conferring cardioprotection. Here, we summarize the role of autophagy in AMI and HF as a potential target for cardioprotection, highlighting studies that focus on the development of new therapies that take advantage of autophagy modulation to prevent or delay the pathogenesis of AMI and progression to HF.
Keywords: autophagy, cardioprotections, acute myocardial infarction, heart failure, cell death
Introduction
Acute myocardial infarction (AMI) and heart failure (HF) contribute to high morbidity and mortality in terms of global health (Mozaffarian et al., 2016; Roth et al., 2017). Timely restoration of coronary flow of the occluded coronary artery with thrombolytic therapy and/or percutaneous coronary intervention (PCI) is the treatment of choice in AMI patients (Ginks et al., 1972; Yellon and Hausenloy, 2007; Heusch, 2020). However, clinical outcomes following restoration of blood flow to ischemic tissue in AMI need to be improved. The reperfusion process itself begets cardiomyocyte (CM) death through a combination of oxidative stress, Ca2+ overload, inflammation and mitochondrial dysfunction – a phenomenon called acute myocardial ischemia-reperfusion injury (IRI) (Yellon and Hausenloy, 2007; Szummer et al., 2017; Ong et al., 2018; Heusch, 2020).
CM death is an important contributor to HF following AMI, and represents a challenge for researchers to preserve the viability of CMs. In this sense, autophagy has emerged as a mechanism enabling CMs to eliminate redundant and damaged proteins (Reimer et al., 1993; Nishida et al., 2008; Kanamori et al., 2011; Gao et al., 2020a). The tight regulation of protein turnover is essential to prevent the irreversible loss of CMs and maintain cellular homeostasis in AMI and HF (Sciarretta et al., 2014; Farah et al., 2020). In this review, we will focus on the importance of autophagy in cardioprotection, and highlight the potential therapeutic approaches that target autophagy to reduce myocardial injury and prevent adverse myocardial remodeling in AMI patients at risk of developing HF.
Autophagy and mitophagy
Autophagy is a catabolic process describing the engulfment, degradation, and recycling of dysfunctional or damaged cellular components, and plays a crucial role in maintaining CM homeostasis. Given its role in reducing CM stress, autophagy generally functions as a pro-survival process. However, some evidence suggests that, under certain circumstances, autophagy may beget cell death. In the context of acute myocardial ischemia-reperfusion injury (IRI), it remains unclear if autophagy-dependent cell death results from excessive autophagy or a qualitative change in the nature of the autophagy process itself (Galluzzi et al., 2018). According to the pattern of cargo delivery to the lysosome and its physiological function, autophagy has been classified into different types including microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy. The most extensively studied form of autophagy is macroautophagy, which degrades both intracellular organelles and cytoplasmic proteins (Ma et al., 2015). In this article, the term autophagy refers to macroautophagy, unless otherwise specified.
Autophagy is regulated by specific autophagy (Atg) genes, which encode for proteins that regulate the initiation and formation of autophagosomes, as well as the fusion of autophagosomes with lysosomes to form autolysosomes. The process of autophagy consists of at least four steps: 1) induction and nucleation, 2) formation and maturation, 3) fusion, and 4) degradation (Figure 1).

In a new window | Download PPT
Figure 1. Schematic diagram of phases of autophagy in cardiomyocytes. (1) Induction and nucleation, (2) formation and maturation, (3) fusion, and (4) degradation. Autophagy-activating kinase 1, ULK1; protein of 200kDa, FIP200; autophagy-related protein, Atg; endoplasmic reticular, ER; phosphatidylethanolamine, PE; phosphatidylinositol 3-phosphate, PIP3; 1 light chain 3, LC3; adenosine monophosphate, AMP; adenosine triphosphate, ATP; mammalian target of rapamycin, mTOR.
Induction and nucleation
The induction of autophagosome formation is regulated by the macromolecular complex unc51-like kinase 1 (Ulk1), comprising Atg13, Ulk1/2 (Jung et al., 2009), RCB1-inducible coiled-coil 1/focal adhesion kinase family interacting protein of 200 kDa (RB1CC1/FIP200) (Ganley et al., 2009) and chromosome 12 open readin gframe 44 (C12orf44)/Atg101 (Hosokawa et al., 2009). The most dominant pathway of autophagy in CMs is the mammalian target of rapamycin (mTOR) pathway. mTOR is a serine/threonine kinase that acts through two distinct multiprotein complexes. When mTOR is complex-associated, it phosphorylates Ulk1/2 and Atg13, inactivating them. However, when CMs are treated with rapamycin, a classic autophagy inducer, mTOR dissociates from the induction complex, resulting in dephosphorylation of Ulk1/2 and Atg13, leading to the induction of autophagy (Jung et al., 2009). During autophagosome formation, the Ulk complex activates a macromolecular protein assembly consisting of Beclin-1, Atg14L, vacuolar protein sorting (Vps) 34, and Vps15 (Furuya et al., 2005; Yan et al., 2009). Ulk1 phosphorylates Beclin-1 at Ser14, thereby activating the Vps34 complex, which is critical for the formation of autophagosome through the formation of phosphatidyl inositol 3-phosphate (PIP3) (Itakura and Mizushima, 2010).
Formation and maturation
In mammals, there are two ubiquitin-like (UBL) systems that contribute to the expansion of the phagophore. The first UBL is the Atg12-Atg5 complex, which promotes the elongation of the maturating phagophore through interaction with Atg16 that acts as an E3-like enzyme to promote lipidation of microtubule- associated protein 1 light chain 3 (LC3); the second UBL system involves phagosome expansion (Mizushima et al., 2003). The conjugated LC3, called LC3-II, is incorporated into the phagophore, mediating its final maturation into an autophagosome. The conjugation and subsequent deconjugation of LC3 from phosphatidylethanolamines are regulated by the cysteine protease, Atg4. Subsequently, the autophagosome fuses with the lysosome to form an autophagolysosome that digests macromolecules by action of lysosomal hydrolases. Once digestion is completed, the degraded components are recycled to the cytoplasm to regenerate the cellular building blocks (Kabeya et al., 2000). The expanding phagophore must eventually mature and close to form a complete autophagosome, which traffics to and fuses with a lysosome, forming an autolysosome.
Fusion
After its formation, the autophagosome undergoes a stepwise maturation process, including fusion events with multivesicular endosomes and lysosomes. Fusion with endosomal compartments depends on Vps4/suppressor of K+ transport growth defect 1 (SKD1) and Rab11 (Nara et al., 2002). The fusion with lysosomes necessary for the complete degradation of segregated cytoplasm depends on microtubules, the microtubule motor dynein, the small GTP-binding protein RAB7, components of HOPS (homotypic fusion and vacuole protein sorting) complex (Itakura et al., 2012), VTI1B (Atlashkin et al., 2003), (Jager et al., 2004; Szatmari and Sass, 2014) and the lysosomal membrane protein lysosome associated membrane protein 2 (LAMP2) (Eskelinen, 2005). After fusion, the cytosolic cargo is broken down by the acidic lysosomal hydrolases in the autophagolysosome. Agents such as the anti-malarial drug chloroquine (CQ) and its derivatives are used experimentally to inhibit autophagy (Mauthe et al., 2018). Attenuation of the excessive increase in autophagy during reperfusion could a priori be expected to confer protection, highlighting chloroquine as a potential therapeutic strategy to limit IRI (Chaanine et al., 2015). However, CQ administration (60 mg/kg, i.p 1 h before ischemia and daily chloroquine injection for up to 2 days) reduced IRI injury at 0.5 and 6 h, but aggravated liver injury at 24 and 48 h after reperfusion (Fang et al., 2013). In the early phase, the protective effect of CQ treatment was associated with a reduction of inflammatory cytokine production. In contrast, in the late phase of reperfusion, chloroquine treatment was associated with autophagy inhibition and induction of apoptosis. Chloroquine could be expected to be protective in other conditions that enhance autophagy flux, such as cardiac hypertrophy, and it also appears to confer protection against IRI in diabetic cardiomyopathy (Yuan et al., 2016; Jeong et al., 2018). There are many case reports of rapidly developing cardiovascular effects following intoxication by CQ/hydrochloroquine (HCQ), ranging from bradycardia and hypotension to eventual cardiac arrest (Mubagwa, 2020). Consequently, this dual effect should be considered in future clinical trials where CQ or HCQ could be used in the treatment of myocardial IRI.
Degradation
Little is known about what happens during degradation. The breakdown within the vacuole allows recycling of the hydrolyzed cargo. Efficient degradation is dependent on proteinase B, lumenal acidification, and the cytoplasm to vacuole targeting protein 17 (Cvt17) protein, a candidate lipase that may degrade the autophagic body (Kim et al., 1997).
Mitophagy
The selective removal of damaged mitochondria by autophagy, termed mitophagy, shares most of the molecular machinery of autophagosome formation but also possesses unique molecular mechanisms, including the PTEN-induced putative protein kinase1 (PINK1)/E3 ubiquitin ligase Parkin pathway (Lazarou et al., 2015; Saito and Sadoshima, 2015). When mitochondria are depolarized, PINK1 is stabilized and accumulates on the outer mitochondrial membrane (OMM), where it phosphorylates mitofusin 2 (Mfn2) at Thr111 and Ser442, which in turn induces Parkin translocation to the OMM. Parkin then ubiquitinates OMM proteins, marking the depolarized mitochondria for autophagosomal engulfment. The autophagosome then fuses with the lysosome, leading to degradation of the mitochondria (Lazarou et al., 2015).
This highlights the role of Mfn2, a mitochondrial fusion protein, as a receptor for Parkin. In addition to Parkin recruitment, Mfn2 has been shown to play an essential role in the fusion of autophagosomes with lysosomes. Zhao et al. (2012) showed that Mfn2 mediates the maturation of autophagy in the heart by serving as an adaptor protein recruiting RAB7 (the small GTP-binding protein needed for the final maturation of late autophagic vacuoles) to the autophagosomal membrane (Yoo and Jung, 2018; Hernandez-Resendiz et al., 2020).
Autophagy in acute myocardial ischemia-reperfusion injury
Autophagy can either enhance survival or accelerate CMs death. While several studies support the notion that enhanced autophagy is cardioprotective, there are also substantial reports that support the hypothesis that increased autophagy is detrimental to CMs (Davidson et al., 2020). Nonetheless, autophagy can be activated in prolonged ischemia, acute IRI, and HF (Zhu et al., 2007; Tannous et al., 2008).
Under basal conditions, Ulk1 is phosphorylated at Ser757 by mTOR1, leading to the inhibition of autophagosome formation. In response to ischemia, the activity of mTOR is suppressed, and autophagy becomes activated (Nishida et al., 2008). Autophagy activation has been observed within 20 min of ischemia in in vivo murine hearts (Matsui et al., 2007). During myocardial ischemia, a rapid drop in ATP levels (i.e. increased AMP/ATP ratio) and glucose deprivation (Zhang et al., 2017) lead to the activation of adenosine monophosphate- activated protein kinase (AMPK) (Matsui et al., 2007). AMPK phosphorylates and activates tuberous sclerosis complex 1/2 (TSC1/2) and regulatory associated protein of mTOR (Raptor), which in turn inhibits Ras, an activator of mTOR. Beclin-1 and Atg5, members of the classical autophagy pathway and essential for autophagosome formation, are found to increase in ischemic hearts (Gustafsson and Gottlieb, 2009). Myocardial ischemia also inactivates the GTP-binding protein Rheb, a protein that promotes cell survival and mediates cellular response to energy derivation. Consequently, inactivation of Ras homolog enriched in brain (Rheb) protects CMs during ischemia through activation of autophagy (Sciarretta et al., 2012). In a direct way, AMPK disassociates Ulk1 from mTOR by phosphorylating Ulk1 at Ser313 and Ser777 (Kim and Guan, 2011; Kim et al., 2011; Laker et al., 2017). Another pathway that triggers autophagy is through the induction of Bcl2-interacting protein 3 (Bnip3) via hypoxia and acidosis. Overexpression of Bnip3 in adult CMs has been associated with a significant increase in autophagy activity. Additionally, as a pro-apoptotic member of the Bcl-2 family, Bnip3 has been involved in upregulation of autophagy in myocardial reperfusion (Hamacher-Brady et al., 2006). Although the precise mechanism through which Bnip3 induces autophagy is poorly understood, it has been proposed that Bnip3 might compromise the integrity of mitochondria such that autophagy is initiated to dispose of the damaged organelles (Figure 2).

In a new window | Download PPT
Figure 2. Signaling regulation of autophagy during acute myocardial IRI and potential cardioprotective therapies. (A) Ischemia increases the AMP/ATP ratio and activates AMPK. AMPK has a dual action to promote autophagy via inhibition of mTOR and activation of Ulk1 via phosphorylation at Ser 377 and Ser 777. AMPK indirectly inhibits mTOR via its phosphorylation at Ser 722 and Ser 792. (B) In contrast, during reperfusion, activation of AMPK is no longer observed. Instead, autophagosome formation during reperfusion is mediated by Beclin-1-dependent mechanisms.
Reactive oxygen species (ROS) generated during ischemia can also contribute to autophagy activation. Hydrogen peroxide (H2O2) regulates autophagosome formation through activation of atg4 by oxidation of an essential cysteine residue, which leads to accumulation of LC3-II on the phagophore membrane and the formation of autophagosomes (Scherz-Shouval et al., 2007). During acute myocardial IRI, ROS damages organelles, cytosolic proteins, and causes lipid peroxidation in the mitochondria, all of which exacerbate autophagy. Thioredoxin- interacting protein (TXNIP), a pro-oxidative molecule, is known to contribute to IRI. TXNIP has been shown to increase autophagosome formation but inhibits autophagosome clearance during myocardial reperfusion (Penna et al., 2013; Gao et al., 2020a).
Another potent inducer of autophagy during ischemia is elevated intracellular Ca2+ due to the sodium-calcium exchanger during prolonged ischemia (Karmazyn and Moffat, 1993; Hoyer-Hansen et al., 2007). AMPK can be phosphorylated and activated by tumour suppressor liver kinase B1 (LKB1) at low energy levels and by Ca2+/calmodulin-dependent protein kinase kinase-β (CAMKK-β) in response to increased cytosolic Ca2+ (Hawley et al., 2005). Another critical molecule that is activated by low oxygen conditions is hypoxia-inducible factor 1 alpha (HIF-1α). In vivo studies have shown that HIF-1α mediates mitochondrial autophagy as an adaptive metabolic response under hypoxia conditions (Zhang et al., 2008); however, the correlation between HIF-1α and autophagy during myocardial ischemia has not been demonstrated.
Autophagy has been reported to be further enhanced during myocardial reperfusion and is associated with the accumulation of autophagosomes. Impaired autophagosome clearance mediated in part due to ROS-induced downregulation of LAMP2 and upregulation of Beclin-1 contributes to increased CM death (Tanaka et al., 2000; Ma et al., 2012). Since AMPK is rapidly inactivated during reperfusion, it is unlikely that the increased autophagy seen in reperfusion is mediated by AMPK-dependent mechanism. In the study by Matsui et al. (2008) reperfusion induced a 7-fold increase in autophagosome abundance despite the absence of further AMPK activation or mTOR1 inhibition, suggesting that autophagosome formation during reperfusion is mediated by Beclin-1-dependent mechanisms. Importantly, it has been shown that Beclin-1 increases within 30 min of reperfusion (Matsunaga et al., 2009), highlighting the importance of Beclin-1 in mediating the autophagy process during myocardial reperfusion. Apoptosis and the size of the myocardial infarct during reperfusion are significantly attenuated in heterozygous Beclin-1+/- mice, suggesting that autophagy during reperfusion may be detrimental (Matsui et al., 2007). Conversely, depletion of Beclin-1 by siRNA transfection attenuated autophagy activity in CM during reperfusion (Valentim et al., 2006). In vitro studies have also shown that ROS induces autophagy through Beclin-1 overexpression during reperfusion (Hariharan et al., 2011). Additionally, H2O2 produced (Matsui et al., 2007) during reperfusion could oxidize the Atg4 cysteine contributing to LC3 lipidation and autophagy initiation (Scherz-Shouval et al., 2007). It is also likely that the deleterious effects of autophagy may be attributed to the crosstalk between autophagy and apoptosis. Beclin-1 mediated autophagy activation during reperfusion is associated with Bcl-2 downregulation (Brady et al., 2007), and the decreased expression of Bcl-2 could contribute to apoptotic cell death.
Autophagy in heart failure
Large myocardial infarcts can lead to HF due to adverse remodeling of the left ventricle (LV). This is characterized by LV dilatation and diminished cardiac contractile function. Adverse cardiac remodeling refers to the structural cardiac alterations that occur in response to hemodynamic load and cardiac injury in association with neurohormonal activation (Cohn et al., 2000). Dead and dying cardiomyocytes showing characteristics of autophagy have been reported in HF patients. Analysis of hearts from patients with end-stage HF show CMs with cytoplasm replaced almost entirely with autophagosomes (Takemura et al., 2006), suggesting autophagic cell death to be the most prominent mechanism contributing to cell death in HF (Knaapen et al., 2001; Kostin et al., 2003). It is still undetermined whether an abnormal increase in autophagy causes CM death in animal models of HF. It is possible that autophagic CM death is simply the result of a failed compensatory mechanism or dysfunction of the autophagy process as seen in the HF associated with Danon disease (a genetic defect in the lysosomal protein LAMP2) (Nishino et al., 2000; Tanaka et al., 2000). Recently, Akazawa et al. (2004) reported that autophagic CM death plays a pathogenic role in a mouse model of diphtheria toxin-induced HF. In a study where HF was induced in rats using an established model of Adriamycin administration, abundant autophagosomes were detected. The administration of 3-methyladenin (3MA), an autophagy inhibitor, strongly suppressed the formation of autophagosomes, reduced mitochondrial injury, and inhibited autophagic CM death (Lu et al., 2009). Similarly, four weeks after transverse aortic constriction (TAC), Atg5-deficient mice showed significant increase in LV dimensions, decreased fractional shortening, accumulation of ubiquitinated proteins, and increased CM death (Nakai et al., 2007).
Targeting autophagy for cardioprotection
Acute myocardial ischemia-reperfusion injury
Several reports support the notion that the enhancement of autophagy is cardioprotective. On the other hand, there are studies showing that autophagy may exacerbate myocardial injury. Matsui et al. (2008) have shown that, in the case of myocardial ischemia, autophagy led to cell survival, whereas excessive autophagy could promote CM death during reperfusion. The AMPK-mTOR pathway is regarded as an important regulator of autophagy in response to acute myocardial IRI, although AMPK is no longer activated during reperfusion (Matsui et al., 2007). With increasing evidence verifying the cardioprotective effects of AMPK-mediated autophagy during reperfusion, multiple pharmacological drugs targeting AMPK activation have been evaluated as potential therapeutic agents. The numerous agents that promote myocardial autophagy during reperfusion identified include the classic autophagy inducer, rapamycin (Wu et al., 2014), metformin (Solskov et al., 2008), some Chinese herbs with cardioprotective effects, and therapies like ischemic postconditioning (Hao et al., 2017), remote ischemic postconditioning (Han et al., 2014), electroacupuncture (Xiao et al., 2020) and engineered nanoparticles (Fu et al., 2018).
Wang et al. (2015) demonstrated that rapamycin exerts a strong cardioprotective effect against IRI in CMs, with rapamycin inducing cardioprotection through autophagy activation via the PI3K/Akt signalling pathway. Similarly, in vivo and in vitro experiments have shown that metformin administration reduced infarct size and attenuated left ventricular post-AMI dysfunction through the induction of AMPK phosphorylation during reperfusion (Solskov et al., 2008; Gundewar et al., 2009; Wang et al., 2019). It has also been described that pramipexole (PPX), an important L-DOPA treatment for Parkinson’s disease (Frampton, 2014), induced cardioprotection through AMPK activation following myocardial IRI (Mo et al., 2016). Mesenchymal stem cells (MSCs), a type of multipotent progenitor cell, also improved cardiac function through autophagy activation during reperfusion (Pfister et al., 2014). MSC-derived exosomes are able to enhance autophagy and reduce the extent of myocardial infarction through AMPK/mTOR1 and Akt/mTOR signalling (Liu et al., 2017). Geniposide (GP), an extract from a traditional Chinese herb Gardenia jasminoides, reduced autophagosome accumulation and protected against IRI through Akt/mTOR signalling activation (Luo et al., 2020). Coenzyme Q10 (CoQ10), which is structurally similar to vitamin E and vitamin K, has beneficial effects in the prevention and treatment of acute myocardial IRI and HF. Liang et al. (2017) demonstrated that CoQ10 preconditioning (3 days before ischemia) reduced IRI and improved cardiac function by upregulation of Beclin-1, ATG5, and LC3-II to LC3-I ratio. The cell-permeable Tat- Beclin 1 has been shown to promote cell survival and CM autophagy in vivo (Shirakabe et al., 2016). Histone deacetylases (HDACs) inhibition has been shown to increase autophagy flux in the border zone, where active cell death is taking place. The FDA-approved HDAC inhibitor, superoylanilide hydroxamic acid (SAHA), reduced myocardial infarct size in large animal models (Xie et al., 2014). Remote ischemic postconditioning (three cycles of 5 min single hindlimb ischemia followed by 5 min reperfusion) induced cardioprotection through the upregulation of myocardial autophagy, and has been shown to induce the upregulation of LC3-II/LC3-I ratio and Beclin-1 level, and downregulation of p62 3 h after reperfusion, demonstrating that autophagy is activated at the early stage of reperfusion in an IRI murine in vivo model (Han et al., 2014). However, Kis et al. (2003) demonstrated that administration of mTOR inhibitors before the onset of ischemia diminished the cardioprotective effect of preconditioning. In contrast to this study, the administration of RAD, an mTOR inhibitor, after ischemia, as is the case in clinical settings, prevented LV remodeling and limited infarct size (Buss et al., 2009).
Electroacupuncture (EA) preconditioning has cardioprotective effects against IRI by modulating the mTOR/ Ulk1 pathway (Xiao et al., 2020). Visnagin, which is extracted from Ammi visnaga, has been evaluated as a cardioprotective agent (Fu et al., 2018). Engineered nanoparticles (NPs) have been mass-produced and widely applied to the development of nanotechnology and material science (Ong et al., 2017). Fu et al. (2018) encapsulated visnagin in NIPAAm-MAA (N-isopropylacrylamide – methacrylic acid) nanoparticles (NP-visnagin) and injected them in an IRI rat model. The NP- visnagin specifically targeted the IRI myocardium through the induction of autophagy and the inhibition of apoptosis (Fu et al., 2018). The protective effect of NP-visnagin could be related to the aryl hydrocarbon receptor (AHR), an upstream protein of the Beclin-1 and Bcl-2 complex (Fu et al., 2018).
Heart Failure
Autophagy activation as a protective mechanism to prevent adverse LV remodeling post-AMI is promising, and the beneficial roles of both activation and inhibition of autophagy have been demonstrated. Rapamycin and its analogues, including everolimus, temsirolimus, deforolimus, and zotarolimus are inducers of autophagy through mTOR- dependent and independent pathways. Although only a few of the autophagy modulators have been tested in HF, many of them may have therapeutic potential. Treatment with everolimus for the first three days post-AMI has been reported to preserve LV function and attenuate AMI-induced remodeling (Buss et al., 2009). Rapamycin was shown to reduce pressure overload cardiac hypertrophy and protected against myocardial IRI (McMullen et al., 2004). Gao et al. (2020b) recently reported that rapamycin treatment for four weeks reduced CM apoptosis and promoted CM autophagy, by regulating crosstalk between the mTOR and endoplasmic reticulum (ER) stress pathway in chronic HF. Treatment with granulocyte colony- stimulating factor (G-CSF) significantly improved survival, cardiac function, and prevented remodeling in an animal model of HF. The cardioprotective effect was accompanied by the production and degradation of autophagosomes (Takemura et al., 2006) (Table 1). The central role of autophagy in HF development has been demonstrated across multiple studies where inhibition of autophagy such as miR-22 prevented post- AMI adverse remodeling along with improved cardiac function (Gupta et al., 2016). Traditional Chinese medicine such as Yangxinkang Tablet (YXK) has demonstrated cardioprotective effects through the inhibition of excessive autophagy. The YXK treatment for four weeks improved cardiac function and reduced cardiac fibrosis in an in vivo post-AMI model. YXK was shown to induce cardioprotection through the abrogation of excessive autophagy by reducing LC3-II and Beclin-1 expression during IRI (Ren et al., 2020) (Figure 2, Table 1).
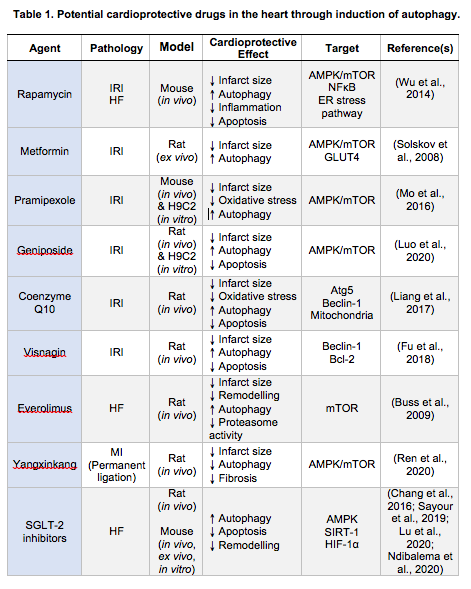
In a new window | Download PPT
With the perpetual high energy demands of the heart (Page and McCallister, 1973; Schaper et al., 1985; Aubert et al., 2016), mitochondrial dysfunction plays a major role in the development of HF (Brown et al., 2017). Decreased mitophagy along with mitochondrial dysfunction were reported in HF patients and in a HF murine model (TAC-induced). The decreased mitophagy was accompanied by an isoform shift of AMPK – from AMPKα2 to AMPKα1 (Kim et al., 2012; Wang et al., 2018). The overexpression of AMPKα2 in murine hearts halted the development of TAC-induced HF through increased mitophagy and mitochondrial function whereas genetic ablation of AMPKα2 led to exacerbation of TAC- induced congestive heart failure development, highlighting the important role of AMPKα2. Further investigation found that AMPKα2 interacts specifically with phosphorylated PINK1 at Ser495 in isolated adult mouse CMs following phenylephedrine stimulation; after which, the phosphorylated PINK1 recruits Parkin to the depolarized mitochondria, initiating the PINK1/Parkin mitophagy pathway (Wang et al., 2018). The consequent increased cardiac mitophagy results in elimination of damaged mitochondria and improvement in mitochondrial function. Interestingly, S495A mutation of PINK1 partially suppressed AMPKα2 overexpression-induced mitophagy while S495D mutation of PINK1 promoted mitophagy following phenylephedrine stimulation. Taken together, the study has shown a central role of AMPKα2 in the modulation of cardiac mitophagy (Wang et al., 2018). In the landmark Empagliflozin Cardioascular Outcome Event Trial in Type 2 Diabetes Mellitis Patients – Removing Excess Glucose (EMPA-REG OUTCOME) clinical trial, empagliflozin, a sodium-glucose co-transporter-2 (SGLT-2) inhibitor was shown to reduce cardiovascular-related death and hospitalization for HF, but the mechanisms underlying this cardiovascular protective effect remain undetermined (Zinman et al., 2015). Other clinical studies have reported similar cardioprotective effects using different SGLT-2 inhibitors – overall ~25% to 40% reductions in the risk of hospitalization for HF have been observed (Zinman et al., 2015; Neal et al., 2017; Perkovic et al., 2019; Wiviott et al., 2019). Interestingly, experimental studies have shown that SGLT-2 inhibitors cause the activation of AMPK, SIRT-1, and HIF-1α (Chang et al., 2016; Sayour et al., 2019; Lu et al., 2020; Ndibalema et al., 2020), and whether modulation of myocardial autophagy contributes to the cardioprotective effects of SGLT-2 inhibitors is unclear.
Summary and future perspectives
Autophagic cell death plays a crucial role in the pathogenesis of AMI and the progression to HF, and therefore presents a potential therapeutic target for cardioprotection. Despite substantial advances in our understanding of the molecular aspects of autophagy, the role of this catabolic process in determining the fate of AMI and HF remains incompletely defined. The importance of autophagy dysregulation in acute myocardial IRI and subsequent post-AMI LV remodeling has now become apparent. Recent findings have revealed that specific autophagic processes may operate in cardiomyocytes but their contribution to the pathogenesis of AMI and HF requires further investigation. Although not conslusive, most experimental studies have reported therapies that upregulate autophagy exert cardioprotection against acute myocardial IRI. However, in terms of preventing HF, the studies have been mixed with therapeutic inhibiton or activation of autophagy both reported as being beneficial. In summary, autophagy modulators have the therapeutic potential to confer cardioprotection by reducing myocardial infarct size and preventing HF, and elucidation of the role of autophagy in these cardiac conditions should result in the discovery of new treatments for modulating autophagy as a strategy for improving outcomes in AMI patients.
Acknowledgement
Sang-Ging Ong is supported by National Institutes of Health grantR00 HL130416 and R01 HL148756. Sauri Hernandez- Resendiz is supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund- Young Individual Research Grant (OF-YIRG)–[NMRC/ OFYIRG/0078/2018]. Derek Hausenloy is supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA- SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/ CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU- CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
References
Jasper Chua1,2
1Cardiovascular and Metabolic Disorder Programme, Duke-NUS Medical School, Singapore. 2National Heart Research Institute Singapore, National Heart Centre, Singapore.
Gustavo E. Crespo-Avilan1-3
1Cardiovascular and Metabolic Disorder Programme, Duke-NUS Medical School, Singapore. 2National Heart Research Institute Singapore, National Heart Centre, Singapore. 3Department of Biochemistry, Medical Faculty, Justus Liebig-University, Giessen, Germany.
Yap En Ping1,2
1Cardiovascular and Metabolic Disorder Programme, Duke-NUS Medical School, Singapore. 2National Heart Research Institute Singapore, National Heart Centre, Singapore.
Sang-Ging Ong4,5
4Department of Pharmacology, University of Illinois College of Medicine, Chicago, Illinois, United States of America. 5Division of Cardiology, Department of Medicine, University of Illinois College of Medicine, Chicago, Illinois, United States of America.
Sauri Hernandez-Resendiz*1,2
1Cardiovascular and Metabolic Disorder Programme, Duke-NUS Medical School, Singapore. 2National Heart Research Institute Singapore, National Heart Centre, Singapore.
Derek J. Hausenloy*1,2,6-8
1Cardiovascular and Metabolic Disorder Programme, Duke-NUS Medical School, Singapore. 2National Heart Research Institute Singapore, National Heart Centre, Singapore.6Yong Loo Lin School of Medicine, National University Singapore, Singapore. 7The Hatter Cardiovascular Institute, Institute of Cardiovascular Science, University College London, UK.8Cardiovascular Research Center, College of Medical and Health Sciences, Asia University, Taiwan.
*Joint senior authors.
Corresponding author:
Derek J. Hausenloy
Email: derek.hausenloy@duke.nus.edu.sg
In a new window | Download PPT
Figure 1. Schematic diagram of phases of autophagy in cardiomyocytes. (1) Induction and nucleation, (2) formation and maturation, (3) fusion, and (4) degradation. Autophagy-activating kinase 1, ULK1; protein of 200kDa, FIP200; autophagy-related protein, Atg; endoplasmic reticular, ER; phosphatidylethanolamine, PE; phosphatidylinositol 3-phosphate, PIP3; 1 light chain 3, LC3; adenosine monophosphate, AMP; adenosine triphosphate, ATP; mammalian target of rapamycin, mTOR.
In a new window | Download PPT
Figure 2. Signaling regulation of autophagy during acute myocardial IRI and potential cardioprotective therapies. (A) Ischemia increases the AMP/ATP ratio and activates AMPK. AMPK has a dual action to promote autophagy via inhibition of mTOR and activation of Ulk1 via phosphorylation at Ser 377 and Ser 777. AMPK indirectly inhibits mTOR via its phosphorylation at Ser 722 and Ser 792. (B) In contrast, during reperfusion, activation of AMPK is no longer observed. Instead, autophagosome formation during reperfusion is mediated by Beclin-1-dependent mechanisms.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11296 | 29 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA