Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Remote Ischemic Preconditioning and Blood Flow Restriction Training: Biochemical and Immunological Mechanisms of Neuroprotection in Stroke
Time:2021-04-05
Number:11770
Author Affiliations

Conditioning Medicine 2021. 4(1):15-27.
Abstract
Abstract Stroke is a deadly and destructive disease with limited treatment options. Despite this, novel therapeutic interventions have stagnated in recent years. This review highlights the recent biochemical and immunological evidence underpinning the mechanisms of a novel intervention, remote ischemic preconditioning, which has shown promising effects in human and in experimental stroke studies (e.g. middle cerebral artery occlusion). Blood flow restriction training, a conceptually related intervention, is also discussed to highlight a potentially viable alternative therapeutic approach for stroke patients that has not yet gained traction for use with this population.
Keywords: Stroke, Neuroprotection, Immnology, Remote ischemic preconditioning, Blood flow restriction training
Abstract
Abstract Stroke is a deadly and destructive disease with limited treatment options. Despite this, novel therapeutic interventions have stagnated in recent years. This review highlights the recent biochemical and immunological evidence underpinning the mechanisms of a novel intervention, remote ischemic preconditioning, which has shown promising effects in human and in experimental stroke studies (e.g. middle cerebral artery occlusion). Blood flow restriction training, a conceptually related intervention, is also discussed to highlight a potentially viable alternative therapeutic approach for stroke patients that has not yet gained traction for use with this population.
Keywords: Stroke, Neuroprotection, Immnology, Remote ischemic preconditioning, Blood flow restriction training
Introduction
Cerebrovascular disease has consistently been one of the top three leading causes of mortality in Australia over the past 50 years, claiming more than 10,000 lives (6.3% of deaths) in 2017 (Australian Bureau of Statistics, 2018). Similarly, stroke is diagnosed in 795,000 people in the USA yearly, with an event occurring about every 40 seconds, and is currently ranked first as a cause of serious long-term disability. In 2014, the total cost (direct and indirect expenses) of stroke in the USA was $45.5 billion, with direct medical costs expected to more than double from 2015 ($36.7 billion) to 2035 ($94.3 billion) (Benjamin et al., 2019; Benjamin et al., 2018).
The deaths, sequelae, and costs associated with stroke stem from cellular death within the brain. This cellular death can be caused by thrombus formation and subsequent lodgement within the brain vasculature or rupture of the brain’s vasculature, both ultimately leading to reduced blood supply and therefore oxygen to the brain’s tissue (Sekerdag et al., 2018). A review article by Sekerdag et al., (2018) provides an excellent and detailed explanation of the cell death types and mechanisims that occur following stroke. Briefly, excitotoxicity is cause by decreased ATP production that leads to an uncontrolable calcium influx (Sekerdag et al., 2018). This subsequently results in increased glutamate release from the presynaptic neuron leading to increased depolarization of the postsynaptic neuron and subsequently increases calcium concentrations (Sekerdag et al., 2018). This can lead to mitochondrial dysfunction and increase free radical production as well as proteases and enzymes that can also upregulate cellular death cascades (Sekerdag et al., 2018). Furthermore, apoptosis is caused by the previously mentioned mitochondrial dysfunction, resulting in cytochrome C release, which ultimately leads to cellular death through DNA fragmentation caused by caspases. Interestingly, stroke can also lead to pyroptosis (cellular death caused by inflammation) and necropoptosis (see the review by Sekerdag et al., (2018) for more information).
Current recommended treatments for ischemic stroke fall into two categories: thrombolysis, where medications are used to dissolve the clot, and thrombectomy, where the clot is retrieved using a mechanical device (Powers et al., 2018). Although considered safe and effective when administered to patients within about 4 hours of the onset of ischemia (Lisboa et al., 2002; Powers et al., 2018; Prabhakaran et al., 2015; Smith et al., 2005), there are currently no neuroprotective agents recommended as adjuvant treatments of ischemic stroke (Powers et al., 2018). However, there are promising novel passive and exercise interventions that may be able to bridge this current treatment gap (Keep et al., 2014).
Much of the research into the mechanisms behind novel, passive interventions have revolved around the induction of ischemic tolerance within an organ through a technique termed ‘ischemic conditioning’ (IC), or ‘ischemic preconditioning’ (IP), when preformed prior to an injurious event. This technique of creating ischemic tolerance requires the sub-lethal occlusion of blood flow to an organ thus resulting in adaptations that make the organ more resistant to future periods of damaging ischemia (Fairbanks et al., 2010; Meller et al., 2015). Thus, developing ischemic tolerance with IP is not a feasible option except under certain circumstances, such as prior to surgery. More recently, a new method of creating ischemic tolerance, remote ischemic conditioning (RIC), a form of passive peripheral blood flow restriction, has been developed and is showing promising results (Figure 1).
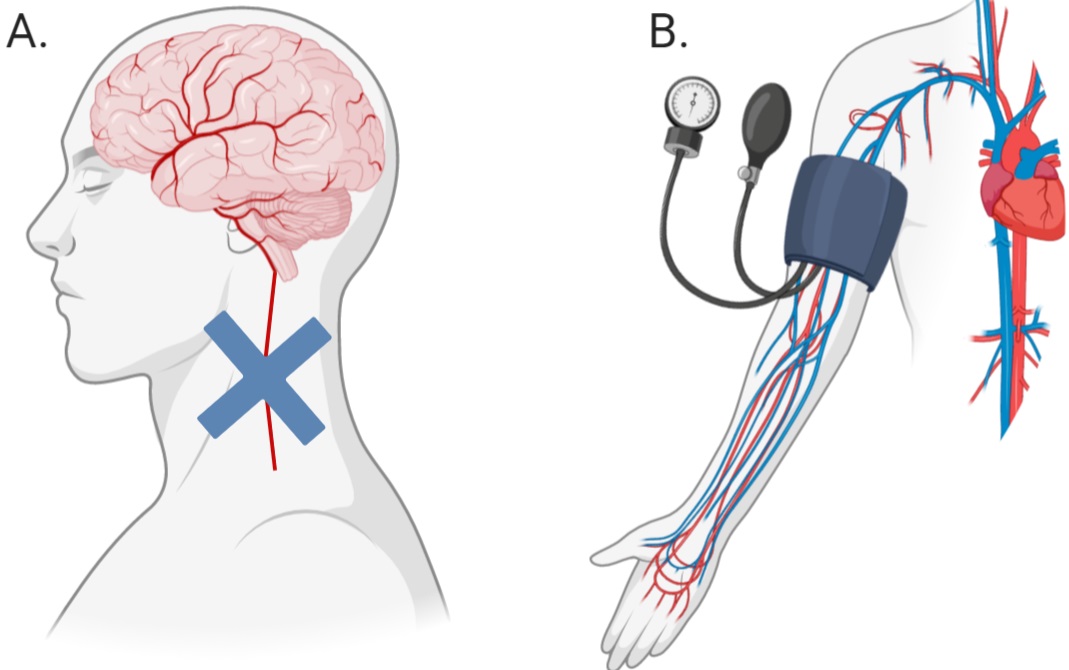
In a new window | Download PPT
Figure 1: Difference between ischemic conditioning (A) and remote ischemic conditioning (B). Blue cross (A) indicates the application of a device to occlude blood flow.
There is increasing interest in the mechanisms and effects of RIC in relation to stroke outcomes (Keep et al., 2014). RIC applied before stroke is known as remote IP (RIPreC), applied during stroke is known as remote ischemic per-conditioning (RIPerC), and applied after stroke is known as remote ischemic post-conditioning (RIPostC) (Figure 2) (Pan et al., 2016). The most studied form of RIC, RIPreC, has been shown to be safe and well tolerated, having no adverse effects in high risk populations such as octogenarians and nonagenarians after stroke (Meng et al., 2015), critically ill patients with subarachnoid haemorrhage (Gonzalez et al., 2014; Koch et al., 2011), or unilateral middle cerebral artery stenosis (Li Sijie et al., 2015). Although there has been less research into the safety of RIPerC and RIPostC, there are no reported adverse events in humans (England et al., 2017; Hougaard et al., 2014).
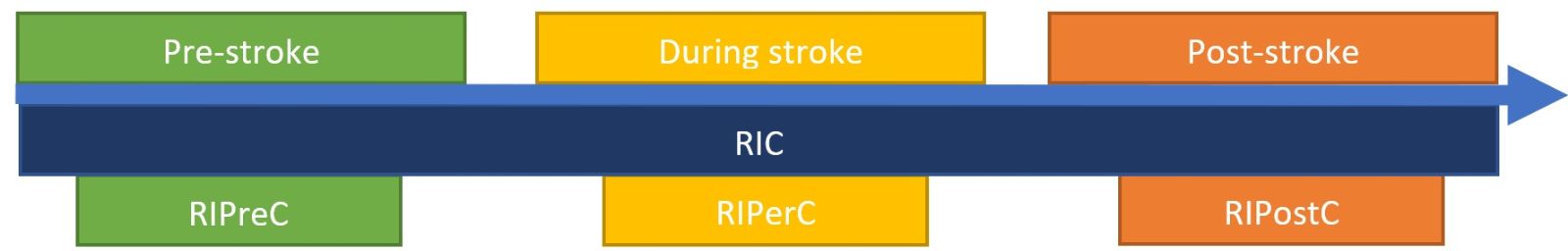
In a new window | Download PPT
Figure 2: Diagram explaining the various types of RIC in relation to stroke onset.
RIC has demonstrated neuroprotective effects on the brain against ischemia/reperfusion (I/R) injury (Pan et al., 2016). In human studies, RIPreC improves cerebral perfusion, reduces inflammatory mediators, improves functional recovery following stroke, and decreases recurrence (Meng et al., 2012; Meng et al., 2015). Additionally, RIPerC and RIPostC have been reported to potentially limit an event to a transient ischemic attack (TIA), and to reduce stroke and TIA recurrence, and inflammatory biomarker levels (Hougaard et al., 2014). With this said, further, larger studies should be conducted with all RIC paradigms before any final conclusions should be made as to their efficacy.
While, the mechanisms by which RIC has a positive impact on stroke remain unclear, what is known arises from studies of myocardial protection and ischemic conditioning of the heart and brain (Hess et al., 2015). Studies have extensively investigated the presence and impact of humoral and neural pathways for myocardial infarction (Basalay et al., 2018). For instance, evidence suggests that humoral factors, which are yet to be completely identified, increase in the blood following RIC and are associated with reduced neurological deficits, increased cerebral blood flow, and reduced infarction volume (Pan et al., 2016; Shan et al., 2013). The use of nerve blocking drugs and transection of nerves diminished protection following RIPreC in animal models, while transferring blood from human or animal subjects that have undergone RIPreC to an isolated heart or untreated subject leads to reduced infarct volume (Hess et al., 2015; Meller et al., 2015). For instance, the ganglion blocker hexamethonium (Malhotra et al., 2011) leads to the abolishment of neuroprotection following RIC in animal models (Pan et al., 2016). Furthermore, alterations in immune cell numbers and migration to the infarct area has been associated with improvements in neurological function and reduced infarction volume in animals (Liu et al., 2016).
RIPreC has two periods of protection (Moskowitz et al., 2011). The first occurs 30-60 minutes after occlusion, lasting for 2-3 hours. This window is likely due to triggering of intracellular signaling cascades and the alteration of pre-existing proteins (Meller et al., 2015; Moskowitz et al., 2011). The second window of protection occurs from 12-24 hours to 72-96 hours post-occlusion (Moskowitz et al., 2011; Pan et al., 2016; Zhao et al., 2018), although the extinction time point is not known with certainty (Meller et al., 2015). It is likely that this second window of protection is caused by de novo synthesis of proteins and gene expression/repression (Moskowitz et al., 2011; Sprick et al., 2019). Due to the existence of two protective windows, exercising with RIC (known as blood flow restriction (BFR) training) could be an innovative way of maximizing the benefit from RIC (Meller et al., 2015).
Although the RIPreC and BFR training is conceptually similar, there are important differences in how the two interventions are applied. In one of the few human studies investigating the effects of RIPreC on stroke recurrence and severity, a protocol of five cycles consisting of five minutes of occlusion (200 mmHg) with a pneumatic cuff and five minutes of reperfusion twice per day was used. This resulted in significantly reduced recurrence rates and stroke severity while being safe in the at risk stroke population (Meng et al., 2015). BFR training, on the other hand, is a training methodology typically implemented in elite sports and rehabilitation settings to facilitate improvements predominantly in muscle hypertrophy. Lighter weights (~30% of 1 repetition maximum) are used with this type of intervention, however hypertrophy still occurs as the load stimulus for hypertrophy in normal resistance training (≥ 60% of 1 repetition maximum) is replaced by a metabolic stimulus (Hwang et al., 2019). The metabolic stimulus is achieved through the application of pneumatic cuffs, belts, or wraps to occlude all venous blood flow out of the limb, while still allowing for some arterial blood flow into the limb. A 7/10 subjective rating of pressure has been recommended for practical reasons (Wilson et al., 2013). Here, we aim to provide an up-to-date review of what is currently known about the biochemical and immune cell alterations following RIPreC and BFR training.
Literature search method
In April of 2020, a literature search was conducted on PubMed with the following search string: ((blood flow restriction training) OR (blood flow restriction exercise) OR (BFR training) OR (BFR exercise) OR (kaatsu) OR (occlusion cuff training) OR (occlusion cuff exercise) OR (remote ischemic conditioning) OR (remote ischemic preconditioning) OR (remote ischemic preconditioning) OR (remote ischemic postconditioning) OR (remote limb conditioning) OR (remote limb preconditioning) OR (remote limb preconditioning) OR (remote limb postconditioning) OR (RIC) OR (RIPC) OR (RIPreC) OR (RIPerC) OR (RIPostC)) AND ((stroke) OR (cerebrovascular disease) OR (cerebral infarct) OR (neuroprotection)), with the results limited to the past 10 years. Results were initially evaluated based on their title for inclusion into the current review, which if deemed appropriate by the authors at that stage, where then read in full before a final list of articles were produced for inclusion into this review. During this process, the authors agreed that the only paradigm of RIC that should be included in this review was that of RIPreC. This decision was reached based on the similarities between BFR training and RIPreC in regards to the timepoint of application (pre-stroke, see Figure 2), as well as publishing requirements relating to word count.
Biochemical, cytokine, and inflammatory alterations resulting from RIPreC
To our knowledge, there has only been two human studies investigating the biochemical alterations following RIPreC, one from the perspective of neuroprotection while the other is from the perspective of inflammation and coagulation. There have been other RIC studies in human populations, however, they were applied shortly after the event, which classifies the intervention as RIPostC, and therefore outside the scope of this review (An et al., 2020; Appleton et al., 2020; England et al., 2017). In 50 healthy young (34.5 ± 12.0 years) male and female participants, it was found that at 1 hour post-intervention, circulating levels of glial cell derived neurotrophic factor (GDNF), vascular endothelial growth factor (VEGF-A), transforming growth factor-β1 (TGF-β1), leukemia inhibitory factor (LIF), matrix metalloproteinase-9 (MMP-9), and tissue inhibitor of metalloproteinase-1 (TIMP-1) were significantly increased compared to baseline, while the MMP-9/TIMP-1 ratio was not significantly different from baseline (Guo et al., 2019). MMP-2 and MMP-3 did not significantly change from baseline (Guo et al., 2019). The conclusion from this study was that the biochemical alterations observed in the blood may partly explain the beneficial effect of the intervention.
The second study consisted of 80 to 95 year-olds with symptomatic intracranial atherosclerotic stenosis that had an ischemic stroke or transient ischemic attack in the past seven days (Meng et al., 2015). The intervention group (n = 30, 83.5 ± 2.3 years) not only had significantly reduced C-reactive protein levels as compared to the control group (n = 28, 84.2 ± 1.6 years) at 15 and 30 days of bi-daily intervention, but also had significantly decreased fibrinogen after 30 days of intervention, reduced plasminogen activator inhibitor-1 levels after 15 and 30 days of intervention, as well as increased tissue plasminogen activator after 15 and 30 days of intervention as compared to the control group (Meng et al., 2015). These findings indicate that the long term use of RIPreC may beneficially alter coagulation tendencies and reduce inflammation in stroke patients (Meng et al., 2015).
Animal studies have provided more information and indicate that RIPreC-mediated reductions in infarct volume and/or improved neurological and behavioral outcomes are a result of activation of multiple non-redundant biochemical pathways (Table 1). For instance, several studies found that when specific pathways were inhibited, the protective effect was reduced to the point of being statistically equivalent to ‘no treatment’ cohorts (Du et al., 2020; Hu et al., 2012; Liang et al., 2019; Wei et al., 2012; Yang et al., 2018; Zhao et al., 2019). This suggests that multiple pathways are involved in RIPreC and that each is integral to its neuroprotective effect.
Table 1: Methodologies and significant findings from animal studies that investigated the biochemical alterations behind the beneficial effects of RIPreC.
.jpg)
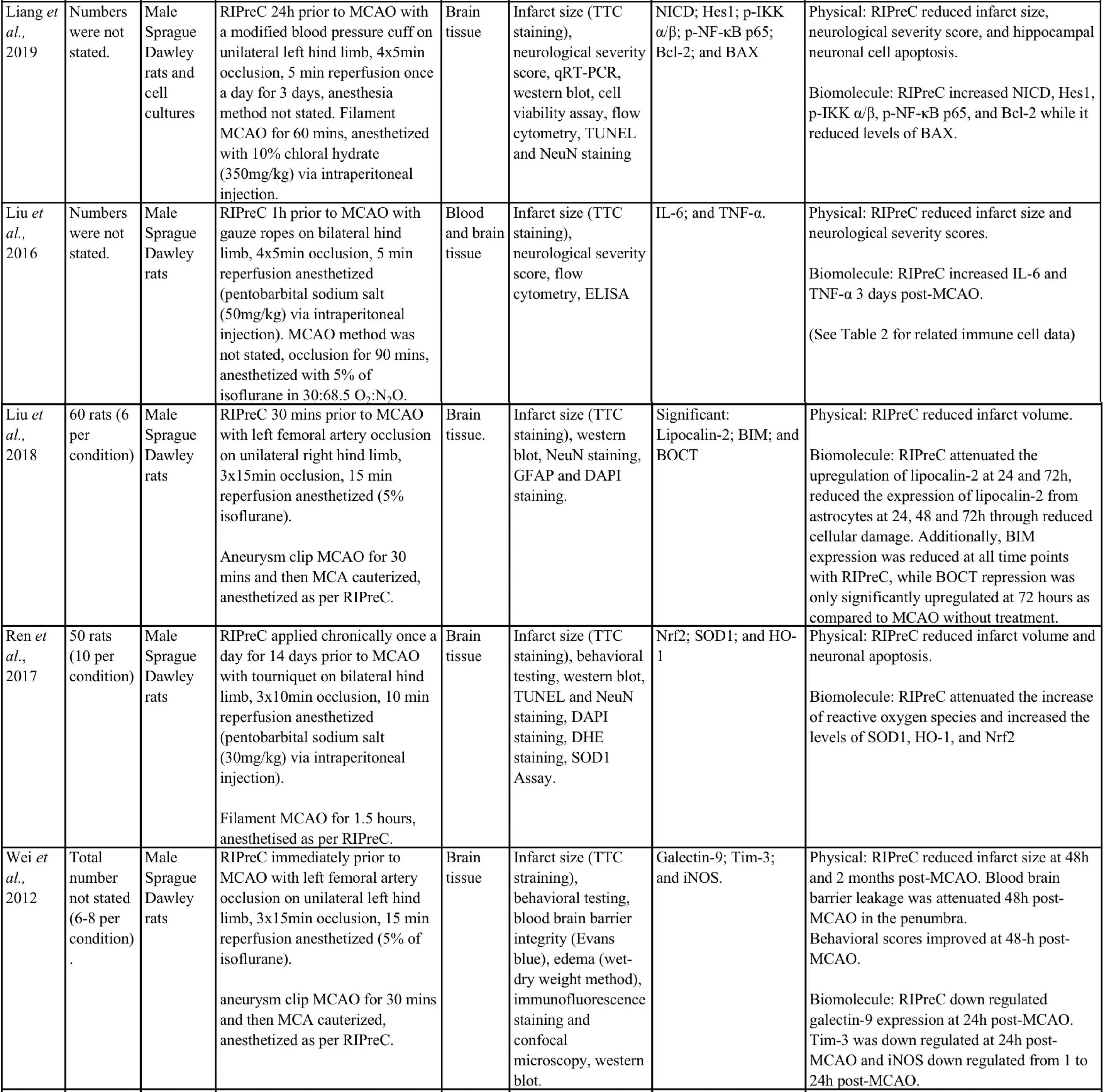
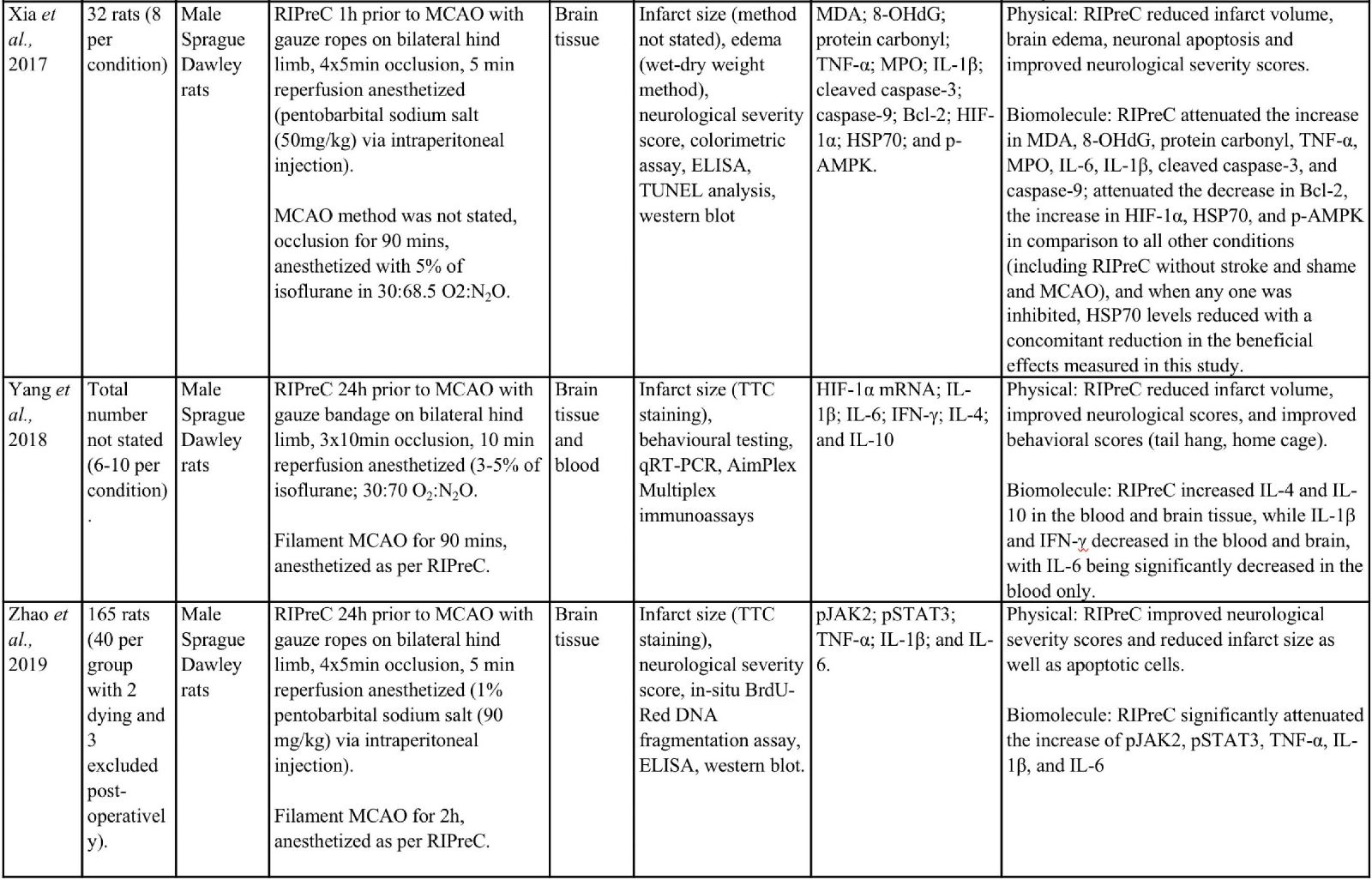
8-OHdG = 8-hydroxy-2' -deoxyguanosine; AIF = Apoptosis Inducing Factor; Akt = Protein kinase B; AMPK = 5' adenosine monophosphate-activated protein kinase; BAX = bcl-2 associated X protein; Bcl-2 = B-cell lymphoma 2; BIM = bcl-2-interacting mediator of cell death; BOCT = brain-type organic cation transporter; BrdU = Bromodeoxyuridine; COX IV = cytochrome c oxidase subunit IV; DAPI = 4′,6-diamidino-2-phenylindole; DHE = Dihydroethidium; GFAP = glial fibrillary acidic protein; GSH = Glutathione; GSSH = oxidized glutathione; H2AX = H2A histone family member X; Hes-1 = hairy and enhancer of split-1; HIF = hypoxia inducible factor; HO-1 = Heme oxygenase-1; HSP = heat shock protein; IFN = interferon; IKK = inhibitor of nuclear factor kappa B kinase; IL = interleukin; iNOS = Inducible nitric oxide synthase; JAK2 = Janus Kinase 2; MCAO = middle cerebral artery occlusion; MDA = Malondialdehyde; MPO = Myeloperoxidase; NeuN = Neuronal Nuclei; NF-κB = nuclear factor kappa B; NICD = notch intracellular domain; Nrf2 = nuclear factor erythroid 2–related factor 2; PAR = Protease-activated receptor; SOD = super oxide dismutase; STAT3 = Signal transducer and activator of transcription 3; Tim-3 = T-cell immunoglobulin and mucin domain-3; TNF = tumor necrosis factor; TTC = 2,3,5-Triphenyltetrazolium chloride; TUNEL = Terminal deoxynucleotidyl transferase dUTP nick end labelling.
Hypoxia inducible factor-1 (HIF-1)
Known as one of the most important molecules mediating the body’s response to hypoxia and/or ischemia, HIF-1 is comprised of two subunits from the basic helix-loop-helix family of transcription factors. Formed from HIF-1α and HIF-1β, these subunits dimerize at the N-terminals resulting in the formation of the functional protein (Bárdos et al., 2005; Hellwig-Bürgel et al., 2005; Shi, 2009; Ziello et al., 2007). HIF-1 modulates a wide variety of pathways important in ischemic tissues (Bárdos et al., 2005). Constitutively expressed HIF-1α is unstable during normoxic conditions due to hydroxylation of proline residues at Pro-402 and Pro-564 on the oxygen-dependent degradation domain of the dimer (Bárdos et al., 2005; Hellwig-Bürgel et al., 2005). During hypoxic conditions however, the proline residues of HIF-1α are not hydroxylated as the reaction with prolyl hydroxylase domain (PHD) enzymes cannot occur without molecular oxygen (Bárdos et al., 2005). In addition to the modulating effect of PHD, there are several other molecules and pathways that can positively or negatively regulate HIF-1α through degradation or stabilization, up or downregulation, or prevention of transcriptional activation (Bárdos et al., 2005; Hellwig-Bürgel et al., 2005; Xia et al., 2017; Zhang et al., 2018). Importantly, oxygen is depleted during RIC due to reduced blood flow, thus reducing the rate of HIF-1α degradation and allowing intracellular levels to rise. Following this increase, HIF-1α translocates to the nucleus and binds with HIF-1β to form HIF-1, which then binds to DNA and upregulates transcription (Bárdos et al., 2005; Sprick et al., 2019).
Once HIF-1 is formed in the nucleus, it binds to hypoxia responsive elements (HRE) in the promoter regions of DNA (Hellwig-Bürgel et al., 2005; Zhang et al., 2018). Through this binding, and the following transcription, gene expression for numerous proteins are upregulated leading to beneficial effects for hypoxic tissue. HIF-1 plays a role in the regulation of blood vessel growth, production of red blood cells, modulating vascular tone, and improved ATP production. This is facilitated by the transcription of VEGF, erythropoietin (EPO), endothelial nitric oxide synthase (eNOS), and increased expression of glucose transporter 1 (GLUT 1) and glycolytic enzymes (Hellwig-Bürgel et al., 2005; Zhang et al., 2018; Ziello et al., 2007). HIF-1-mediated gene transcription can also drive matrix and barrier functions and inflammation through TIMP-1 and CD18; proliferation and apoptosis regulation by the B-cell leukemia /lymphoma-1 (Bcl-2) and p21/p27 mediated pathways; increased oxygen delivery via erythropoiesis, iron metabolism, angiogenesis and vascular tone; and modulate oxygen consumption through anaerobic metabolism promotion and tricarboxylic acid cycle inhibition (Kanehisa, 2019; Kanehisa et al., 2000).
It is believed that over accumulation of HIF-1α in the nucleus leads to the stabilization of p53, which may lead to increased rates of apoptosis in vitro. Additionally, it has also been observed that through inhibition of HIF-1, there is a decrease in VEGF and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) levels affording neuroprotection through a decrease in mitochondrial dysfunction (Shi, 2009; Zhang et al., 2018).
Importantly, evidence suggests that RIC-mediated generation of HIF-1α and therefore HIF-1, is neuroprotective. For instance, the HIF-1α/5’adenosine monophosphate-activated protein kinase (AMPK)/heat shock protein (HSP) 70 pathway was a significant contributor to the neuroprotective effects mediated by RIPreC in a rodent study that specifically examined RIPreC and HIF pathway activation (Xia et al., 2017). RIPreC reduced neuronal apoptosis, attenuated the cerebral inflammatory response (i.e. myeloperoxidase (MPO), tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β), increased levels of Bcl-2, and reduced levels of Caspase-3 and -9 in brain tissue following medial cerebral artery occlusion (MCAO). Most importantly, it was observed that treatment with RIPreC resulted in a significant increase in brain tissue levels of HIF-1α, AMPK, and HSP70, and when any one of these were inhibited, HSP70 levels reduced with a concomitant reduction in the beneficial effects observed (Xia et al., 2017).
Additionally, in a study using young male Sprague-Dawley rats in an ischemia/reperfusion (I/R) MCAO model, RIPreC did not affect the levels of the pro-inflammatory cytokines IL-1β, IL-6. and interferon (IFN)-γ in rats without MCAO, but the circulating levels of all three factors were reduced by RIPreC after MCAO (Yang et al., 2018). RIPreC reduced both IL-1β and IFN-γ in brain tissue of rats post-MCAO but had no effect on the levels of IL-6 in brain. The study also demonstrated that HIF-1α mRNA expression increased with RIPreC in healthy rat brains compared to brains in rats not treated with RIPreC and that the anti-inflammatory cytokines IL-4 and IL-10 were significantly increased in peripheral blood and brain tissue following RIPreC. Furthermore, pharmacologic upregulation of HIF-1α using dimethyloxalylglycine (DMOG) resulted in a significant reduction of infarction, whereas inhibition of HIF-1α with acriflavine hydrochloride (ACF) resulted in no appreciable difference. These data were reinforced by the findings that both RIPreC and DMOG treatment significantly improved performance in behavioral tests (Yang et al., 2018). Du et al., (2020) reported similar findings using the same method but in aged rats (Du et al., 2020). They also found that RIPreC resulted in a significantly reduced infarct volume of 25.4% to 38.8%, reduced the blood levels of IL-1β, IL-6, and IFN-γ with RIPreC compared with no treatment, and decreased the levels of IL-1β and IFN-γ in brain tissue. RIPreC also improved neurological and functional scores. Furthermore, they also found that inhibition of HIF with ACF resulted in an increased infarct volume following RIPreC and worse outcomes on neurological and functional tests. In addition, ACF inhibition of HIF with RIPreC led to either similar or increased pro-inflammatory cytokines and decreased anti-inflammatory cytokines, when compared to RIPreC alone (Du et al., 2020).
Adenosine A1 receptors
Adenosine A1 receptors are significantly implicated in the effects of RIPreC, requiring nanomolar concentrations of adenosine for activation and being found in high concentrations throughout various regions of the brain (Hu et al., 2012; Paul et al., 2011; Stockwell et al., 2017). These membrane-bound receptors have long been implicated in neuroprotection, with high levels of adenosine generated during stroke either intracellularly or extracellularly from the degradation of adenosine monophosphate (AMP) by two enzymes: 5’-nucleotidase or ecto-5’nucleotidase (Paul et al., 2011). Following intracellular degradation of AMP, adenosine can accumulate extracellularly following transport through equilibrative nucleoside transporters on the cellular membrane. When located extracellularly, adenosine can be transported to other parts of the body to interact with adenosine receptors in distal organs, such as the brain (Paul et al., 2011). Adenosine A1 receptors actively inhibit neuronal activity through a cascade of interactions resulting in reduction of neurotransmitter release when activated pre-synaptically. Furthermore, adenosine A1 receptors decreases excitability of neurons when the receptor is activated post-synaptically, effectively reducing the metabolic load on the neuron leading to improved neuronal survival (Cunha, 2008; Stockwell et al., 2017).
In rat studies, RIPreC, vehicle with RIPreC, and administration of 2-chloro-N6-cyclopentyladenosine (CCPA), an agonist of adenosine A1 receptors, all resulted in significantly lower neurological deficit scores and reduced infarction volumes compared to sham RIPreC controls. Additionally, CCPA and RIPreC resulted in decreased circulating TNF-α and nitric oxide levels compared to cerebral I/R injury with no treatment (Hu et al., 2012). Further to this, glutathione (GSH), superoxide dismutase (SOD) and manganese SOD levels increased while oxidized glutathione (GSSH) decreased in the blood and brain with RIPreC. When groups that received CCPA or RIPreC were treated with 8-cyclopentyl-1,3-dipropulxanthine (DPCPX), an antagonist of A1 receptors, the beneficial effects on infarct volume, physical, and biochemical outcomes were abolished. This study indicates that adenosine A1 receptors constitute an integral pathway through which RIPreC delivers it beneficial effects (Hu et al., 2012).
Nuclear factor E2-related factor 2 (Nrf2)
Nrf2 is a basic leucine zipper transcription factor normally found in the cytosol attached to Kelch-like ECH-associated protein 1 (Keap1) that plays a vital role in the transcription of antioxidant- and cytoprotective-related genes following RIPreC (Baird et al., 2011; Li et al., 2019; Paunkov et al., 2019; Ren et al., 2017). Keap1 inhibits Nrf2’s translocation to the nucleus and facilitates its ubiquitin-mediated degradation via an interaction with the Cullin3/RING box protein 1-based E3-ubiquitin ligase complex (Li et al., 2019). Nrf2 dissociates from Keap1 and stabilizes in response to specific biochemical “inducers” that result from oxidative or electrophilic stress (Li et al., 2019). In a homeostatic manner, inducers upregulate the activity of Nrf2, eventually leading to the release of antioxidants and cytoprotective biomolecules, thus decreasing the levels of oxidative and electrophilic stress on the cell (Baird et al., 2011).
Reactive oxygen species (ROS) and electrophiles oxidize cysteine residues found on Keap1, which act like sensors, leading to the dissociation of Nrf2 (Li et al., 2019; Yamamoto et al., 2018). Additionally, p62, acting in a positive feedback loop, competitively binds to Keap1 marking it for autophagy, thus stabilizing Nrf2, which can then translocate to the nucleus (Li et al., 2019). Once in the nucleus, Nrf2 binds to the p62 gene antioxidant response element (ARE) where it promotes the transcription of p62 and other antioxidant genes (Baird et al., 2011; Li et al., 2019). Furthermore, the phosphoinositide 3-kinase (PI3K)/Akt pathway is able to upregulate Nrf2 through the phosphorylation and inhibition of glycogen synthase 3-beta (GSK-3β), preventing GSK-3β from phosphorylating Nrf2, leading to Nrf2’s destruction by ubiquitination (Li et al., 2019). Additionally, the p65 and p50 subunits of nuclear factor kappa B (NF-κB) can upregulate the transcription of Nrf2 through binding to the gene promoter of Nrf2 (Li et al., 2019). However, the resulting antioxidant generation from the activation of Nfr2 reduces oxidative stress and subsequent generation of NF-κB (Li et al., 2019).
Ren et al., (2017) showed that RIPreC increased both Nrf2 levels and that of some of its downstream effectors, including SOD1 and heme oxygenase 1 (HO-1) (Ren et al., 2017). SOD1 reduces oxidative stress and the subsequent DNA damage and neuronal apoptosis through the scavenging and dismutation of superoxide anions (Mondola et al., 2016). The role of HO-1 is not as well understood but it is believed that during HO-1-mediated degradation of heme molecules to biliverdin, there is a release of carbon monoxide, which leads to mitogen-activated protein kinase (MAPK) pathway activation, resulting in upregulation of cytoprotective molecules (Waza et al., 2018). Interestingly, the combination of atorvastatin, a cholesterol lowering drug, and RIPreC together significantly improves all outcomes outlined above, including neuronal and biochemical outcomes, when compared to atorvastatin or RIPreC alone (Ren et al., 2017).
Notch signaling and NF-κB
Liang et al., (2019) found that the Notch1 and NF-κB pathways were also involved in RIPreC-mediated neuroprotection (Liang et al., 2019). RIPreC resulted in statistically significant reduction of infarct volume, improvements in the neurological deficit score, and reduced hippocampal neuronal cell apoptosis as compared to no treatment. Importantly, RIPreC increased the levels of Notch intracellular domain (NICD), Hes1, phospho-IκB kinase (p-IKK) α/β, p-NF-κB p65, and Bcl-2 while reducing levels of Bcl2-associated X protein (BAX), indicating that the Notch and NF-κB signaling pathways were activated by RIPreC. When Notch1 signaling is inhibited, the beneficial effects induced by RIPreC were no longer observed. Additionally, NICD and Hes1 expression was suppressed, although not significantly as compared to the no treatment condition, while p-IKK α/β, p-NF-κB p65, and Bcl-2 remained unchanged (Liang et al., 2019).
The notch signaling pathway is responsible for cell survival, differentiation, and proliferation (Baron, 2003). The extracellular portion of Notch receptors (Notch extracellular domain, NECD) interact with membrane bound ligands, such as delta-like 1, which is activated by ubiquitination via mind bomb and Jagged 1. Mind bomb and Jagged 1 are cell membrane-bound ligands, which result in a juxtacrine-induced conformational change of the Notch receptor on adjacent cells (Imayoshi et al., 2011; Steinbuck et al., 2018). This results in a disintegrin and metalloprotease (ADAM)-mediated cleavage of the extracellular portion of the Notch receptor and subsequent γ-secretase mediated proteolysis of the NICD, allowing it to translocate to the nucleus (Imayoshi et al., 2011). NICD forms a complex with recombining binding protein suppressor of hairless (RBPj) that acts as a transcription activator, modulating gene transcription of Hes1 and Hes5, which are known to inhibit neuronal differentiation (Imayoshi et al., 2011).
NF-κB is a member of the Rel family of proteins and plays a complex role in inflammatory and apoptotic processes (Lawrence, 2009). With several different heterodimers constituting different forms of NF-κB, the p65 (RelA) and p50 heterodimers are the most extensively studied and most abundantly found in the brain (Shih et al., 2015). According to the canonical pathway, cytosolic NF-κB is initially inhibited by IκB, which when phosphorylated, induces the release of NF-κB and its translocation to the nucleus (Lawrence, 2009; Shih et al., 2015). Phosphorylation and subsequent activation of the IκB kinase complex occurs following the stimulation of, among others, the IL-1 and TNF receptors, inducing ubiquitination and degradation. Following translocation to the nucleus, NF-κB facilitates the transcription of genes by binding to the κB gene promoter (Lawrence, 2009; Shih et al., 2015). Suppression of the NF-κB signaling pathway with a Notch1 pathway inhibitor indicated that Notch1 activation is at least partly responsible for NF-κB pathway activity in RIPreC and this activity is important for the beneficial effects of RIPreC-mediated neuroprotection (Liang et al., 2019).
Galectin-9/ T-cell immunoglobulin domain and mucin domain 3 (Tim-3) pathway
Wei et al., (2012) found that RIPreC modulated the galectin-9/Tim-3 pathway (Wei et al., 2012). Tim-3 is a cell membrane receptor that exists on various immune cells and can be differentially modulated by several ligands including galactin-9, which inhibits T cell activity (Gorman et al., 2014; Han et al., 2013). RIPreC downregulated galectin-9 expression after 24 hours, but not 4 hours following MCAO, whereas iNOS was downregulated from 1-24 hours post-MCAO (Wei et al., 2012). Interestingly, the decrease in galectin-9 seems contradictory to what one would expect to happen with a neuroprotective treatment. Increased T cell numbers appear to be detrimental in stroke (Ao et al., 2018). One might expect the level of galectin-9 to increase with a neuroprotective treatment, as this would cause T cell apoptosis and therefore decreased numbers (John et al., 2016), which have been observed in other RIPreC studies (Chen et al., 2018; Liu et al., 2016). Despite the incongruent finding, this study, like many others, demonstrated a significant decrease in infarction, blood brain barrier (BBB) leakage, and brain edema, as well as an observed reduction of functional deficits (Wei et al., 2012). This study also observed through the inhibition of peripheral afferent nerves with capsaicin, as well as inhibition of the dorsal root ganglion with hexamethonium, that the positive effects of RIPreC was decreased, although no investigation was conducted into the direct inhibition of the galectin-9/Tim-3 pathway.
Janus kinase 2/signal transducers and activators of transcription 3 (JAK2/STAT3) pathway
The JAK2/STAT3 pathway has an interesting relationship to inflammation and neural recovery following stroke. To date, the literature provides conflicting data as to whether upregulation or downregulation of this pathway is most beneficial following ischemic damage (Raible et al., 2014). This indicates the likelihood of highly complex interactions with several pathways leading to variable outcomes. Currently, it is known that following the release of various molecules associated with stroke, JAK2 is phosphorylated through interactions with a cell membrane receptor (Raible et al., 2014). Once phosphorylated, JAK2 in turn phosphorylates STAT3, which translocates to the nucleus where it binds to the promoter region of γ-activated sequence containing genes. This upregulates the expression of genes that are involved in cell survival, differentiation, and proliferation (Raible et al., 2014).
In a study by Zhao et al., (2019) the JAK2/STAT3 pathway was also identified to convey significant effects to the outcome of RIPreC (Zhao et al., 2019). Investigation of RIPreC and the JAK2 inhibitor, AG490, found that both RIPreC and AG490 had similar effects in that they both significantly improved the neurological deficit scores, reduced infarct size, neuronal apoptosis, and levels of TNF-α, IL-1β, and IL-6. An increase in these inflammatory markers have been observed in other studies (Liu et al., 2016; Xia et al., 2017), but this is not a consistent finding in all RIPreC studies (Yang et al., 2018). It is believed that both treatments elicited positive effects though the reduced phosphorylation of JAK2. Subsequently, STAT3 was not phosphorylated, leading to decreased translocation to the nucleus and therefore reduced expression of pro-inflammatory cytokines and initiation of the post-stroke inflammatory process (Zhao et al., 2019).
Cell death
Significant injury to the brain, such as stroke, can induce parthanatos, a biochemically and morphologically distinct process of cell death (Andrabi et al., 2008). Parthanatos is a caspase-independent, poly [ADP-ribose] polymerase 1 (PARP-1)-dependent pathway of cellular death that occurs after significant DNA damage (Andrabi et al., 2008). Through the use of nicotinamide adenine dinucleotide (NAD+) as a substrate, protease-activated receptor (PAR)-1, in concert with poly (ADP-ribose) glycohydrolase (PARG), another PAR family member, is able to generate PAR within the nucleus (Andrabi et al., 2008). PAR translocates to mitochondria via the cytosol where it activates calpain-1 to liberate apoptosis-inducing factor (AIF), which then translocates to the nucleus and interacts with the histone H2AX to promote DNA damage and fragmentation, inevitably leading to cell death (Andrabi et al., 2008; Artus et al., 2010; Cheng et al., 2018; Norberg et al., 2010). RIPreC downregulates PAR and inhibits truncation and translocation of AIF to the nucleus. Moreover, AIF/H2AX intranuclear interactions were reduced leading to decreased neuronal parthanatos and chromatolysis (Andrabi et al., 2008; Artus et al., 2010; Jin et al., 2016).
RIPreC has been observed to produce neuroprotection by reducing oxidative DNA damage to neurons and amelioration of neuronal parthanatos (Jin et al., 2016). RIPreC reduced the number of 8-hydroxy-2’-deoxyguanosin (8-OHdG) positive cells, a biomarker of oxidative DNA damage. Furthermore, neuronal death was significantly reduced at 24 and 48 hours, with a larger percentage of neurons spared in the penumbra compared to the ischemic core following RIPreC. Neuronal DNA fragmentation was also reduced in the penumbra and ischemic core (Jin et al., 2016).
Similarly, RIPreC exhibited beneficial effects on mitochondrial damage and subsequent mitochondrial-mediated apoptosis (Jing et al., 2020). RIPreC resulted in a reduced infarction, as well as increased glucose metabolism, decrease neurological deficits, and improved functional outcomes as compared to the untreated group. For instance, RIPreC was found to increase cytochrome C oxidase IV (COX IV) and decrease HSP60, AIF, and EndoG translocation from the mitochondria to the nucleus. Furthermore, there was an increase in mitochondria-derived vesicles with concomitant reduction in mitochondrial deformity (Jing et al., 2020). This indicates mitochondrial adaptation, which could lead to a decrease in apoptosis. Others have found that RIPreC attenuated an increase of lipocalin-2 and BCL2-interacting mediator of cell death (BIM), resulting in modulation of the mitochondrial-mediated apoptosis pathway and reduction of neuronal apoptosis (Liu et al., 2018). Normally expressed in minute amounts in the healthy brain, lipocalin-2 expression is upregulated following stroke, with astrocytes being the main contributor (Chia et al., 2015). In the brain, lipocalin-2 interacts with its receptor, brain type organic cation transporter (BOCT) located on the membrane of neurons, which subsequently results in the binding of intracellular iron to a siderophore-iron complex within the neuron and an upregulation of BIM. Subsequently, BIM either directly or indirectly interacts with BAX and Bcl-2 homologous antagonist/killer (BAK) leading to the release of pro-apoptotic molecules, cytochrome C, second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO), and 5-hydroxytryptamine receptor 2A (HtrA2) into the cytosol from the mitochondria. This induces the production of caspase-9 and ultimately neuronal apoptosis (Liu et al., 2018; Sionov et al., 2015).
Immune cell alterations resulting from RIPreC
As compared to the biochemical outcomes investigated following the application of RIPreC, relatively little research has been conducted regarding immune cell alterations with RIPreC and stroke as compared to preconditioning via exposure to systemic hypoxia (Monson et al., 2014; Selvaraj et al., 2017; Stowe et al., 2012). To the author’s knowledge, two studies have reported immune cell alterations in the context of stroke and RIPreC (Table 2). These studies indicated that RIPreC led to altered immune cell population composition following stroke in the spleen (Chen et al., 2018; Liu et al., 2016).
Table 2: Methodologies and significant findings from animal studies that investigated immune cell alterations behind the beneficial effects of RIPreC.
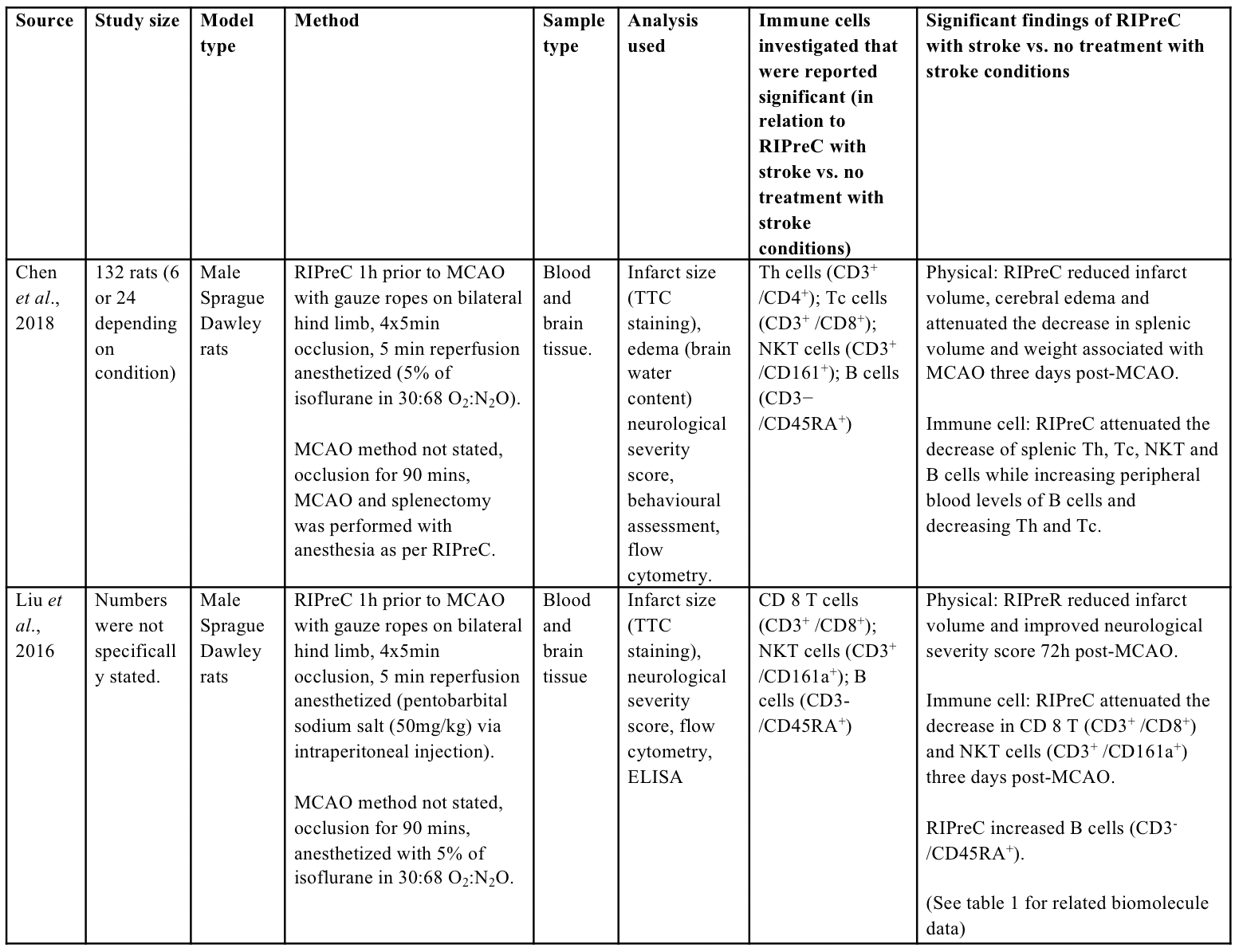
Th = helper T cell, Tc = cytotoxic T cell, NKT = natural killer T cell
The spleen appears to play a crucial role in immune cell alterations that underlie the neuroprotective effect of RIPreC, as splenectomy abolished neuroprotection (Chen et al., 2018). In addition, RIPreC increased the release of B cells (CD3-CD45RA) from the spleen, but reduced the release of helper T cells (Th; CD3+CD4+), cytotoxic T cells (Tc; CD3+CD8+) and natural killer T cells (NKT; CD3+CD161a+) (Chen et al., 2018). Similarly, others found that RIPreC attenuated the decrease in Tc and NKT cells while increasing B cells numbers in the blood following MCAO (Liu et al., 2016). The attenuated decrease in Tc and NKT cells in the peripheral blood with RIPreC despite a decreased release from the spleen is potentially due to alterations in immune cell infiltration into the brain. With RIPreC attenuating the infiltration of Tc and NKT cells into the damaged brain (Chen et al., 2018), this would proportionally increase the number of immune cells in the blood, leading to an attenuated decrease in peripheral blood as observed in the previous study (Liu et al., 2016), although this has not been simultaneously assessed.
Alteration of circulating immune cells and subsequent infiltration are also known to affect humans following stroke (Wang et al., 2017). T cells, particularly Th and Tc cells, are associated with increased inflammation and infarct volumes as well as poorer neurological outcomes (Ao et al., 2018). Specifically, lower peripheral T cells numbers and increased infiltration to the brain appear to be particularly detrimental in humans (Ao et al., 2018). In contrast to T cells, the literature regarding B cells is sparse and inconsistent, with outcomes dependent on the time post-stroke and the type of stroke model used. In rodent MCAO studies it has been determined that up to 24 hours post-MCAO, B cells do not play an important role in the pathophysiology (Tanabe et al., 2019). However, by 48 hours post-MCAO, the increase of IL-10 secreted from B cells is associated with decreased infarct volumes and neurological deficits, while at one week a decrease in B cells is associated with reduced cognitive impairment (Tanabe et al., 2019). As literature continues to emerge regarding the alterations of B cell number, type, and timing of such alterations, there will likely be new insights into novel immunological-based mechanisms for RIPreC-induced neuroprotection.
How does BFR training compare biochemically and immunologically to RIPreC?
To the best of the authors’ knowledge, there have been no reported BFR training studies conducted on stroke patients, nor has there been any analysis specifically investigating the biochemical applicability of BFR training to improve stroke severity or outcomes. Consequently, there is currently a lack of solid evidence to support BFR training as an adjunct therapy for stroke patients. With this in mind however, if BFR training activates the same pathways and generates similar biomolecules as RIPreC, which is conceptually a passive form of BFR training, an argument could be made in support of BFR training in a stroke patient population. This is an important argument as one could speculate that a physical intervention such as BFR training could lead to additional benefits compared to a passive intervention, such as RIPreC. This section will present what is currently known regarding similarities between both therapies, with the aim of presenting an argument for the potential neurological benefit of BFR training to stroke patients.
BFR and RIPreC-induced biochemical signaling
HIF appears to be highly significant in the neuroprotective effect of RIPreC. This is not only supported by the inhibition studies previously described but also through observation of the effects of biomolecules downstream of the HIF pathway, such as VEGF, on stroke and recovery (Geiseler et al., 2018). In several human studies, HIF-1α mRNA was upregulated following BFR training compared to control conditions, which included either no intervention or exercise without BFR (Ferguson et al., 2018; Larkin et al., 2012). In addition, it was found that transcription of genes downstream of HIF-1α were also upregulated, including VEGF (Ferguson et al., 2018; Gustafsson et al., 2005; Larkin et al., 2012), endothelial nitric oxide synthase (eNOS) (Ferguson et al., 2018), neuronal nitric oxide synthase (nNOS) (Larkin et al., 2012), and inducible nitric oxide synthase(iNOS) (Larkin et al., 2012). Furthermore, on review of the available literature, no significant alteration (Larkin et al., 2012), and significant increases (Takano et al., 2005) in serum VEGF levels were observed following BFR training compared to exercise without BFR. Interestingly, to the author’s knowledge, there are currently no studies investigating the effects of BFR training on EPO production, another downstream biomolecule of the HIF pathway that has shown mixed results on stroke outcomes (Chan et al., 2017).
Unfortunately, there are no published studies investigating the generation of adenosine or the activation of adenosine A1 receptors following BFR training. Despite the lack of specific literature around adenosine and BFR training, one might speculate that while RIPreC conveys neuroprotection potentially through adenosine A1 receptor activation, BFR training might also lead to the generation of adenosine and the activation of adenosine A1 receptors given it is well established that adenosine is generated during muscular contractions (Marshall, 2007; Simpson et al., 1992). This hypothesis is conceived with the understanding that although some research has been conducted regarding adenosine A1 receptors recently, large gaps remain in the literature, regarding RIPreC, exercise, or BFR training (Stockwell et al., 2017).
Much like the evidence for adenosine A1 receptors, there is little direct evidence to support the notion that Nrf2 is generated during BFR training despite this being a reasonable hypothesis. Indeed, Nrf2 is known to be activated during exercise, and muscular levels of SOD1 are increased following BFR training (Christiansen et al., 2019; Done et al., 2016; Vargas-Mendoza et al., 2019). Taken together, it is likely that Nrf2 and downstream effectors are also activated during BFR training and could contribute to neuroprotection from ROS during stroke, such as that observed following RIPreC. Similarly, there is no published evidence indicating that Notch signaling or NF-κB is upregulated during BFR training. Like other pathways, there is some tentative evidence to suggest that both pathways may be activated. It is well established that TNF-α levels can increase during BFR training (Rossi et al., 2018). As previously mentioned, TNF-α can facilitate the intracellular release of NF-κB through membrane bound TNF receptors. With regard to evidence in the exercise literature, a single mouse study showed an association between exercise and Notch signaling (Mackenzie et al., 2013). Furthermore, there is good evidence for the activation of NF-κB with resistance exercise. Not only does TNF-α and IL-1 increase following strenuous exercise (Pedersen, 2000), it has been shown that NF-κB activity is also upregulated by resistance exercise (Vella et al., 2012).
Finally, as with other pathways, no published studies directly support the activation of the JAK2/STAT3 signaling pathway following BFR training. There is, however, evidence that the pathway is activated in muscle tissue following exercise. For example, it has been observed that phosphorylation and translocation of phosphorylated STAT3 to the nucleus occurs maximally two hours post exercise with cardiovascular and resistance exercise respectively, as determined by muscle biopsy (Fuentes et al., 2012; Trenerry et al., 2007). This evidence, with that provided by RIPreC rat studies, indicates a potential for the JAK2/STAT3 pathway to be activated during BFR training.
BFR and RIPreC-induced immune cell alterations: At the time this review was written, only one study was identified that investigated the alterations of T lymphocytes following the application of BFR training. The authors found that BFR training resulted in an increase in both Th and Tc cells (Souza et al., 2019). This finding is contrary to what has been observed in RIPreC studies, and therefore could indicate a potentially maladaptive immune response for implementation in stroke populations. With everything considered, further studies are required to resolve the effects of BFR training on B lymphocyte numbers.
Conclusion
In conclusion, there is much still to uncover about BFR training. Overall, we know that RIPreC appears to be a promising, safe, adjuvant and preventative treatment, potentially able to reduce detrimental outcomes while also seemingly improving recovery following stroke (Figure 3). With this said, the main issue with RIPreC is that we still know relatively little about the activation of different pathways with this treatment compared to exercise, as demonstrated by a difference in the abundance of literature. There is also a significant knowledge gap around how BFR training impacts human physiology and pathophysiology. Being an active form of RIPreC, the potential for further neurologically beneficial adaptations resulting from BFR training could influence the way that we think about and implement exercise interventions and treatments for stroke populations. Hidden within this lack of knowledge, however, could be either negative or positive outcomes for people living with chronic disease, such as stroke. A cautious approach should be taken with physical interventions, such as through initial trials in healthy populations and the use of animal models, to identify biochemically and immunologically efficacious interventions that could be used in at-risk populations as well as following stroke. This should be done to determine if BFR training activates beneficial pathways known to generate a protective and regenerative effect before being directly applied to clinical populations.
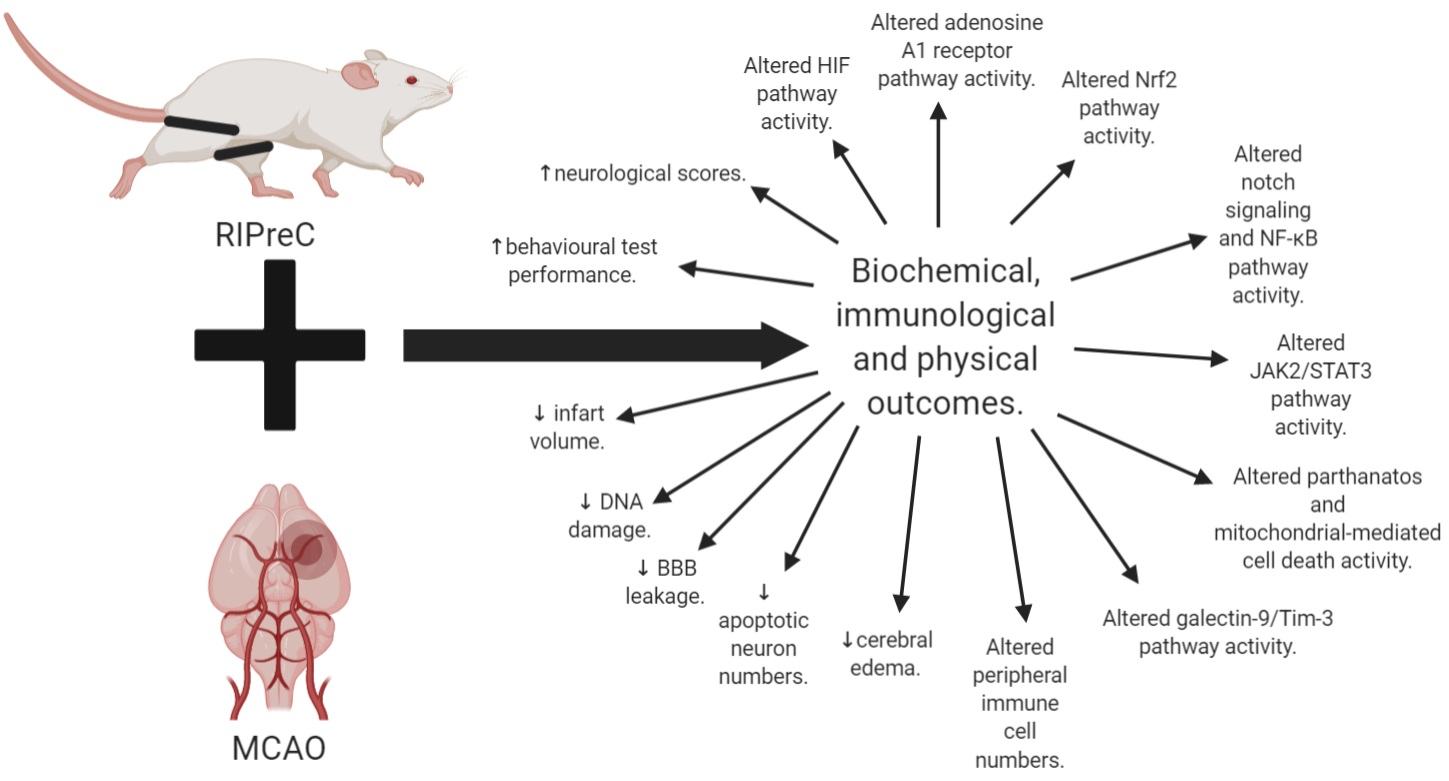
In a new window | Download PPT
Figure 3: Summary of all the biochemical, immunological, and physical outcomes of RIPreC with MCAO as compared to MCAO without RIPreC.
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
Luke J. Schmidt1
1Queensland University of Technology, Tissue Repair and Translational Physiology Group, School of Biomedical Sciences, Faculty of Health, Brisbane, Queensland, Australia.
Daniel A. Broszczak1#
1Queensland University of Technology, Tissue Repair and Translational Physiology Group, School of Biomedical Sciences, Faculty of Health, Brisbane, Queensland, Australia.
Gregory J. Bix2,3
2Clinical Neuroscience Research Center, Department of Neurosurgery, Tulane University School of Medicine, New Orleans, Louisiana, USA. 3Tulane Brain Institute, Tulane University, New Orleans, Louisiana, USA.
Ann M. Stowe4,5#
4Department of Neurology, University of Kentucky, Lexington, Kentucky, USA. 5Department of Neurology and Neurotherapeutics, Peter O’Donnell Jr. Brain Institute, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
Tony J. Parker1
1Queensland University of Technology, Tissue Repair and Translational Physiology Group, School of Biomedical Sciences, Faculty of Health, Brisbane, Queensland, Australia.
#These authors contributed equally to this work.
Corresponding author:
Luke Schmidt
Email: l2.schmidt@qut.edu.au
In a new window | Download PPT
Figure 1: Difference between ischemic conditioning (A) and remote ischemic conditioning (B). Blue cross (A) indicates the application of a device to occlude blood flow.
In a new window | Download PPT
Figure 2: Diagram explaining the various types of RIC in relation to stroke onset.
In a new window | Download PPT
Figure 3: Summary of all the biochemical, immunological, and physical outcomes of RIPreC with MCAO as compared to MCAO without RIPreC.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11770 | 60 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA