Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Hypoxia in the CNS: a focus on multiple sclerosis
Time:2021-04-05
Number:12203
Author Affiliations

Conditioning Medicine 2021. 4(1):3-14.
Abstract
Abstract Multiple sclerosis (MS) is the most common neurological disease affecting the young-middle age population in the Western hemisphere. Pathologically, it is a chronic inflammatory disease in which autoreactive T lymphocytes cross the blood-brain barrier to destroy the myelin insulating sheaths around axons. While accumulating evidence suggests a potential role for hypoxia in disease pathogenesis, interestingly, recent studies in the mouse MS model experimental autoimmune encephalomyelitis (EAE) have demonstrated the protective effect of hypoxic conditioning, both in the form of pre-conditioning and more importantly, when used to treat pre-existing disease. In this review we summarize the findings of these studies with a focus on: (i) the molecular mechanisms underlying the protective effect of hypoxic conditioning, and (ii) the potential clinical implications of these findings.
Keywords: multiple schlerosis, hypoxia, blood vessels, inflammation, blood-brain barrier integrity, angiogenesis
Abstract
Abstract Multiple sclerosis (MS) is the most common neurological disease affecting the young-middle age population in the Western hemisphere. Pathologically, it is a chronic inflammatory disease in which autoreactive T lymphocytes cross the blood-brain barrier to destroy the myelin insulating sheaths around axons. While accumulating evidence suggests a potential role for hypoxia in disease pathogenesis, interestingly, recent studies in the mouse MS model experimental autoimmune encephalomyelitis (EAE) have demonstrated the protective effect of hypoxic conditioning, both in the form of pre-conditioning and more importantly, when used to treat pre-existing disease. In this review we summarize the findings of these studies with a focus on: (i) the molecular mechanisms underlying the protective effect of hypoxic conditioning, and (ii) the potential clinical implications of these findings.
Keywords: multiple schlerosis, hypoxia, blood vessels, inflammation, blood-brain barrier integrity, angiogenesis
Introduction
1. Multiple sclerosis is a heterogenous disease
Multiple sclerosis (MS) is the most common neurological disease affecting the young and middle-aged in the Western hemisphere, with a prevalence of 1.4 in 1000 in North America and 1.1 in 1000 in Europe. It has a gender preference, being 2-3 times more common in women than men (Wingerchuk and Carter, 2014; Brownlee et al., 2017; Doshi and Chataway, 2017). At the clinical level, it presents with a broad spectrum of signs and symptoms including visual disturbance, loss of sensation, weakness, paralysis, and incontinence, as well as cognitive defects, which together, reflect disrupted neural transmission in the visual, motor, sensory, and autonomic pathways. At the pathological level, MS is a chronic inflammatory disease that leads to stripping of the insulating myelin sheath that wraps around axons, a process known as demyelination. If the axons are left unmyelinated for extended periods of time, axonal degeneration ultimately ensues, resulting in permanent disability (French-Constant, 1994; Lassmann, 1998; Compston and Coles, 2008). MS is a highly complex disease, with patients showing tremendous heterogeneity not just in the way their disease manifests, but also in how it progresses and responds to different medications (Wingerchuk and Carter, 2014; Brownlee et al., 2017; Doshi and Chataway, 2017).
To try and understand why so much variation exists between different MS patients, more than twenty years ago, a series of studies examined a large number of MS lesions at the histopathological level and this revealed some insightful findings. Interestingly, while all active MS lesions were characterized by a chronic inflammatory demyelinating process mediated by T lymphocytes and macrophages, the immunological and cell-specific structural components varied significantly between different patients. By careful analysis, four basic patterns were described (Lucchinetti et al., 1996; Lucchinetti et al., 1999, 2000). In the first (pattern I), the demyelinating lesion included inflammatory T cells and macrophages wrapped around blood vessels but showed relative preservation of myelin-producing cells (oligodendrocytes) and no antibody involvement (Brück et al., 1995; Lucchinetti et al., 1996; Gay et al., 1997; Babbe et al., 2000). Pattern II showed similar characteristics to pattern I but also included the accumulation of antibody and complement on degenerating myelin, suggesting the presence of an underlying antibody-mediated pathogenic process (Prineas and Graham, 1981; Storch et al., 1998a; Storch et al., 1998b; Diaz-Villoslada et al., 1999; Genain et al., 1999). Pattern IV was unique in showing oligodendrocyte death within the focus of the demyelinating lesion, but no antibody deposition, suggesting that this type of lesion is caused by a defect in oligodendrocytes, i.e., is a primary oligodendrogliopathy (Lucchinetti et al., 1996). Pattern III was also highly distinct in that it showed little to no inflammation but was described as a “dying back” oligodendrogliopathy, whereby oligodendroglial proteins located at the distal end of oligodendrocyte processes, such as myelin associated glycoprotein (MAG) and cyclic nucleotide phosphodiesterase (CNPase) were completely absent, while myelin proteins concentrated at a more central position on oligodendrocytes (in the compact myelin), such as myelin basic protein (MBP) and proteolipid protein (PLP), remained intact (Itoyama et al., 1980; Ludwin and Johnson, 1981; Lucchinetti et al., 2000). With progression of this type of lesion, the destruction appeared to spread more centrally, leading ultimately to oligodendroglial apoptosis. Because of the progressive nature of type III lesion pathology, it was initially thought that this may represent an early evolutionary stage of all types of MS lesions (Lassmann, 2003; Barnett and Prineas, 2004; Barnett et al., 2009) but later studies that demonstrated the persistence of individual-based different types of MS lesion over time suggests that the type III lesion is a distinct entity in its own right (König et al., 2008; Metz et al., 2014). In light of the unique nature of this “dying back” oligodendrogliopathy, subsequent studies began to analyze whether this type of demyelinating lesion is also present in animal models of MS or in other human neurological diseases. This revealed that while it is not observed in the most widely used animal model of MS, experimental autoimmune encephalomyelitis (EAE), it is a characteristic feature of oligodendrocyte degeneration in the cuprizone demyelination model, in which the toxin cuprizone interferes with energy metabolism within oligodendrocytes, perhaps providing the first clue that deficient energy metabolism might be a potential cause of this “dying back” oligodendrogliopathy (Ludwin and Johnson, 1981). In other human diseases, distal oligodendrogliopathy was also demonstrated in some cases of virus-induced white matter inflammation, including herpes simplex and cytomegalovirus-induced encephalomyelitis (Rodriguez, 1992), as well as the often fatal condition, progressive multifocal leukoencephalopathy (PML) (Itoyama et al., 1982). Most interestingly, distal oligodendrogliopathy is also a robust and consistent feature of acute white matter stroke, raising the possibility that ischemic damage may be the primary trigger resulting in the dying back of oligodendrocytes in type III MS lesions (Aboul-Enein et al., 2003). Further analysis revealed that amongst the different types of MS lesions, only type III lesions show high expression levels of the master regulatory transcription factor, hypoxia-inducible factor (HIF)-1α (Aboul-Enein et al., 2003). Taken together, these findings prompted the suggestion that hypoxia may play a central role in driving white matter inflammation and oligodendrocyte death in MS, at least in the type III subset of MS lesions.
2. Is hypoxia a contributory factor in the pathogenesis of demyelinating disease?
While MS is traditionally described as a chronic inflammatory disease of autoimmune origin in which autoreactive T lymphocytes cross the blood-brain barrier (BBB) to destroy myelin, the precise sequence of events that triggers the autoimmune attack are still unknown. Over the last thirty years, a growing number of studies have provided evidence supporting the idea that hypoxia may be an etiological factor in MS, either as the initial trigger or in maintaining the ongoing inflammatory attack. This was first suggested by the similarity between the dying back oligodendrogliopathy common to type III MS lesions and acute white matter stroke, suggesting that a hypoxia-driven pathogenic mechanism may underlie this type of MS lesion (Lucchinetti et al., 1999, 2000). This concept was supported by the finding that HIF-1α expression was found in oligodendrocytes and other cell types in type III lesions but not in other patterns of MS lesions (Aboul-Enein et al., 2003). Subsequent studies confirmed hypoxic-like injury in the brains of MS patients as well as implicating the inflammatory mediators, reactive oxygen species (ROS) and nitric oxide (NO) in mediating mitochondrial dysfunction (Redford et al., 1997a; Aboul-Enein and Lassmann, 2005). Other studies using microarray analysis demonstrated that MS tissue showed upregulation of genes involved in ischemic pre-conditioning, such as HIF-1α and its target, vascular endothelial growth factor receptor (VEGFR)-1, as well as those involved in mediating neuroprotective responses against oxidative stress, while another described increased levels of nitric oxide synthetase (NOS), NO, and adjacent oligodendrocyte damage, as well as induction of heme oxygenase-1 (Graumann et al., 2003; Zeis et al., 2009). Furthermore, marked suppression of mitochondrial respiratory chain complexes was observed in degenerating axons in MS lesions (Dutta et al., 2006). Based on these observations, it was hypothesized that during progression of the demyelinating lesion, inflammatory cells release ROS and NO, which then inhibit mitochondrial function in demyelinated axons, resulting in an energy-deficient metabolic crisis. This situation was described as “virtual hypoxia,” implying not a specific lack of oxygen per se, but rather an energy-deficient state caused by NO and ROS-induced mitochondrial dysfunction (Trapp and Stys, 2009). These findings have since been substantiated in recent studies and it is now widely accepted that “virtual hypoxia” is an important pathogenic component of the demyelinating lesion (Mahad et al., 2015).
In addition to the contribution of “virtual hypoxia” to MS pathogenesis, during the last decade, a series of studies examining oxygen levels both in the brains of the EAE animals and in living MS patients have definitively demonstrated that a state of true hypoxia, or oxygen deficiency, exists within demyelinating lesions. In the spinal cord of EAE rats, Davies et al. (2013) demonstrated robust hypoxia using implanted oxygen probes and pimonidazole labeling. Interestingly, the level of hypoxia closely correlated with the degree of neurological deficit, with spinal cord pO2 levels falling from 35 mm Hg in control disease-free animals to an average of 20 mm Hg in animals with hindlimb paralysis. Of note, pO2 levels returned to normal during clinical remission but spinal cord tissue became hypoxic again during clinical relapse. This study also made the important observation that pO2 levels in spinal cord white matter were always lower than in gray matter, in keeping with the lower levels of vascularity and greater hypoxic vulnerability demonstrated in these regions (Hassler, 1966; Turnbull et al., 1966; Tveten, 1976; Koyanagi et al., 1993a, b; Halder et al., 2018b).
In follow-up studies, Desai et al. (2016) devised an approach to model the molecular events underlying the early development of an inflammatory demyelinating lesion by focal injection of lipopolysaccharide into the spinal cord. In this model, they detected transient hypoxia at an early timepoint of disease progression within the white-gray matter border and dorsal white column (watershed) areas of the spinal cord and found that this was associated with increased ROS and NO, which over time, led to marked oligodendrocyte loss and demyelination (Desai et al., 2016). These observations led the authors to suggest a model in which activation of innate immune cells within the inflamed spinal cord triggers transient hypoxia in vascular watershed areas, culminating in a state of energy deficiency that leads to oligodendrocyte death and demyelination.
These observations have been substantiated by several key studies from the Dunn laboratory using a variety of novel techniques to demonstrate true hypoxia in demyelinating lesions both in the central nervous system (CNS) of EAE animals and MS patients. By using susceptibility-weighted imaging (SWI), an iron-sensitive magnetic resonance imaging (MRI) method, that is typically used to detect iron deposition in tissues, Nathoo et al. (2013) modified this technique to estimate the levels of deoxyhemoglobin in blood vessels. This showed that SWI lesions were present in the spinal cord and cerebellum of EAE mice but importantly, many of these lesions disappeared following perfusion (removal of blood), indicating that these lesions represent deoxyhemoglobin (i.e., poorly oxygenated blood) within blood vessels (Nathoo et al., 2013). In a follow-up study, the same group showed that by increasing the level of inspired oxygen (from 30 to 100%), the SWI lesions in EAE mice largely disappeared (Nathoo et al., 2015). This implies that the majority of SWI lesions were not due to iron deposition within tissue per se, but rather due to increased levels of deoxyhemoglobin within blood vessels, because when oxygen supply was increased, the oxygen deficit within blood vessels disappeared. This reinforces the concept that a state of true oxygen deficiency exists in blood vessels within demyelinating lesions and suggests that SWI might provide a useful therapeutic tool to estimate the level of deoxyhemoglobin (and thus hypoxia) in MS patients. Additional studies using novel pO2 sensors implanted in the cerebral cortex and cerebellar gray matter of EAE rats demonstrated a strong correlation between the degree of white matter autoimmune inflammation and degree of hypoxia (Johnson et al., 2016). Consistent with these findings, a recent study used frequency domain near-infrared spectroscopy (fdNRS) to measure microvascular hemoglobin oxygen saturation (StO2) in the cerebral cortex of MS patients. This revealed that compared to control subjects, StO2 levels in MS patients were markedly decreased, and a significant relationship between StO2 and clinical disability was demonstrated, strongly supporting the concept of hypoxia as a pathogenic contributor in MS progression (Yang and Dunn, 2015). Taken together, these combined data demonstrate that a true state of hypoxia exists in MS lesions, but also suggest that this may be substantially augmented by a state of virtual hypoxia as a result of ROS and NO-mediated mitochondrial dysfunction.
3. Potential causes of hypoxia in the developing MS lesion
If hypoxia is a potential trigger of MS, then it is important to first understand more about the factors that could lead to this state of hypoxia (see table 1).
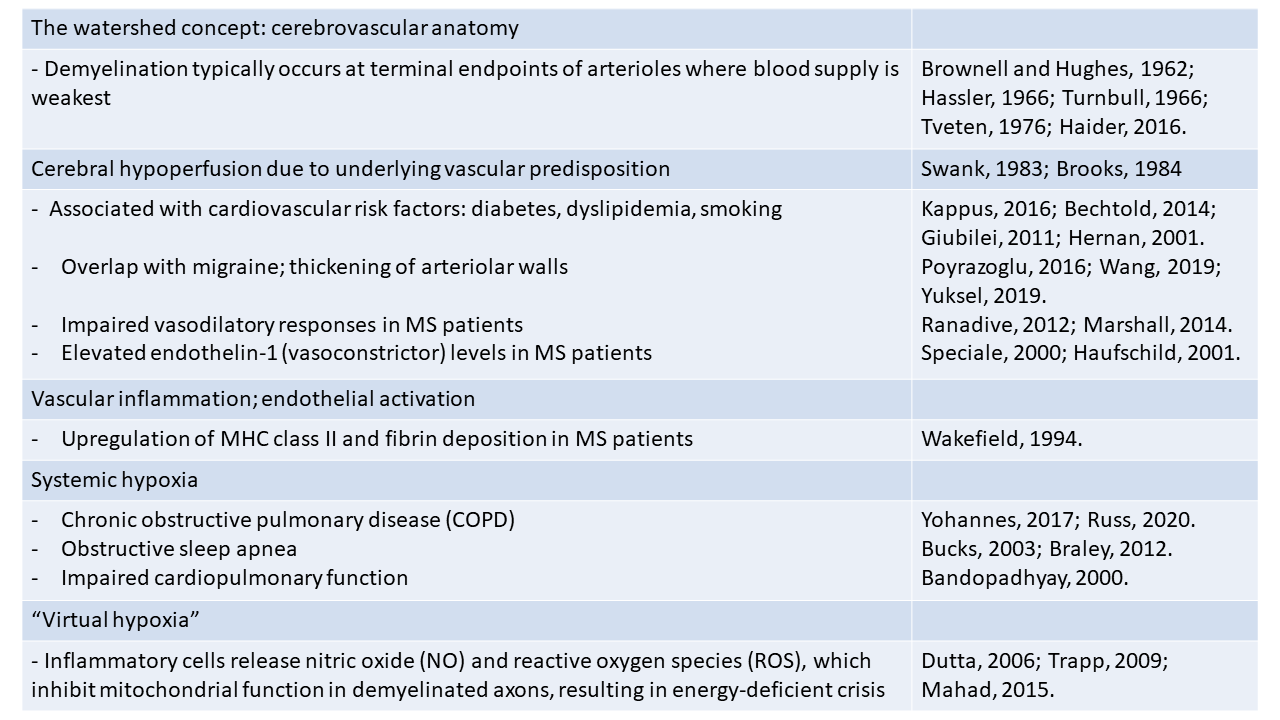
3.1 The watershed concept
Almost sixty years ago, in a study of MS lesions in periventricular regions, Brownell and Hughes (1962) noted that the majority of demyelinating lesions in the brain occur in watershed areas between the anterior, middle, and posterior cerebral arteries, areas that are located at the terminal endpoints of the arterial branches and where the blood supply is at its lowest level (Brownell and Hughes, 1962; Haider et al., 2016). Equally, in the spinal cord, several studies have shown that demyelinating lesions tend to occur in white matter tracts or in regions around the gray-white matter border zone, areas that have the poorest blood supply, i.e., the lowest density of blood vessels (Hassler, 1966; Turnbull et al., 1966; Tveten, 1976; Koyanagi et al., 1993a, b; Halder et al., 2018b). More recently, an MRI study noted that lesions are most often found in cerebral regions that have relatively poor perfusion (Holland et al., 2012). Other studies have reinforced this concept by showing that cerebral perfusion in the brain of MS patients is markedly reduced compared to normal controls (Law et al., 2004; Adhya et al., 2006; Varga et al., 2009; Papadaki et al., 2012). Taken together, these findings support the concept that cerebral hypoperfusion may predispose to the development of MS lesions, and that specific watershed areas in the CNS may be more susceptible because of the inherent limitations of their blood supply. Thus, under normal circumstances, blood supply would be adequate to meet metabolic demands; but, when the blood (or oxygen) supply is reduced, these vulnerable regions would be the first to manifest hypoxic distress. Thus, lack of adequate blood supply predisposes to the development of focal demyelinating lesions.
3.2 Cerebral hypoperfusion due to vascular dysfunction
Strong evidence supports the notion that cerebral blood flow (CBF) is reduced in MS patients. This was first demonstrated more than 30 years ago by a cluster of studies using a combination of single-photon emission computed tomography and positron emission tomography (PET) (Swank et al., 1983; Brooks et al., 1984; Lycke et al., 1993; Sun et al., 1998). More recently, the development of improved imaging analysis using dynamic susceptibility contrast-enhanced perfusion magnetic resonance imaging (DSC-MRI) has enabled investigators to distinguish between demyelinating lesions, normal appearing white matter (NAWM), and grey matter. Importantly, this revealed that CBF is reduced both in NAWM and in deep grey matter in patients with all different types of MS, including relapsing-remitting (R-R), primary progressive, and clinically isolated syndrome (Law et al., 2004; Adhya et al., 2006; Varga et al., 2009; Papadaki et al., 2012). These observations support the concept that cerebral hypoperfusion may be a universal early event in the pathogenesis of all types of MS. A recent MRI study confirmed similar reductions in cervical blood flow in MS patients (El Sankari et al., 2013). This raises the fundamental question: if MS is triggered by reduced CBF, then what is the cause of this reduced blood flow? It seems unlikely to be a result of reduced energy demand due to axonal degeneration because reduced CBF is an early event, being detected well before the relatively late appearance of axonal degeneration (D'Haeseleer et al., 2015).
In light of recent studies highlighting the high degree of comorbidity between vascular disease and MS, and elevated levels of cardiovascular risk factors in MS patients, one possibility is that disruption of vascular function may be a cause (Christiansen et al., 2010; Karmon et al., 2012; Tettey et al., 2014; Marrie et al., 2015; Kappus et al., 2016). In one study of children and teenagers, the relative risk of developing MS was found to be much higher in patients with preexisting type 1 diabetes than a control group (Bechtold et al., 2014). Dyslipidemia is also implicated by the finding that elevation of both total cholesterol and low-density lipoprotein cholesterol are strongly associated with both the first clinical episode of MS as well as a worsening of clinical status (Giubilei et al., 2002; Weinstock-Guttman et al., 2011; Weinstock-Guttman et al., 2013). Other reports have shown that another cardiovascular risk factor, cigarette smoking, is strongly associated with increased risk of MS in a dose-dependent manner (Hernán et al., 2001; Hedström et al., 2013; O'Gorman et al., 2014). Importantly, MS patients show a higher incidence of ischemic stroke (Capkun et al., 2015; Tseng et al., 2015; Hong et al., 2019). Taken together, these combined data support the idea that vascular pathology may be an important pre-disposing factor in MS pathogenesis.
In an alternative explanation, it’s interesting to compare MS with another neurological disease, migraine, which also shows a female:male incidence ratio of 3:1, as well as predominantly affecting the young-middle age population (Burch et al., 2018). Interestingly, several studies have pointed to the clinical overlap between migraine and MS (Elliott, 2007; Applebee, 2012), with the incidence of migraine 3-fold higher in MS patients, and the presence of migraine being a predictor of a more symptomatic MS course (Kister et al., 2010). Other studies have suggested that migraine with aura may be an early symptom of MS and/or an indicator of exacerbation of MS symptoms (Tabby et al., 2013). In addition, migraineurs have an elevated risk of developing MS compared to the control population (Kister et al., 2012). In migraine pathophysiology, a long-held belief is that defective autoregulation of CBF may be responsible (Blau, 1978; Lauritzen, 1987) while other studies have described intimal thickening in the cerebral arterioles of migraine patients (Poyrazoglu et al., 2016; Wang et al., 2019). So, could the reduced CBF in MS patients be the result of abnormalities in structure or function of cerebral blood vessels? In support of this concept, studies have demonstrated intimal thickening of cerebral arteries (Yuksel et al., 2019) as well as stiffer retinal arterioles in MS patients (Kochkorov et al., 2009), while at the functional level, impaired vasodilatory responses in cerebral arterioles and reduced levels of reactive hyperemia have been described in MS patients (Ranadive et al., 2012; Marshall et al., 2014).
At the molecular level, one mechanism that may link neuroinflammation and reduced blood flow is based on the finding that MS patients show impaired vasodilatory responses in cerebral arterioles (Marshall et al., 2014). Endothelin-1 (ET-1) is a vasoconstrictive peptide that is normally produced by endothelial cells but not glial cells (Piechota et al., 2010). Interestingly, ET-1 levels in peripheral blood and cerebrospinal fluid (CSF) are elevated in MS patients (Speciale et al., 2000; Haufschild et al., 2001), and ET-1 expression, normally absent in resting astrocytes, is upregulated in reactive astrocytes (Nie and Olsson, 1996; Ostrow et al., 2000). In support of this concept, a recent study showed that CBF in MS patients was reduced by approximately 20% in many brain regions and importantly, pharmacological blockade of the ET-1 receptor restored CBF to control levels (D'Haeseleer et al., 2013). Based on these findings, one mechanism that has been proposed is that early activation of astrocytes in MS lesions triggers ET-1 expression, which enhances arteriolar constriction, thus reducing CBF (D'Haeseleer et al., 2013).
3.3 Vascular inflammation
The idea that vascular dysfunction may be an early pivotal trigger of MS pathogenesis is further supported by the key observation that in acute demyelinating MS lesions, endothelial cell activation, as indicated by upregulation of MHC class II and fibrin deposition, is observed prior to parenchymal reaction or demyelination (Wakefield et al., 1994). Endothelial activation leads to upregulation of adhesion molecules such as selectins, vascular cell adhesion molecule (VCAM)-1, and intercellular adhesion molecule (ICAM)-1, which promote T lymphocyte adherence to the luminal side of endothelial cells (Engelhardt et al., 1994; Lee and Benveniste, 1999). This in turn, results in aggregation of lymphocyte clusters within the lumen of microvessels, that secrete a spectrum of pro-inflammatory cytokines and chemokines that further promote leukocyte extravasation and amplify the inflammatory cascade. Infiltrated leukocytes also secrete proteases, such as matrix metalloproteinase-9 (MMP-9), which can strip endothelial cells of critical cell surface proteins necessary for maintaining tight barrier integrity, e.g., tight junction proteins and adherens proteins (Redford et al., 1997b; Milner et al., 2007; Boroujerdi et al., 2015). In addition, the inflammatory environment can also promote release of vasoactive peptides, which impact vascular function by inhibiting homeostatic vasodilatory responses, leading to constriction of blood vessels and reduced blood flow. A good example is the vasoconstrictive peptide endothelin-1 (ET-1), which is strongly upregulated by reactive astrocytes, and whose vasoconstrictive action reduces CBF (D'Haeseleer et al., 2013; D'Haeseleer et al., 2015). Blood vessels within inflammatory regions can also be targeted for damage and destruction by several different components of the immune system (cytokines, antibodies or CD8+ T lymphocytes), or by virtue of their heightened activation state, inflamed blood vessels can trigger local activation of the clotting cascade resulting in thrombotic occlusion and ischemia (Tomasson et al., 2009; Emmi et al., 2015). This is because inflammation promotes a pro-thrombotic state as a result of downregulation of anti-thrombotic proteins such as thrombomodulin and upregulation of thrombotic factors, such as tissue factor and thrombin (Kopp et al., 1997; Esmon, 2005, 2012; Foley and Conway, 2016).
Furthermore, at the earliest stage of demyelinating lesion development, the BBB is disrupted (McDonald and Barnes, 1989; Kermode et al., 1990; Gay and Esiri, 1991; Colosimo et al., 1992; Kirk et al., 2003) resulting in extravascular leakage of serum proteins into the CNS, which perturbs tissue fluid homeostasis in the local environment. The ensuing localized edema compresses cerebral microvessels, resulting in ischemia and hypoxic tissue damage. In the clinical setting, BBB disruption is evaluated using gadolinium-enhanced MRI, which provides one of the earliest markers of impending tissue damage and is currently the best prognostic tool for monitoring the appearance of new active MS lesions (McDonald and Barnes, 1989; Colosimo et al., 1992; McDonald, 1994; Compston and Coles, 2008).
3.4 Systemic hypoxia
In addition to being the result of reduced blood flow, tissue hypoxia can also occur when blood flow is relatively normal, but the body is in a state of systemic hypoxia. Many different medical conditions can lead to a hypoxic state, including obstructive sleep apnea, several types of lung disease, including acute (bronchitis, pneumonia) and chronic (asthma, pulmonary fibrosis, emphysema) as well as reduced cardiac function (Bandopadhyay and Selvamurthy, 2000; Yaffe et al., 2011; Bucks et al., 2013; Leng et al., 2017; Yohannes et al., 2017; Russ et al., 2020). When systemic hypoxia occurs, areas of the CNS with the poorest blood supply (watershed areas) will be most vulnerable and will therefore be the first to display signs of hypoxia. Interestingly, epidemiological studies have highlighted a strong association between sleep apnea and MS, in that sleep apnea is much more common in MS patients compared to control subjects (Braley et al., 2012), prompting the question: what accounts for the increased incidence of sleep apnea in MS patients? However, considering the emerging evidence implicating hypoxia as a potential trigger of MS, it might be equally insightful to ask the reverse question.
4. The protective effect of hypoxic conditioning in animal models of demyelinating disease
Inspired by overwhelming evidence demonstrating the strong neuroprotective influence of hypoxic pre-conditioning in animal models of ischemic stroke (Dowden and Corbett, 1999; Miller et al., 2001; Stowe et al., 2011), the Dore-Duffy lab was the first to examine whether hypoxic pre-conditioning might also reduce the clinical severity of EAE. Using a chronic progressive EAE model (MOG35-55 peptide immunogen in C57BL6/J mice), they demonstrated that mice maintained under chronic mild hypoxic conditions (10% O2) from the time of immunization (i.e., before EAE was manifest), showed reduced levels of neurological disability, which correlated with reduced levels of leukocyte infiltration into the spinal cord (Dore-Duffy et al., 2011). In a subsequent study, the same group found that chronic mild hypoxia (CMH) reduced the degree of CD4+ T lymphocyte infiltration into the spinal cord and this correlated with a delayed Th17-specific cytokine response. Conversely, spinal cords of CMH-treated mice contained increased numbers of CD4+/CD25+/FoxP3+ regulatory T cells (Tregs) as well as higher levels of the anti-inflammatory cytokine IL-10 (Esen et al., 2016). Based on these findings, the authors concluded that CMH triggers a number of endogenous adaptations that promotes the establishment of an anti-inflammatory milieu within the CNS.
In a previous series of studies, we demonstrated that CMH (8-10% O2) promotes favorable changes in cerebrovascular properties that includes a marked angiogenic remodeling response, resulting in enhanced vessel density as well as marked upregulation of endothelial tight junction proteins, suggestive of increased vascular integrity (Milner et al., 2008; Li et al., 2010; Boroujerdi and Milner, 2015; Halder et al., 2018b). Based on these findings, we hypothesized that CMH might confer protection from EAE, in part via its positive influence on vascular integrity. In contrast to the Dore-Duffy lab, we chose to perform our hypoxic conditioning studies in the relapsing-emitting (R-R) EAE model (PLP139-151 peptide immunogen in SJL/J mice) for two reasons; first, this model is more representative of the most common form of MS seen in patients (85% get the R-R form), and second, this model facilitates analysis of an intervention not just on peak disease but also on the extent of remission, and on rates of relapse. Strikingly, we found that when started at the same time as EAE induction, CMH significantly reduced the severity of peak disease (by more than 50%), with protection maintained for the duration of the experiment (7 weeks) (Halder et al., 2018a). This was matched at the histological level by reduced levels of BBB breakdown, leukocyte infiltration, and demyelination. Tissue analysis revealed that hypoxia-enhanced BBB stability correlated with reduced expression of the endothelial activation leukocyte-docking molecules, vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1, as well as increased expression of the endothelial tight junction proteins occludin and ZO-1. CMH also triggered increased levels of the extracellular matrix (ECM) protein laminin-111 in the vascular basement membrane, which has been shown to inhibit leukocyte transmigration (Sixt et al., 2001). Based on these findings, we concluded that the protective effect of hypoxic pre-conditioning in EAE is underpinned, at last in part, by several different changes in blood vessel properties that contribute to enhanced vascular integrity (Halder et al., 2018a).
More recently, we investigated the clinically important question of whether CMH can impact the progression of pre-existing EAE. Using the R-R EAE model, we found that when mice with significant neurological disability were treated with CMH, while this had no impact on the peak disease score, CMH markedly accelerated clinical recovery during the remission phase of disease, resulting in long-term stable reductions in neurological deficit (Halder and Milner, 2020). At the histological level, this correlated with marked reductions in vascular disruption, leukocyte accumulation and demyelination within the spinal cord. Mechanistically, we found that CMH reduced endothelial expression of VCAM-1 while promoting faster re-expression of the tight junction proteins occludin and ZO-1 following their disappearance during the peak phase of disease. Most interestingly, while CMH did not influence the level of peak clinical disease or the degree of inflammatory leukocytosis at peak disease (3 days after CMH started), apoptotic removal of infiltrated leukocytes during the remission phase was profoundly enhanced, correlating with marked clinical improvement. Within the EAE spinal cord, HIF-1α was strongly expressed in infiltrated monocytes and the number of HIF-1α-positive monocytes was greatly increased by CMH. Taken together, these findings are consistent with our previous data in suggesting that CMH confers protection from EAE, in part by enhancing different mechanisms contributing to vascular integrity. They also make the important point that CMH greatly accelerates monocyte apoptotic removal within acute inflammatory lesions by amplifying the hypoxic stress manifest within these cells.
5. Which molecular mechanisms account for the protective effect of hypoxic conditioning?
The studies described to date have identified three potential mechanisms that may underlie the protective effect of hypoxic conditioning on inflammatory demyelinating disease: (i) modulation of the peripheral immune system, (ii) enhanced BBB integrity, and (iii) accelerated apoptotic removal of infiltrated monocytes (see Figure 1).

In a new window | Download PPT
Figure 1: Summary of the proposed molecular mechanisms underlying the protective influence of hypoxic conditioning in MS. These include: (1) promoting vascular remodeling, which will accelerate vascular repair following leukocyte-induced breakdown as well as increase removal of waste products via increased vascularity, (2) enhanced vascular integrity due to upregulation of tight junction proteins and vascular basal lamina ECM protein expression (e.g., laminin), (3) promoting an anti-inflammatory environment within the CNS by shifting the balance between pathogenic Th17 encephalitogenic T cells and immunosuppressive regulatory T cells, and (4) promoting apoptosis of infiltrated monocytes.
5.1 The influence of hypoxia on the immune system
In their first study, documenting the protective effect of CMH on the progression of EAE, the Dore-Duffy lab correlated reduced clinical disease with reduced leukocyte infiltration into the spinal cord (Dore-Duffy et al., 2011). In their second study, they correlated this protection with reduced numbers of CD4+ lymphocyte infiltration into the spinal cord, which was associated with a decreased destructive Th17-specific response, but interestingly, they also noted increased numbers of immuno-suppressive regulatory T cells (Tregs) and increased levels of the anti-inflammatory cytokine interleukin (IL)-10 within the spinal cord (Esen et al., 2016). Based on these observations, the authors concluded that by tilting the Th17/Treg balance in favor of Tregs, CMH triggers an anti-inflammatory environment within the CNS. When considering how this might be achieved, three possible explanations spring to mind; first, that CMH exerts an anti-inflammatory effect on the whole immune system and what occurs in the spinal cord is simply a reflection of what is happening elsewhere in the body, second, that CMH alters the ability of the BBB to selectively regulate which types of immune cell enter the CNS, and third, that the balance between Th17 and Treg cells is regulated at the local level within the CNS.
The influence of hypoxia on cells of the immune system is complex and depends on several different factors, including the immune cell type, severity and duration of hypoxia, as well as the inflammatory context. Broadly speaking, hypoxia stimulates the function of many innate immune cell types such as monocyte and neutrophils, as seen by increased chemotaxis and phagocytosis, and enhanced production of ROS and pro-inflammatory cytokines such as IL-6, which all serve to promote clearance of pathogens (Scannell et al., 1993; Klausen et al., 1997; Hartmann et al., 2000; Wang and Liu, 2009; Kiers et al., 2016). However, the impact of hypoxia on lymphocyte function is less clear. One study demonstrated that hypoxia drives T cell differentiation towards the Th17 effector pathway and away from Treg differentiation, an effect that was substantiated by the finding that transgenic mice with HIF-1-deficient T cells are resistant to EAE induction due to defective generation of Th17 effector cells (Dang et al., 2011). On the other hand, another group studying these events in intestinal mucosa showed that hypoxia tilts the balance in favor of Tregs, and that HIF-1α deletion resulted in reduced numbers of Tregs and a failure to control T cell-mediated inflammatory colitis (Clambey et al., 2012). Interestingly, Esen et al. (2016) showed that in the EAE model, systemic hypoxia had no impact on MOG-specific T cell sensitization or proliferation, suggesting that T cell antigen-specific functions per se are not altered by hypoxia. Clearly, more studies are required to clarify how hypoxia modulates the Th17/Treg balance during EAE and to understand whether this is a direct effect on T cells or an indirect effect via the influence of hypoxia on other CNS-resident cell types.
5.2 The hypoxic influence on vascular remodeling and vascular integrity
It is well established that CMH induces a robust vascular remodeling response in the CNS, resulting in enhanced vascularity as well as upregulation of several components of the BBB (Milner et al., 2008; Li et al., 2010; Boroujerdi and Milner, 2015; Halder et al., 2018b). Interestingly, in our recent study demonstrating the therapeutic benefit of hypoxic treatment of pre-existing EAE, we showed that the hypoxia-induced angiogenic response proceeds, even in the middle of a full-blown inflammatory response seen in EAE (Halder and Milner, 2020). Surprisingly, this vascular remodeling did not lead to more BBB disruption as might be predicted when endothelial cells uncouple from their neighbors during the angiogenic process, but instead resulted in enhanced vascular integrity. So how can vascular remodeling be beneficial in EAE? We think three potential reasons might account for this. First, one possibility is that when EAE occurs on a pro-angiogenic background, because the angiogenic process is already up and running, any blood vessels disrupted because of leukocyte infiltration, will be more quickly repaired and sealed. This idea is supported by our recent finding that transgenic mice with endothelial deletion of the angiogenic integrin α5β1 (α5-EC-KO mice), which show a delayed angiogenic response to CMH (Li et al., 2012), manifest earlier clinical disease along with greater BBB disruption in the EAE model (Kant et al., 2019).
Second, it’s plausible that the increased vessel density improves the perfusion of affected tissue as well as speeding the removal of any waste/myelin breakdown products or inflammatory cytokines that are facilitating the inflammatory cycle. This idea is consistent with previous studies in MS patients, which demonstrated that enhanced vasodilation and increased blood flow triggered by factors such as histamine and amyl nitrate provided short-term clinical relief of symptoms, described as “relief by flush” (Jonez, 1948; Brickner, 1955; Brickner, 1958). It would also be in keeping with the “clean up” hypothesis, whereby it is suggested that angiogenesis is promoted under inflammatory conditions in part, as an attempt to remove tissue debris (Manoonkitiwongsa et al., 2001).
The third possibility is that CMH promotes BBB integrity. In a series of studies in non-EAE conditions, we have shown that CMH enhances both endothelial expression of tight junction proteins on CNS blood vessels, as well as increasing expression of the leukocyte-inhibitory ECM protein laminin within the vascular basal lamina (Li et al., 2010; Boroujerdi and Milner, 2015; Halder et al., 2018b; Halder et al., 2018c). Our recent work demonstrates that CMH triggers the same mechanisms during EAE progression (Halder et al., 2018a; Halder and Milner, 2020). In addition, CMH also reduces expression of the endothelial activation molecules VCAM-1 and ICAM-1 (Halder et al., 2018a; Halder and Milner, 2020), suggesting that CMH acts to dampen the vascular inflammatory response induced by EAE, while simultaneously reducing leukocyte extravasation across the BBB. Indeed, in our recent study describing the protective influence of CMH on pre-existing EAE, the impact on suppressing endothelial VCAM-1 expression was the earliest detectable event in CMH-treated mice (Halder and Milner, 2020). These findings are consistent with the marked impact of CMH on reducing vascular disruption and preventing leukocyte infiltration into the CNS.
5.3 Hypoxia accelerates the apoptosis of infiltrated leukocytes
One of the most notable findings from our recent EAE study was that hypoxic treatment of pre-existing disease strongly enhanced apoptotic removal of infiltrated monocytes from the spinal cord during the remission phase (Halder and Milner, 2020). Interestingly, in comparison to CD4+ T cells, which were evenly distributed throughout the entire spinal cord, monocytes accumulated in high-density cellular aggregates within the white matter and displayed the highest levels of HIF-1α expression and apoptotic cell death. This is consistent with the idea that after entering the CNS parenchyma, infiltrated monocytes continue to proliferate and transform into activated, highly metabolic cells, to create high-density aggregates where hypoxia is most severe (Le Moan et al., 2015; Halder and Milner, 2020). Based on this, it is logical to assume that CMH will greatly enhance the hypoxia already experienced by these monocytes, and as suggested by previous studies, will trigger apoptotic pathways (Biju et al., 2004; Krick et al., 2005; Luo et al., 2006). This concept is supported by the observation that inhibition of caspase-mediated apoptosis prevents clinical remission in EAE (Okuda et al., 2000). Taken together, this data suggests that approaches designed to enhance monocyte hypoxia or apoptosis by other means (e.g., activating caspase-3 activation) could offer new ways forward in promoting clinical recovery in MS.
6. Does hypoxic conditioning hold therapeutic potential in MS?
As animal EAE studies have demonstrated the clinical efficacy of hypoxic conditioning, not just in pre-conditioning, but also more recently in the treatment of pre-existing disease (Esen et al., 2016; Halder et al., 2018a; Halder and Milner, 2020), how might this be exploited for therapeutic potential? Two possible approaches are worth considering. First, a growing number of reports in other neurological diseases including ischemic stroke, Alzheimer’s disease, spinal cord injury and epilepsy, have demonstrated the therapeutic potential of intermittent hypoxic training (IHT), which consists of regular, short hypoxic exposures (Fuller et al., 2003; Golder and Mitchell, 2005; Manukhina et al., 2010; Stowe et al., 2011; Zhen et al., 2014). Because CMH promotes long term adaptive changes in vascular (enhanced vascularity and vascular integrity) (Milner et al., 2008; Li et al., 2010; Boroujerdi and Milner, 2015; Halder et al., 2018b) and immunological properties (Esen et al., 2016), it’s possible that these changes may provide benefit in treating MS. Before this is ready to test in MS patients however, it will first be necessary to demonstrate proof of the IHT concept in EAE studies, something that has not yet been examined, as well as define an optimal IHT protocol that would provide maximum benefit. Second, rather than using hypoxia itself, drugs called hypoxia mimetics could be used to stimulate hypoxic signaling pathways. These drugs inhibit the prolyl hydroxylase domain (PHD) group of enzymes, which prevents hydroxylation of HIF-1α, thus raising HIF-1α levels (Jain et al., 2016). Interestingly, recent studies have demonstrated that the cannabinoid VCE-004.8, which has hypoxic mimetic activity, reduces the severity of EAE (Navarrete et al., 2018), suggesting promising clinical potential for this therapeutic approach.
7. Conclusions
Recent studies have demonstrated that hypoxic conditioning is effective in reducing clinical severity in the EAE animal model of MS, not just as pre-conditioning, but more importantly, when used to treat pre-existing disease. Mechanistic studies have suggested that this protection is mediated by beneficial adaptive changes in the properties of blood vessels and the immune system. More studies are now needed to further clarify these changes and define the key intracellular signaling pathways responsible. At the same time, current data suggests it may be fruitful to explore the therapeutic potential of this phenomenon by examining the impact of intermittent hypoxic training (IHT) and hypoxia mimetics in EAE.
References
Sebok K. Halder 1
1San Diego Biomedical Research Institute, 10865 Road to the Cure, Suite 100, San Diego, CA 92121, USA.
Richard Milner 1
1San Diego Biomedical Research Institute, 10865 Road to the Cure, Suite 100, San Diego, CA 92121, USA.
Corresponding author:
Dr. Richard Milner
Email: rmilner@sdbri.org
In a new window | Download PPT
Figure 1: Summary of the proposed molecular mechanisms underlying the protective influence of hypoxic conditioning in MS. These include: (1) promoting vascular remodeling, which will accelerate vascular repair following leukocyte-induced breakdown as well as increase removal of waste products via increased vascularity, (2) enhanced vascular integrity due to upregulation of tight junction proteins and vascular basal lamina ECM protein expression (e.g., laminin), (3) promoting an anti-inflammatory environment within the CNS by shifting the balance between pathogenic Th17 encephalitogenic T cells and immunosuppressive regulatory T cells, and (4) promoting apoptosis of infiltrated monocytes.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 12203 | 36 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA