Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Mitochondria as the memory of preconditioning
Time:2021-08-21
Number:7920
Author Affiliations

Conditioning Medicine 2021. 4(3): 151-160.
Abstract
Preconditioning is such a paradigm that a stimulus below the threshold of causing harm makes the brain stronger and resilient to subsequent injury. Preconditioning affords a vigorous tolerance to the brain against neurodegeneration. Numerous efforts have tried to identify the molecular targets involved in preconditioning-induced protective responses and interestingly many of those diverse mechanisms posit mitochondria as a master regulator of preconditioning. Therefore, in this review, we will critically discuss recent and emerging evidence for the involvement of mitochondria within the preconditioning paradigm. We will introduce the crucial targets and signaling cascades by which mitochondria exert preconditioning with a focus on white matter mitochondria and whether and how mechanisms for preconditioning differ in neurons and glial cells. In this aspect, we will evaluate the role of mitochondrial shaping proteins to establish structure-function interdependence for fusion-fission balance, motility, ATP production, Ca+2, and ROS scavenging. We will also discuss how aging impacts mitochondria and the consequences of mitochondrial aging on preconditioning mechanisms. We will concentrate on the regulation of mitochondrial DNA content and quantification specifically for its value as a biomarker to monitor disease conditions. The identification of these mitochondrial preconditioning mechanisms can be translated to potential pharmacological interventions to increase intrinsic resilience of the brain to injury and to develop novel approaches to neurodegenerative diseases. Moreover, mitochondria dynamics can be used as a memory or biomarker of preconditioning.
Keywords: Mitochondria, White matter, Aging, Preconditioning, Biomarker
Abstract
Preconditioning is such a paradigm that a stimulus below the threshold of causing harm makes the brain stronger and resilient to subsequent injury. Preconditioning affords a vigorous tolerance to the brain against neurodegeneration. Numerous efforts have tried to identify the molecular targets involved in preconditioning-induced protective responses and interestingly many of those diverse mechanisms posit mitochondria as a master regulator of preconditioning. Therefore, in this review, we will critically discuss recent and emerging evidence for the involvement of mitochondria within the preconditioning paradigm. We will introduce the crucial targets and signaling cascades by which mitochondria exert preconditioning with a focus on white matter mitochondria and whether and how mechanisms for preconditioning differ in neurons and glial cells. In this aspect, we will evaluate the role of mitochondrial shaping proteins to establish structure-function interdependence for fusion-fission balance, motility, ATP production, Ca+2, and ROS scavenging. We will also discuss how aging impacts mitochondria and the consequences of mitochondrial aging on preconditioning mechanisms. We will concentrate on the regulation of mitochondrial DNA content and quantification specifically for its value as a biomarker to monitor disease conditions. The identification of these mitochondrial preconditioning mechanisms can be translated to potential pharmacological interventions to increase intrinsic resilience of the brain to injury and to develop novel approaches to neurodegenerative diseases. Moreover, mitochondria dynamics can be used as a memory or biomarker of preconditioning.
Keywords: Mitochondria, White matter, Aging, Preconditioning, Biomarker
Introduction
Since its first introduction in 1964 (Janoff, 1964) the concept of preconditioning in the brain has been widely accepted as a small dose of noxious stimulus to afford neuroprotection responses against subsequent injury (Dirnagl et al., 2003). In other words “Was mich nicht umbringt macht mich starker” (Friedrich Nietzsche’s Twilight of the Idols 1888), “what does not kill me makes me stronger. In fact, hypoxia, ischemia, small doses of endotoxins, intermittent fasting, caloric restriction, exercise, stem cells, and drugs have all been shown to evoke tolerance to future injuries (Cadet and Krasnova, 2009; Bernstock et al., 2017; Li et al., 2017; Zhang et al., 2019; Cozene et al., 2020; Kinoshita et al., 2021; Vemuganti and Arumugam, 2021). Surprisingly adaptation to one stimulus provides resilience to the other, which underlies the essence of “cross-tolerance”. Depending on the onset of the effect, three different phases of preconditioning are de ned as: a) immediate preconditioning occurs within minutes after the preconditioning stimulus and involves cellular changes related to the activity of function of the enzymes, secondary messengers, and ion channels, b) delayed preconditioning takes several hours or days to generate new gene expression and protein synthesis (Barone et al., 1998; Kirino, 2002; Gidday, 2006) and a recent addition is c) chronic conditioning, which is characteristic of remote ischemic conditioning (RIC). Alternatively, depending on the timing of the treatment, three types of preconditioning protection can be described: 1) preconditioning when applied before the onset of the injury, 2) per-conditioning when the treatment with sub lethal ischemia occurs during ischemia and before the reperfusion (Hahn et al., 2011), and finally 3) post conditioning if the conditioning occurs during the reperfusion (Zhao et al., 2003). Trials in stroke in which single neuroprotective pathways were targeted for activation have been unsuccessful, so alternative strategies that target multiple pathways are attractive, and preconditioning the brain with RIC is one such feasible approach. Ideally, the conditioning stimulus provides protection not only when triggered before ischemia but also when applied during ischemia or reperfusion after ischemia. Note that, preconditioning may be limited to clinical conditions in which ischemia is predictable such as before surgery, high-risk patients with a history of transient ischemic attack, or cardiac problems prone to emboli. However, the protective effects of ischemic preconditioning spans across many species and organs including heart, lung, brain, intestines, and kidneys (Murry et al., 1986; Kitagawa et al., 1991; Ates et al., 2002; Waldow et al., 2005; Chen et al., 2008; Abu-Amara et al., 2011).
Almost after 60 years (Janoff, 1964), our information on the complex molecular mechanisms for the induction and maintenance of preconditioning induced brain tolerance remains largely undefined. However, mitochondrial-mediated mechanisms seem to mediate an important portion of the preconditioning response. Increasing evidence support the proposal that brief exposure to physiological or pathological stimuli, cellular events, pharmacological applications, and remote interventions induce mitochondrial changes that ultimately protect neurons against a variety of lethal insults (Duchen, 2004; Perez-Pinzon et al., 2005; Hess et al., 2015). A precise sequence of events occur where a change in mitochondrial function leads to energy preservation and manifests itself as preconditioning induced neuroprotection. This sequence of events has been successfully observed in vitro and in vivo cerebral ischemia models (Duchen, 2004; Perez-Pinzon et al., 2005). These experiments demonstrated that antioxidants and mitochondrial adenosine triphosphate (ATP)-sensitive potassium channel (mito-KATP) blockers abolish preconditioning induced protection in cerebral ischemia, and an important role for mitochondrial reactive oxygen species (ROS) generation and mito-KATP channel activation were among the first pathways established in the preconditioning phenomenon (Vanden Hoek et al., 1998; Oldenburg et al., 2002). Indeed, these findings initiated the investigation of numerous approaches to test and characterize preconditioning mechanisms.
Mitochondria proved to be a merging integrator of preconditioning mediated endogenous neuroprotection. Therefore, this review summarizes the structural and functional characteristics of “preconditioning mitochondria” with a focus on necessary vs. sufficient conditions to precondition that were presented and discussed during the 2021 Virtual Conditioning Medicine Workshop by the American Association of Conditioning Medicine. We will also highlight whether preconditioning mitochondria show the location and age- related specificity by comparing neuronal, axonal, and glial preconditioning requirements taking into account the impact of age by considering aging mitochondria. Finally, we will discuss whether preconditioning mitochondria and their mitochondrial DNA content can be used as a biomarker to predict disease onset, prognosis, and the effect of treatment on disease course. We propose that long-lasting changes in mitochondria structure and function serve as a memory of preconditioning and as a result, a better description and understanding of the role of preconditioning mitochondria will help in the development of novel, effective, and more targeted therapeutic strategies against neurodegenerative diseases.
Preconditioning Mitochondria
Mitochondria are dynamic organelles that produce ~90% of the ATP via the tricarboxylic acid cycle and oxidative phosphorylation, regulate intracellular Ca2+ and redox signaling, and command apoptosis (Green and Kroemer, 2004; Beal, 2005; Mattson et al., 2008). Consequently, mitochondria are considered as the “gatekeeper” for cell viability and death. Therefore, it is not surprising that mitochondria play a pivotal role in the preconditioning phenomenon for many pathological conditions, foremost neurodegeneration (Cho et al., 2010) and stroke (Piquereau et al., 2013).
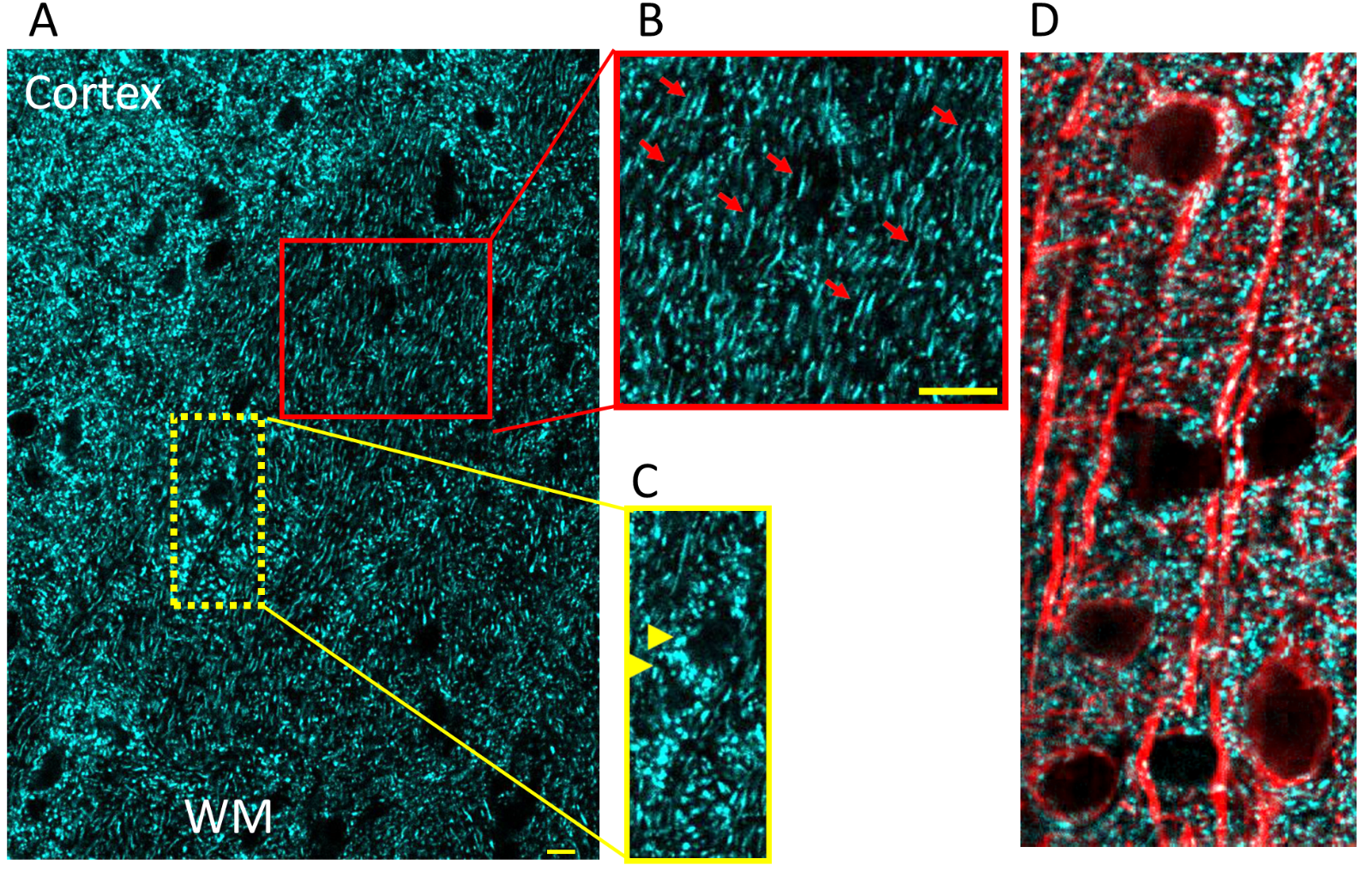
As dynamic organelles, mitochondria continuously undergo fission and fusion while carefully maintaining their structural integrity and motility. Mitochondrial length is determined by the balance between the rates of mitochondrial fission and fusion and is important for controlling the spatiotemporal properties of mitochondrial responses during physiological and pathophysiological processes (Szabadkai and Duchen, 2008). The precise balance between fission and fusion is equally an important process for mitochondria to adapt their shape speci c to cellular compartments within a cell (Figure 1, A to D). For instance, in neurons with their nerve processes mitochondria assume an elongated tubular structure in axons (Figure 1B, red arrows) while in neuronal cell bodies they switch to a rounder smaller shape (Figure 1C, yellow arrows). This rapid transient change in shape is mostly dictated by cellular activity and metabolic demand so that ATP production, Ca2+, and ROS regulation can be efficiently controlled under physiological conditions. The processes of fusion and ssion also contribute to the response of mammalian cells to stress such that fusion is stimulated by energy demand and stress, while ssion generates new organelles and facilitates quality control (Frank et al., 2001; Skulachev, 2001; Tondera et al., 2009). Mitochondrial fission is facilitated by the translocation of dynamin-related protein 1 (Drp-1) from cytosol to mitochondria (Reddy et al., 2011; Hatch et al., 2014; Flippo and Strack, 2017; Bastian et al., 2018), while mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) on the mitochondrial outer membrane, and optic atrophy gene 1 (Opa1) on the mitochondrial inner membrane are essential for mitochondrial fusion. A pathological event such as stroke causes extensive fission and fragmentation, leading to smaller-sized dysfunctional mitochondria (Figure 2) that generate excessive amounts of ROS and Ca2+, with reduced levels of ATP leading to irreversible neuronal and axonal structural and functional loss. Note that mitochondrial structural disintegration is a continuous process that allows an opportunity to interrupt and improve mitochondrial fate (Figure 2). For instance, pharmacological blockade of Drp-1 with mitochondrial division inhibitor 1 (Mdivi-1) is shown to be protective in several tissues such as heart, kidney, retinal ganglion cells, spinal cord, and in cerebral ischemia-reperfusion models (Brooks et al., 2009; Ong et al., 2010; Park et al., 2011a; Grohm et al., 2012; Liu et al., 2015). Therefore, whether regulation of mitochondrial fusion-fission balance underlies mitochondria-mediated preconditioning has been of great interest. Indeed, studies in neuronal cells established the concept that Mdivi-1, which prevents translocation of cytosolic Drp-1 into mitochondria and thus inhibits ssion, (Kim et al., 2017; Valenti et al., 2017) confers preconditioning to neuronal function in gray matter (GM) (Ravati et al., 2000; Ravati et al., 2001; Dirnagl and Meisel, 2008; Jou, 2008; Dirnagl et al., 2009). These studies also set the expectation for the visual description of preconditioning mitochondria that is “dressed for preconditioning” with a size and shape appropriate for the cellular anatomic location.
In a new window | Download PPT
Figure 1: (A, C) Mitochondria in neuronal cell bodies (yellow arrow heads) and (B) nerve processes (red arrows); bar = 10 μm. (D) Combining cyan fluorescent protein (CFP) with microtubule associated protein 2 creates useful images in which to contrast mitochondrial morphology in the perinuclear region of cell bodies as compared with neuronal processes. A "preconditioned mitochondria" is expected to conform to the size and shape of its cellular location.
In a new window | Download PPT
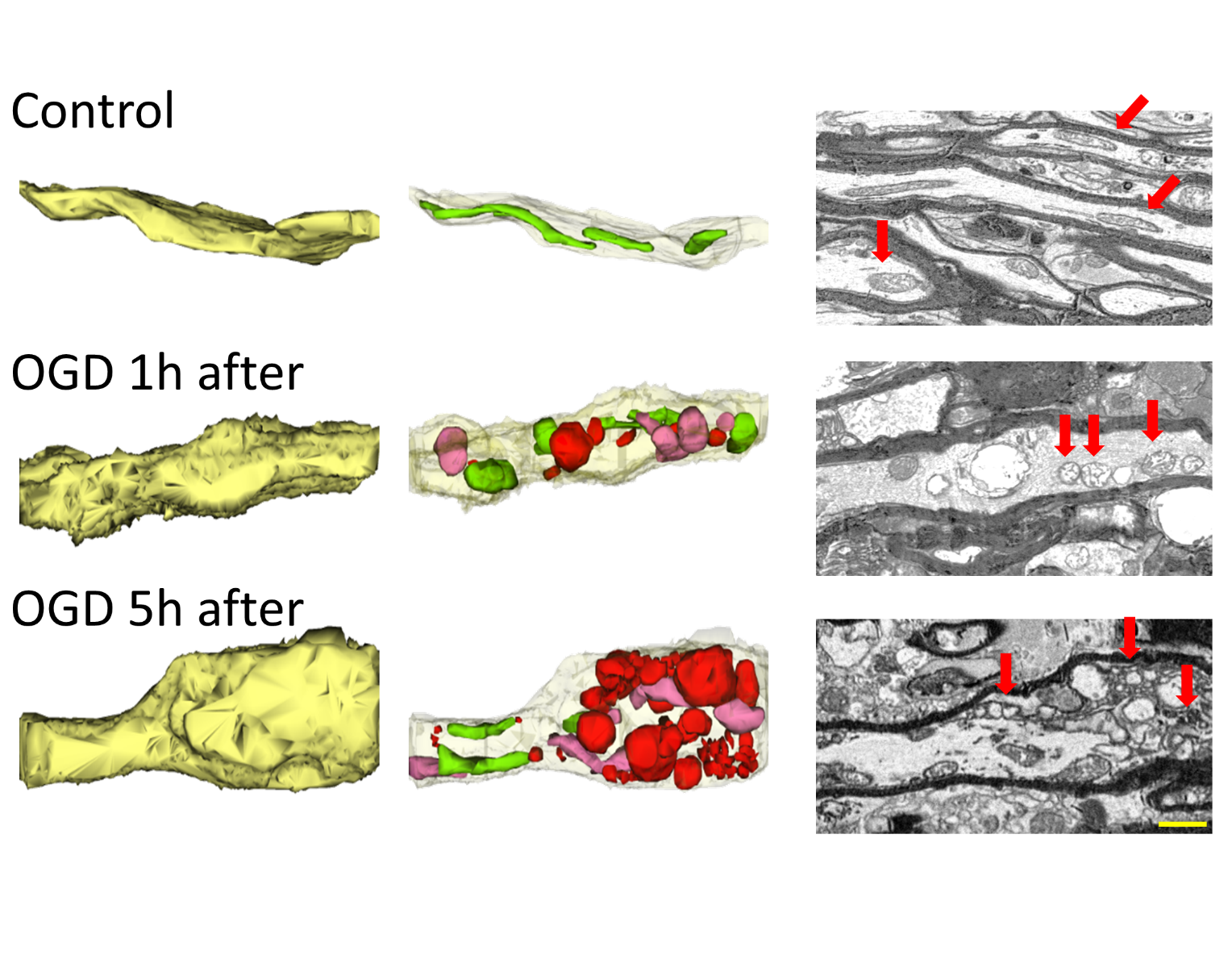
Figure 2: Three-dimensional construction of control axons (yellow, left top) with their elongated green mitochondria (middle top) and the two- dimensional electron micrographs of myelinated axons with numerous elongated mitochondria with their cristea (red arrows). One hour after oxygen-glucose deprivation (OGD 1h after, middle left) axons are swollen and mitochondria are fragmented (midline, middle, in colors) as seen as circular swollen hollow structures with loss of cristae. The OGD induced mitochondrial destruction progresses with time such that axons become visibly deformed with bulging sections (OGD 5h after, bottom left) characterized with mitochondria further disintegrating into smaller fragments (bottom middle and left). A "preconditioned mitochondria" is expected to remain elongated and tubular in axons akin to control conditions. Scale bar = 1 μm.
Identification of recent molecular mechanisms of preconditioning from the cancer and cardiac eld independently confirmed the concept that inhibition of mitochondrial fission is a merging target that underlies preconditioning. For instance, studies by Dr. Jurgen Bernhagen (2021 Virtual Conditioning Medicine Workshop) provided compelling evidence that macrophage migration inhibitory factor (MIF) could be a key player mediating ischemic preconditioning.
Following the expression and localization of mitochondrial dynamics associated proteins after MIF inhibition showed excessive fission due to upregulation of Drp-1 translocation into mitochondria, correlated with robust downregulation of Opa-1 and Mfn-1 levels (De et al., 2018) resulting in extensive mitochondrial fission. These structural alterations resulted in lower levels of ATP production, increased mitochondrial pore opening, and depolarization of transmembrane potential leading to mitochondrial dysfunction.
A more recent paradigm of self-protection proposed by Drs. Xunming Ji, Davis Hess, and Derek Hauser (2021 Virtual Conditioning Medicine Workshop) revealed that remote ischemic conditioning (RIC) can recruit neuroprotective and cardioprotective pathways with high potential for a wide variety of patients suffering from cerebral ischemia, cerebral hemorrhage, and cardiac ischemia. An exciting aspect of this preconditioning paradigm is that the RIC can involve the third phase of chronic conditioning, which implies protection when applied, before during, or after the injury. The leading candidates for this remote signaling to the brain include stromal cell-derived factor (SDF-1), which is activated by hypoxia and involved in stem cell traf cking, interleukin 10 (IL-10), micro ribonucleic acid (miRNA)-144, and nitrite. Most pathways that are triggered by RIC converge on the mitochondria such that blood-borne factors stimulate G-protein coupled receptors on the cell surface, which induce intracellular kinase signaling that leads to the opening of KATP preventing the formation of the mitochondrial permeability transition pore. Subsequently, inhibition of KATP abrogates the RIC-induced preconditioning. The mitochondria are also a subcellular target of nitrite, which is involved in key nitrosylation of key mitochondrial proteins such as complex I and IV. As a result, the generation of mitochondrial ROS declines to protect mitochondrial structure and function. Clinical trials testing the role of RIC in patients with subarachnoid hemorrhage, intracranial atherosclerosis, and ischemic heart conditions will also help identify the targets for pharmacological manipulations for preconditioning (Hess et al., 2015).
Since studies that were performed in white matter (WM) provided interesting results emphasizing the differences in the role of neuronal, axonal, and glial mitochondria in preconditioning (see below), whether RIC offers mitochondria- induced preconditioning in WM components remain to be investigated.
Preconditioning Mitochondria in WM
Mechanisms of ischemic preconditioning have been extensively studied in GM. An ischemic episode affects both the GM and WM portions of the brain (Mohr et al., 2011) and faithful axon conduction is crucial for signaling among neuronal cell bodies to connect GM and WM to achieve and maintain proper function. However, the effect of preconditioning on WM axons, myelin, and glial cells has received much less attention compared to neurons, presumably assuming that preconditioning of neuronal cells bodies may extend to axons and suf ce to protect the entire brain. Although axons are anatomical extensions of neuronal cell bodies, injury mechanisms differ between GM and WM such that approaches for neuronal protection fail to improve, or even impede, axon function recovery after ischemia (Tekkok et al., 2007; Baltan, 2009, 2012; Baltan et al., 2014). In addition, axons are independent of their neuronal soma in terms of their energy status; consequently to assume that preconditioning of neuronal soma spreads and induces similar tolerance in axons is a misleading extrapolation. Inhibition of mitochondrial ssion has been shown to induce preconditioning tolerance to neurons in vivo (Park et al., 2011b; Xie et al., 2013; Zhang et al., 2013a; Zuo et al., 2014; Jin et al., 2016; Kim et al., 2016; Deryagin et al., 2017) and in vitro (Correia et al., 2010; Wang et al., 2014). Ischemic preconditioning has been reported to protect WM against ischemic injury, (Hamner et al., 2015) however whether preservation of mitochondria can precondition WM against ischemic injury remains to be investigated.
The optic nerve is a pure isolated WM tract and provides a compelling in vitro preparation to tests injury mechanisms specific to WM components where axons, myelin, and glial cells in their three-dimensional configuration maintain their cell to cell signaling. This in vitro preparation addresses the concern that rodent brains contain only 10% WM by volume, as opposed to the human brain (50% WM by volume) (Zhang and Sejnowski, 2000) and thus, the response to ischemia is dominated by neuronal injury in rodent brain. Similar to neuronal cell bodies, mitochondria in myelinated axons undergo rapid and profuse fission during oxygen-glucose deprivation (OGD) that is mediated by translocation of cytoplasmic Drp-1 to mitochondria (Bastian et al., 2018). OGD-induced mitochondrial fission correlates with reduced mitochondrial motility and loss of axon function. Mitochondrial fragmentation and loss of motility become permanent during the recovery period (Bastian et al., 2018). Mdivi-1 is a small molecule that is readily permeant through the blood-brain barrier and it provides ischemic tolerance to neurons by maintaining mitochondrial integrity and function. Expectedly, administering Mdivi-1 to WM during OGD preserves mitochondrial shape and motility and promotes axon functional recovery. However, despite the Mdivi-1 effect on preconditioning neuronal cell bodies (Grohm et al., 2012; Zhang et al., 2013b; Wang et al., 2014; Cui et al., 2016), in WM Drp-1 inhibition fails to offer ischemic preconditioning tolerance to myelinated axons and fails to benefit axon function (Bastian et al., 2018). These findings suggest that inhibition of mitochondrial ssion during ischemia is necessary to promote axon functional recovery but is not sufficient to precondition myelinated axons against ischemia. These results raise caution in that approaches to preconditioning neuronal cell bodies may not successfully translate into functional improvement following ischemia.
The Mdivi-1 application also modifies mitochondrial dynamics as evidenced by the enhanced mitochondrial motility in myelinated axons (Bastian et al., 2018). This increase in motility is preserved during OGD, albeit to a smaller extent compared to Mdivi-1 application during ischemia. However, the enhanced motility of mitochondria vanishes during the recovery period. This suggests that mechanisms of fission and mitochondrial motility are interconnected. Mitochondrial motility along microtubules is regulated by protein complexes, of which ATP and the Ca2+-dependent protein mitochondrial Rho GTPase-2 (Miro-2) play a major role (Guo et al., 2005; Saotome et al., 2008; Russo et al., 2009; Melkov et al., 2016) and is intricately linked to the mitochondrial ssion protein Drp- 1 (Saotome et al., 2008). Miro-2 forms complexes with kinesin for anterograde mitochondrial transport (Guo et al., 2005; Russo et al., 2009) and dynein for retrograde mitochondrial transport (Russo et al., 2009; Morlino et al., 2014; Melkov et al., 2016) along microtubules. Miro-2 is increasingly associated with neurodegenerative diseases that are characterized by mitochondrial dysfunction (Tang, 2015). Dysregulation of Miro-2 leads to mitochondrial arrest in movement and clearance (Wang et al., 2011). Miro-2 also affects both anterograde (Macaskill et al., 2009; Wang and Schwarz, 2009) and retrograde motility (Morlino et al., 2014) and the fusion- fission dynamics of mitochondria (Misko et al., 2010; Tang, 2015). Live imaging studies of mitochondria in optic nerves show that axonal mitochondria move bi-directionally, change direction, or become stationary in response to OGD (Bastian et al., 2018). Mitochondria are dynamic organelles whose coordinated motility ensures that metabolically active areas of axons are adequately supplied with ATP. Moreover, injured mitochondria are replaced with healthy ones following injury. Kymograph analysis of mitochondrial motility illustrates that under control conditions, most mitochondria are still, and those that move maintain a stable speed. The onset of OGD stalls mitochondrial motility to 50% of baseline levels both in the anterograde and retrograde directions and preservation of mitochondrial motility against OGD is an important predictor of axon function recovery (Bastian et al., 2018). For instance, electrophysiology and live mitochondrial imaging studies propose that nitric oxide synthase 3 inhibition promotes axon functional recovery by preventing mitochondrial ssion and by preserving mitochondrial structure and motility during ischemia by conserving Miro-2 levels. Based on the interplay between mitochondrial motility and mitochondrial ssion, it is plausible that motile mitochondria are an indicator of better functional recovery. In particular, the extent of mitochondrial motility during recovery guides functional recovery. The downstream molecular mechanisms of this protection are currently under investigation such as whether Miro-2 and Drp-1 interact to precondition mitochondria in WM axons. In summary, conserving mitochondrial structure sustains mitochondrial integrity and motility during ischemia, however, this approach fails to precondition axon function against ischemia, in contrast to its protective effects on neuronal survival. These findings support the notion that manipulations to precondition the brain should consider interventions to be bene cial for both the gray and WM portions of the brain.
The protective effect of astrocytes on neurons has been shown to include mitochondrial transfer, although the protective role of preconditioning astrocytes and astrocyte mitochondria has been reported more recently (Wu et al., 2021). In astrocyte and neuron co-culture systems, prior hypoxic preconditioning of astrocytes improves astrocyte mitochondrial structure and function through peroxisome proliferator-activator receptor 1 alpha (PGC-1α)/hypoxia-inducible factor (HIF) signaling, leading to increased astrocyte viability, reduced oxidative stress, and greater neuroprotection. The proposed mechanism is that the hypoxia-activated PGC-1α/HIF signaling improves mitochondrial biogenesis in astrocytes, which subsequently lowers neuronal apoptosis after OGD (Wu et al., 2021). Another interesting approach to preconditioning astrocytes is the use of resveratrol (RPC), which activates nuclear erythroid 2 related factors (Nrf2). Nrf2 localizes to the outer membrane in astrocyte mitochondria and its activation increases mitochondrial abundance, glycolysis, and mitochondrial respiration efficiency. Therefore, the contribution of Nrf2 to RPC-induced neuroprotection through maintaining astrocyte mitochondrial coupling and anti-oxidant protein expression exerts neuroprotection during cerebral ischemia (Narayanan et al., 2015; Khoury et al., 2019). Expectedly, in transgenic mice without Nrf2 (Nrf2-/-) the loss of neuroprotection correlates with induction of altered supercomplex formation in mitochondria and injury to neurons (Narayanan et al., 2018) while transgenic mice overexpressing Nrf2 is more resilient to neurotoxicity (Calkins et al., 2010). Preconditioning using a combination of mild oxidative stress with glucose deprivation (OSGD), further con rms that astrocyte survival and astrocyte- mediated neuroprotection is activated via the Nrf2 pathway. Nrf2 can enhance IL-10 expression, therefore mild OSGD preconditioning of astrocytes confer neuroprotection, with the participation of IL-10 to manipulate mitochondria and reduce oxidative injury (Segev-Amzaleg et al., 2013).
Nrf2 is also expressed in oligodendrocytes (Licht-Mayer et al., 2015) and ischemic preconditioning (medial cerebral artery occlusion for 12 min) promotes the generation of oligodendrocyte precursors (OPCs) and oligodendrogenesis by differentiation of OPCs in the striatum, corpus callosum, and external capsule, mediated by Nrf2 signaling. Knocking down Nrf2 blocks oligodendrogenesis induced by ischemic preconditioing (Li et al., 2021). In oligodendrocytes, sodium azide treatment inhibits mitochondria metabolism, results in depolarization of mitochondria membrane, decreases metabolic activity, and increases cytotoxicity. Endoplasmic reticulum stress induces Nrf2 hyperactivation, which partially rescues the mitochondrial metabolism inhibition induced by sodium azide. Furthermore, knocking down Nrf2 expression in oligodendrocytes worsens mitochondrial dysfunction regulating oligodendrocyte survival against injury (Liessem- Schmitz et al., 2018). Surprisingly, cell culture studies using primary OPCs reveal that prior exposure of OPCs to sublethal OGD as a mode of preconditioning results in enhanced vulnerability to subsequent excitotoxicity or to OGD mediated by down-regulation of the α-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid receptor subunit glutamate receptor 2 on the cell surface that subsequently increases Ca2+ permeability (Deng et al., 2003). In neonatal rat brains, hypoxic injury reduces the amount of myelin basic protein without affecting the early or late OPCs in the corpus callosum. Interestingly, hypoxic preconditioning prevents the loss of myelin caused by a hypoxic injury without affecting the density of OPCs, suggesting that hypoxic preconditioning directly protects myelin or by promoting the maturation of pre oligodendrocytes to regenerate the damaged myelin (Suryana and Jones, 2014). Further studies investigating preconditioning of oligodendrocytes and their mitochondria and whether preconditioning oligodendrocytes support myelin and axon function are yet to be designed.
Aging Mitochondria and Preconditioning
Age-related reduction of preconditioning was rst demonstrated in the isolated and perfused rat heart from 24-month-old rats subjected to an ischemic preconditioning with a short period of ischemia (2 minutes) followed by 10 minutes of reperfusion. Interestingly, caloric restriction and physical activity can restore preconditioning in aged hearts in both animals and humans (Abete et al., 2001). Reduction of “brain” preconditioning mediated protection in the aged brain may be due to the composite mechanisms that characterize the aged brain, ie, loss in many neurons, impairment in mitochondrial function with an increase in ROS production, alteration in gene expression and metabolic regulation, and alteration in intracellular Ca2+ homeostasis. All these modifications make the organ more susceptible to stress such as ischemia (Shankar, 2010). In particular, a gene expression pro le following middle cerebral anterior occlusion shows reduced transcriptional activity, proapoptotic genes, and downregulation of axonogenesis and neurogenesis in the periinfarct area, which is more pronounced in aged versus young animals (Budas et al., 2007). These ndings suggest that an aging brain is capable of upregulating gene expression, but the response is often blunted and temporally uncoordinated in response to cerebral ischemia (Della-Morte et al., 2012).
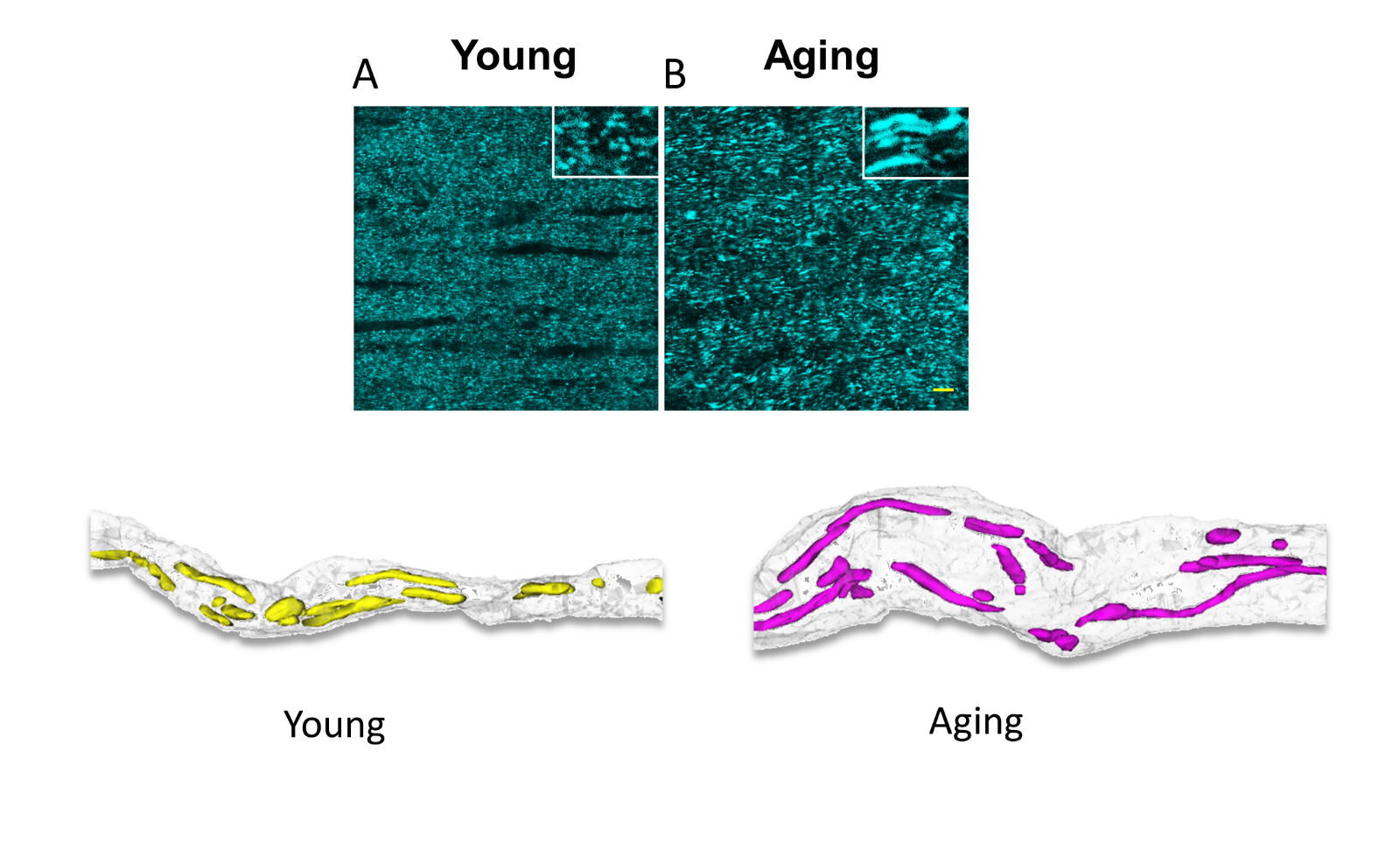
The impact of aging on central nervous system (CNS) WM is of particular interest because it appears that the global effects of aging on myelinated axons are more complex and profound than those in GM (Peters, 1999). Myelinated nerve fibers in WM deteriorate structurally and functionally with age, leading to impaired communication between different parts of the CNS. The underlying reasons for increased susceptibility with age to stroke, dementia, Alzheimer's disease, or other neurodegenerative diseases are just beginning to be explored; therefore, it is important to separate between WM structural and functional changes that are due to natural aging and pathological changes associated with neurodegenerative diseases. Mitochondrial dysfunction and oxidative damage with age are gaining increasing interest as merging targets that underlie increased risk for stroke, AD, PD, and different forms of dementia. Indeed fewer, but longer and larger mitochondria produce increased oxidative stress markers and less ATP in aging axons. The processes of fusion and ssion contribute to mitochondrial quality control and the response of mammalian cells to stress such that fusion is stimulated by energy demand and stress, while ssion generates new organelles and facilitates quality control (Frank et al., 2001; Skulachev, 2001; Tondera et al., 2009). In WM, aging axons have fewer mitochondria than young axons, but they are thicker and longer (Baltan, 2014; Stahon et al., 2016) (Figure 3). This age-dependent expansion of mitochondrial morphology correlates with a mismatch among mitochondrial shaping proteins such that increases in levels of fusion proteins, for example Mfn-1 and Mfn-2, and decreases in fission protein Drp-1 levels result in conditions that favor mitochondrial fusion in aging axons (Baltan, 2014; Stahon et al., 2016). Fused mitochondria is an adaptation in response to the lower ATP levels observed in aging axons and results in shared components, thereby helping to maintain energy output during stress (Westermann, 2010). However, this also results in reduced mitochondrial numbers, which when combined with increased axonal volume results in parts of the axon being without mitochondria (Stahon et al., 2016) (Figure 3). The number of mitochondria directly correlates with the level of cellular metabolic activity. An interruption in mitochondrial dynamics due to a mismatch in mitochondrial shaping proteins results in reduced ATP production in aging axons. In addition, aging axons also show increased levels of oxidative stress markers (4-hydroxynonenal, 3-nitrotyrosine, and nitric oxide), which can lead to mitochondrial impairment and resulting stress (Stahon et al., 2016). Morphological alterations compromise mitochondrial function and the resultant reduced energy production and increased oxidative stress underlie the increased vulnerability of WM to an ischemic attack. Yet, aging axons adapt to transmit signals faithfully under control conditions as reported previously (Baltan et al., 2008; Baltan et al., 2010). Then the question of whether it is possible to precondition the aging brain raises substantial interest, particularly in the face of a rise in the aging population and how to support brain function in health and disease. Agents able to mimic the “cerebral” preconditioning effect may represent a new and powerful therapeutic option for the treatment of acute ischemic stroke in the elderly. Further studies are needed to establish the age- related reduction of brain preconditioning in the aging brain and translate these discoveries to clinical practice.
In a new window | Download PPT
Figure 3: Aging increases the length and thickness of mitochondria as seen in CFP (+) mitochondria in optic nerves obtained from Thy-1 mito-CFP mice (upper two panels). Three-dimensional construction of electron micrograph of young and aging axons demonstrate that age causes a prominent increase in axon diameter correlated with a drastic increase in mitochondrial length, thickness, and volume. Note the areas devoid of mitochondrial coverage in aging axons. A "preconditioned aging mitochondria" is speculated to maintain size and shape comparable to normal aging conditions. Scale bar = 1 μm.
Mitochondria as a Biomarker for Preconditioning
Each mitochondrion can contain several copies of the mitochondrial genome (Veltri et al., 1990; Cavelier et al., 2000; Navratil et al., 2008), and changes in mitochondrial DNA (MtDNA) content are observed in physiological and pathological conditions, for instance in aging brain increase in MtDNA content has been reported (Barrientos et al., 1997). Under normal conditions, the maternally inherited mitochondrial genome in an individual will be the same in all cell types unless it has been damaged (Chinnery, 1993; Moraes et al., 2003). The number of mitochondria in a cell varies depending on the energetic requirements of the cell, for example, a brain cell may have around 2000 mitochondria (Uranova et al., 2001), a white blood cell may have less than a hundred (Selak et al., 2011), whereas oocytes may contain several hundred thousand mitochondria (Piko and Matsumoto, 1976; Duran et al., 2011). The number of mitochondria in a particular cell type can also vary depending on many factors, including the stage in the cell cycle, the environment and redox balance of the cell, the stage of differentiation, and several cell signaling mechanisms (Rodriguez-Enriquez et al., 2009; Michel et al., 2012).
As mitochondria contain their own DNA outside of the nuclear genome, a convenient way to measure mitochondrial DNA content in a cell is to measure mitochondrial versus nuclear genome ratio, called Mt/N (Malik et al., 2009; Malik et al., 2011). This approach is attractive, as the methodology for quantifying nucleic acids with real-time quantitative polymerase chain reaction (RT-qPCR) has become conventional (Gourlain et al., 2003; Andreu et al., 2009; Malik et al., 2011). Changes in MtDNA content could be used as a biomarker to detect mitochondrial dysfunction. To determine if Mt/N is a consistent biomarker of mitochondrial dysfunction, it is important to validate methods for accurate and reproducible measurement of cellular MtDNA content. Previous studies using RT-qPCR have tended to use primers for estimation of nuclear genome content from control genes previously used for mRNA quantification, such as β-actin and 18S rRNA. The rationale for using these genes for mRNA quantification is that they are presumed to have “housekeeping” functions and are expected to be expressed at equal levels in all cells. However, when quantifying copy numbers from genomic DNA these genes are not the best choice as many of the regions being amplified are not single copies (Zhang and Gerstein, 2003; Malik et al., 2011). The currently recommended stable gene that shows a low level of variability is β-2 microglobulin (Malik and Czajka, 2013).
Mt/N is attractive as a putative biomarker because it can be measured in as little as 1 pg of genomic DNA (Malik et al., 2011), thereby requiring very little clinical sample. Subsequently, MtDNA content changes have been reported using DNA isolated from various body fluids such as circulating blood cells, cell-free serum, saliva, sperm, urine, and cerebrospinal fluid and also in tissue samples such as tumor tissue and various organs and biopsy materials indicating common interest in measuring Mt/N in different body uids and tissues in numerous human diseases, and during development, and aging. The use of body fluids as surrogate markers for changes in organs is a feasible option as tissues and organs are not easily accessible. As blood cells are in contact with the whole body and organs they may reflect changes in mitochondrial function/dysfunction.
Despite the growing interest in MtDNA content as a putative biomarker in numerous diseases, there are several conflicting studies and there is little consensus on how to interpret the results due to significant problems associated with the accuracy of measurement of MtDNA with current methodology. Furthermore, the development of assays for measuring the integrity of MtDNA is needed as current methods for Mt/N quantification do not distinguish between intact mitochondrial genomes and damaged MtDNA fragments. Emerging quantification systems such as digital PCR may offer the accuracy needed for Mt/N determination (Baker, 2012). Therefore, in conclusion, MtDNA could be a novel biomarker for mitochondrial dysfunction. In the presence of oxidative stress, ROS would lead to increased mitochondrial biogenesis resulting in increased MtDNA throughout the whole body and this change would be reflected in circulating cells. Accumulation of damaged MtDNA may directly contribute to pathology by eliciting a cellular anti-inflammatory response. Therefore, according to this hypothesis, an increase in MtDNA may precede mitochondrial dysfunction as an adaptive response and could therefore be a predictive biomarker. Hence, utilizing the changes in MtDNA after induction of preconditioning as a biomarker for preconditioning is a novel concept and warrants of further investigation. These studies are yet to be designed.
Conflicts of Interest
The authors declare that they have no con icts of interest.
Acknowledgments
This work was supported by grants from the National Institute of Aging (NIA, AG033720) and the National Institute of Neurological Diseases (NINDS, NS094881) to S.B. We thank Dr. Chris Nelson for help editing this paper.
References
Sarah Zerimech1
1Anesthesiology and Peri-Operative Medicine (APOM), Oregon Health and Science University, Portland, Oregon 97239.
Hung Nguyen1
1Anesthesiology and Peri-Operative Medicine (APOM), Oregon Health and Science University, Portland, Oregon 97239.
Selva Baltan1
1Anesthesiology and Peri-Operative Medicine (APOM), Oregon Health and Science University, Portland, Oregon 97239.
Corresponding author:
Selva Baltan
Email: baltan@ohsu.edu
In a new window | Download PPT
Figure 1: (A, C) Mitochondria in neuronal cell bodies (yellow arrow heads) and (B) nerve processes (red arrows); bar = 10 μm. (D) Combining cyan fluorescent protein (CFP) with microtubule associated protein 2 creates useful images in which to contrast mitochondrial morphology in the perinuclear region of cell bodies as compared with neuronal processes. A "preconditioned mitochondria" is expected to conform to the size and shape of its cellular location.
In a new window | Download PPT
Figure 2: Three-dimensional construction of control axons (yellow, left top) with their elongated green mitochondria (middle top) and the two- dimensional electron micrographs of myelinated axons with numerous elongated mitochondria with their cristea (red arrows). One hour after oxygen-glucose deprivation (OGD 1h after, middle left) axons are swollen and mitochondria are fragmented (midline, middle, in colors) as seen as circular swollen hollow structures with loss of cristae. The OGD induced mitochondrial destruction progresses with time such that axons become visibly deformed with bulging sections (OGD 5h after, bottom left) characterized with mitochondria further disintegrating into smaller fragments (bottom middle and left). A "preconditioned mitochondria" is expected to remain elongated and tubular in axons akin to control conditions. Scale bar = 1 μm.
In a new window | Download PPT
Figure 3: Aging increases the length and thickness of mitochondria as seen in CFP (+) mitochondria in optic nerves obtained from Thy-1 mito-CFP mice (upper two panels). Three-dimensional construction of electron micrograph of young and aging axons demonstrate that age causes a prominent increase in axon diameter correlated with a drastic increase in mitochondrial length, thickness, and volume. Note the areas devoid of mitochondrial coverage in aging axons. A "preconditioned aging mitochondria" is speculated to maintain size and shape comparable to normal aging conditions. Scale bar = 1 μm.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 7920 | 16 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA