Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The underlying mechanisms involved in the protective effects of ischemic postconditioning
Time:2018-02-21
Number:9584
Author Affiliations

Conditioning Medicine, 2018. 1(2):73-79.
Abstract
Cerebral ischemic postconditioning (PostC) refers to a series of brief ischemia and reperfusion (I/R) cycles applied at the onset of reperfusion following an ischemic event. PostC has been shown to have neuroprotective effects, and represents a promising clinical strategy against cerebral ischemia-reperfusion injury. Many studies have indicated that cerebral PostC can effectively reduce neural cell death, cerebral edema and infarct size; improve cerebral circulation; and relieve inflammation, apoptosis and oxidative stress. In addition, several protective molecular pathways such as Akt, mTOR and MAPK have been shown to play a role in PostC-induced neuroprotection. PostC represents an attractive therapeutic option because of its ability to be induced rapidly or in a delayed fashion, as well as being inducible by pharmacological agents. As a potential clinical treatment, PostC is therapeutically translatable as it can be induced remotely. The underlying mechanisms of PostC have been systematically investigated, but still need to be comprehensively analyzed. As most PostC studies to date were conducted preclinically using animal models, future studies are needed to optimize protocols in order to accelerate the clinical translation of PostC.
Key Words: postconditioning; cerebral I/R injury, neuro-protection, mechanism, molecular pathway
Abstract
Cerebral ischemic postconditioning (PostC) refers to a series of brief ischemia and reperfusion (I/R) cycles applied at the onset of reperfusion following an ischemic event. PostC has been shown to have neuroprotective effects, and represents a promising clinical strategy against cerebral ischemia-reperfusion injury. Many studies have indicated that cerebral PostC can effectively reduce neural cell death, cerebral edema and infarct size; improve cerebral circulation; and relieve inflammation, apoptosis and oxidative stress. In addition, several protective molecular pathways such as Akt, mTOR and MAPK have been shown to play a role in PostC-induced neuroprotection. PostC represents an attractive therapeutic option because of its ability to be induced rapidly or in a delayed fashion, as well as being inducible by pharmacological agents. As a potential clinical treatment, PostC is therapeutically translatable as it can be induced remotely. The underlying mechanisms of PostC have been systematically investigated, but still need to be comprehensively analyzed. As most PostC studies to date were conducted preclinically using animal models, future studies are needed to optimize protocols in order to accelerate the clinical translation of PostC.
Key Words: postconditioning; cerebral I/R injury, neuro-protection, mechanism, molecular pathway
1. Introduction
Stroke is one of the leading causes of human disability and mortality in the world. Currently, there are only two effective FDA-approved treatments: i) thrombolytic drugs to dissolve blood clots, and ii) mechanical removal of clots from the blood vessels themselves. These methods result in reperfusion, where the blood flow is restored to the tissue. However, this action can also lead to tissue damage, a phenomenon known as reperfusion injury. Reperfusion injury has been extensively studied, but to date no clinically effective therapeutics exist. Ischemic Postconditioning (PostC) was initially defined in the research field of myocardial ischemia as a series of brief mechanical occlusions and reperfusions (Zhao et al., 2003; Na et al., 1996), and has recently been proven to be effective against ischemia/reperfusion injury in animal stroke models. Since then, its protective effects have been tested in other organs with ischemia-reperfusion (I/R) injury, including cerebral ischemia. Both in vivo (Gao et al., 2008a; Ren et al., 2009) and in vitro (Scartabelli et al., 2008; Leconte et al., 2009) studies have demonstrated the protective effects of PostC against cerebral ischemic injury. The protective effects of PostC were also found to be effective against spinal cord I/R injury in vitro (Li et al., 2017), which closely relates to cerebral ischemia in the central nervous system (CNS). In the past decade, ischemic PostC has been the subject of many research studies. Here, we will briefly review the neuroprotective cellular and molecular mechanisms of ischemic PostC against stroke.
2. PostC types
The concept of ischemic PostC has evolved to include a broad range of PostC types; these can be divided into rapid and delayed PostC based on the onset time, as well as ischemic, pharmacological and remote PostC, according to the methods used for PostC induction (Zhao et al., 2011).
2.1 Rapid PostC
Rapid PostC is induced immediately or a few minutes after reperfusion, and is the method tested in the majority of heart research studies. Several groups tested the protective effects of rapid PostC against focal cerebral ischemia, which is conducted in Middle Cerebral Artery occlusion (MCAo) or distal Middle Cerebral Artery occlusion (dMCAo) models. The initial studies showed that rapid PostC conducted 10 to 30 seconds or a few minutes after reperfusion could reduce infarct size (Zhao et al., 2007; Pignataro et al., 2008; Zhao et al., 2006). In addition, a few groups have also discovered the protective effects of rapid PostC on transient global cerebral ischemia (Rehni and Singh, 2007). For example, Wang et al. showed that rapid PostC applied immediately after reperfusion attenuated neuronal death in both the hippocampus and the parietal cortex after 10 minutes of transient global ischemia (Wang et al., 2008). The protective effects of rapid PostC depend on the number of reperfusion/occlusion cycles, the duration of each cycle, as well as the onset time of PostC (Gao et al., 2008a).
2.2 Delayed PostC
Though rapid PostC, which interrupts early reperfusion after stroke, reduces brain infarction, its extremely short therapeutic time windows may restrict its clinical application. Thus, delayed PostC, tested as late as 3 or 6 hours after ischemia-reperfusion, was found to be protective against stroke (Leconte et al., 2009; Ren et al., 2008). For global cerebral ischemia, the therapeutic time window of PostC can be much longer, as late as 48 hours after reperfusion. Burda and colleagues found that delayed PostC with a single 5-minute ischemia, even induced 2 days after I/R injury, could generate neuronal protection and reduce neuronal death in the hippocampus when measured 7 days after global ischemia, a time point at which neuronal death is considered matured (Burda et al., 2006). The prolonged therapeutic time window for delayed PostC could potentially benefit more stroke patients who often experience delays in treatment secondary to transportation or other issues.
2.3 Pharmacological PostC
Pharmacological PostC refers to the administration of drugs that mimic the actions of ischemic PostC. One of the best candidates for a pharmacological PostC drug is the anesthetic isoflurane or sevoflurane (Li et al., 2013; Zhu et al., 2007). In 2007, Li et al. found that isoflurane PostC generated long-term protective effects against brain I/R injury in rats; this may be attributable to the inhibition of nuclear factor κB (NF-κB) and interleukin 1β (IL-1β) production in the ischemic penumbra (Li et al., 2013). Besides these anesthetic drugs, other pharmacological PostC drugs have been added in the past years. Xie et al. demonstrated that pre- and post-treatments of alpha-lipoic acid (ALA), an endogenous short-chain fatty acid, could provide protective effects against simulated I/R injury in cerebral endothelial cells (CEC) (Xie et al., 2012). Moreover, a recent study suggested that dexmedetomidine PostC could improve neurological outcomes following brain I/R injury in neonatal rats, which may be regulated by inhibiting neuro-inflammation through α2 adrenergic receptors (Ren et al., 2016).
Another form of pharmacological PostC is induced by CO2 or other volatile anesthetics. It was first demonstrated by Leconte and colleagues that chronic, intermittent hypoxia initiated at 5 days after I/R reduced delayed thalamic atrophy (Leconte et al., 2009). Following their study, many reports confirmed this phenomenon (Zhan et al., 2012; Galle and Jones, 2013). In 2014, Fan et al. found that inhalation of 10% or 20% CO2 for 5 minutes, at 5 or 50 minutes after onset of reperfusion, induced a pronounced neuroprotective effect, including decreased infarct size and cleaved caspase-3 expression. This confirmed the neuroprotective and therapeutic strategy of inhalation against cerebral I/R injury (Fan et al., 2014).
2.4 Remote PostC
Remote conditioning is a transient ischemia conducted in a peripheral organ to protect against brain injury after stroke (Ren et al., 2009), and can be divided into Preconditioning (PreC) and PostC. In animal models, most remote conditioning is conducted in a hind limb by occluding the femoral artery for 5 to 10 minutes, followed by reperfusion for 5 to 10 minutes and repeated for 3 to 5 cycles. Ren demonstrated that remote PreC could generate protection at three therapeutic time windows, as robust neuroprotection was found when limb ischemia was conducted immediately, 12 hours, and 24 hours before stroke onset (Ren et al., 2009).
Remote PostC often uses the same parameters as remote PreC, except the limb ischemia is performed after reperfusion. Remote PostC may be better suited for clinical translation since the intervention is performed after stroke. In 2009, Ren et al. conducted remote ischemic PostC in an ipsilateral hind limb to generate protection against brain ischemia and, for the first time, demonstrated that remote PostC markedly reduced brain infarct size (Ren et al., 2009), which provided a potential clinical application of ischemic PostC. Subsequently, a few studies confirmed the protective effects of remote PostC against cerebral I/R injury. Li et al. demonstrated that limb remote PostC can reduce cerebral I/R injury by inhibiting apoptosis (Li et al., 2015a). Zhang et al. demonstrated that the protective effects of limb remote PostC against cerebral I/R injury was through the activation of the AKT pathway (Zhang et al., 2017).
3. The protective mechanism of ischemic PostC
3.1 Increasing the cerebral blood flow after ischemia-reperfusion
Disturbances in cerebral blood flow (CBF) occur throughout the reperfusion period following ischemic stroke. Since rapid ischemic PostC interrupts early reperfusion, its protective effects must be closely associated with CBF changes after ischemia-reperfusion.
As early as 2008, Gao et al. showed that rapid PostC with mechanical interruption results in CBF changes, measured by a laser Doppler probe in the penumbra in rats, and that CBF improved at 30 minutes after reperfusion (Gao et al., 2008a). Wang and colleagues confirmed this effect in a global ischemia model (Wang et al., 2008). After that, many studies showed more evidence that ischemic PostC had a strong ability to increase CBF, especially around the ischemic penumbra (Wu et al., 2015; Hess et al., 2016). In 2014, Han et al. found that ischemic PostC could reduce the structural and functional damage caused by ischemia-reperfusion injury to the neurovascular unit (NVU), which can eventually protect the NVU from injury and improve cerebral circulation (Han et al., 2014). Recently Esmaeeli-Nadimi et al. have reported that ischemic PostC combined with a tissue plasminogen activator (tPA) injection caused a gradual increase in CBF, as well as reduced BBB leakage, brain edema, apoptosis and reactive oxygen species levels (Esmaeeli-Nadimi et al., 2015).
3.2 Anti-oxidative stress
Oxidative stress has an important role in the early stages of ischemia-reperfusion injury (Kahles and Brandes, 2012). When ischemia-reperfusion happens in the ischemic brain, the excessive accumulation of free radicals could lead to oxidative damage by diverse signaling pathways, and exacerbate the ischemic injury. As PostC interrupts reperfusion that leads to oxidative stress, PostC may execute its protective effects via its role of attenuating oxidative stress.
In 2006, Zhao et al. found that rapid PostC profoundly attenuates the amount of superoxide 30 minutes after reperfusion in the dMCAo model (Zhao et al., 2006). Several subsequent studies demonstrated that rapid or delayed PostC had the ability to inhibit oxidative stress by increasing the activity of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase and catalase in different cerebral ischemia models, including focal cerebral ischemia and global brain ischemia in adult rats (Danielisova et al., 2006; Li et al., 2015b). Wang further showed that rapid PostC reduced the release of cytochrome c from the mitochondria to the cytosol, which is a critical cascade for apoptosis induction (Wang et al., 2008). Liang found that the protective role of ischemic PostC in focal I/R injury might be due to decreased mitochondrial ROS production (Liang et al., 2013).
Nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase, especially in neutrophils, was the main source of ROS under ischemic injury conditions (Fan et al., 2017; Fujita et al., 2012). Ischemic PostC could also decrease neutrophil NADPH oxidase activity, and thus generate the protective effects against stroke in vivo (Chen et al., 2016). Taken together, most of the above experiments showed a strong anti-oxidative stress ability of PostC in relation with its protection against cerebral I/R injury.
3.3 Anti-apoptosis of neurons
Apoptosis is a process of cell death that occurs after cerebral ischemia-reperfusion injury, usually accompanied with increased oxidative stress. Thus, preventive or therapeutic approaches targeting ischemia should also focus on apoptosis after I/R (Xing et al., 2008).
In 2006, Zhao et al. had shown that rapid PostC could block the terminal deoxynucleotidyl TUNEL-positive staining, an apoptosis marker, in the penumbra 2 days after cerebral ischemia-reperfusion injury in rats (Zhao et al., 2006). Later, other published papers supported the anti-apoptosis effects of ischemic PostC. Both rapid and delayed PostC could reduce neuronal apoptosis by downregulating the pro-apoptosis proteins of cleaved caspase-3, caspase-6, caspase-9 and Bax, but upregulating the anti-apoptosis protein Bcl-2 (Ding et al., 2012; Cheng et al., 2014; Liang et al., 2014; Saad et al., 2015). In 2014, Xie et al. demonstrated that the protective effects of ischemic PostC against brain ischemia was through the mTOR pathway activation, which also inhibited cell apoptosis (Xie et al., 2014).
Nuclear factor κB (NF-κB), a nuclear transcription factor, is related to transient ischemia-induced neuronal apoptosis (Zhu et al., 2007). Liang noticed that ischemic PostC could attenuate NF-κB/p65 activity in neurons, then reduce the neuronal apoptosis caused by ischemic damage and exert the protective effects against ischemia (Liang et al., 2013).
In remote PostC, anti-apoptosis is strongly related with its protective effects. One study indicated that protection against ischemic damage in the brain was via the p38 MAPK signaling pathway, which could improve neuronal morphological changes in the ischemic penumbra and reduce neuronal cell apoptosis in focal cerebral I/R rat models (Cheng et al., 2014).
Even in cultured spinal cord neurons in vitro, Li et al. found that Bax mRNA and protein levels increased after OGD-reperfusion and were restored by PostC treatment. Moreover, caspase-3 assay found the up-regulation of cleaved 17kd levels after OGD-reperfusion, and PostC could attenuate the increased Cleaved 17kd expression levels to base levels. These results indicated that the protective effects of PostC against OGD-reperfusion injury were via anti-apoptosis (Li et al., 2017).
3.4 Anti-inflammation
Previous studies showed that many inflammatory cytokines increased in expression and activity, and were involved in the pathological processes in cerebral I/R injury (Huang et al., 2006). A series of studies demonstrated that inflammatory inhibition accounted for the protective neuronal effects of rapid PostC. Xing et al. found that rapid PostC could inhibit myeloperoxidase (MPO) activity, an indicator of leukocyte accumulation, and the expression of IL-1β and TNF-α mRNA, and ICAM-1 protein expression in the ischemic cortex at 24 hours after ischemia (Xing et al., 2008). Some studies showed that PostC may reduce the expressions of pro-inflammatory factors such as IL-1b, IL-6, TNF-α, ICAM-1 and iNOS (Zhao et al., 2006; Zhu et al., 2007; Saad et al., 2015; Kong et al., 2013; Teo et al., 2015; Wei et al., 2015), and increase the expression of anti-inflammatory factor TGF-β1 (Wang et al., 2016) in the ischemic brain.
As T cells have been shown to contribute to brain injury (Gu et al., 2012; Xiong et al., 2015), Xie and colleagues sought to address whether ischemic PostC had similar protective effects in T cell-deficient nude rats. They found that PostC did not reduce infarct size measured at 2 days, but reduced brain injury size measured at 30 days post-stroke. These results suggest that T cells are crucial in modulating the acute protective effects of ischemic PostC, and that cell types other than T cells may mediate the long-term protective effects of PostC (Xie et al., 2013b). In 2013, Joo et al. demonstrated that PostC could spare the ischemic penumbra in the cortex and striatum and protect against cerebral ischemia by reducing the infiltration of both innate and adaptive immune cells such as macrophages, T cells, and B cells (Joo et al., 2013).
3.5 Reducing cerebral edema
Disruptions in the energy and material supply of brain tissue during cerebral ischemia-reperfusion, accompanied by the failure of ion pumps in the cell membrane, lead to cerebral edema. AQP4 is a water channel protein localized in the end-feet of astrocytes in the central nervous system. It is an important factor for water hemostasis during the development of cytotoxic and vasogenic edema (Kozlu et al., 2014). Ischemic PostC could alleviate cerebral edema and improve BBB integrity through the down-regulation of AQP4 expression in the early formation of post-reperfusion edema (Han et al., 2015; Yu et al., 2015). Accumulating evidence has revealed that matrix metalloproteinase 9 (MMP-9) was significantly elevated in humans after stroke (Switzer et al., 2012). PostC could suppress the elevation of MMP-9 activity and expression, and thus prevent cerebral edema and BBB disruption (Yang et al., 2013).
3.6 Regulating autophagy
Autophagy is a vital cellular pathway for the degradation of intracellular macromolecules or organelles for subsequent reuse that helps to maintain intracellular homeostasis in physiological conditions. Previous studies have shown that autophagy might be activated by cerebral ischemia-reperfusion, but its role remains controversial. Zhang et al. showed that autophagy inhibition generated protective effects against permanent cerebral ischemia but aggravated brain injury caused by ischemia-reperfusion, suggesting that autophagy played different roles at the cerebral ischemic phase and at the subsequent reperfusion phase (Zhang et al., 2013). In 2012, Gao et al. reported that autophagy inhibition could induce neuroprotection in rapid ischemic PostC (Gao et al., 2012).
4. Cell signaling pathways involved in ischemic PostC
It has been shown that multiple pathways take part in neuronal death and consequent brain damage after ischemic-reperfusion injury, such as the PI3K/Akt, mTOR, and PKC pathways. Thus, the protective effects of PostC against ischemic-reperfusion injury should be related with multiple pathways. What should be taken seriously, however, is that none of these are decisive pathways; it must be acknowledged that the protective effects of ischemic PostC may come from the effects induced by multiple pathways.
4.1 Akt pathway
The PI3K/Akt pathway is a well-known and important mediator in signal transduction, which is primarily involved in cell survival by inhibiting apoptotic processes and cell growth. Akt dysfunction results in apoptosis induction, while Akt activity blocks apoptosis by phosphorylating its substrates, including GSK3β (glycogen synthase kinase 3β), FKHR and Bad (Zhao et al., 2011). Akt activity is considered to be regulated by phosphorylation, which is modulated by upstream molecular signals, such as PTEN and PDK1 (phosphoinositide-dependent protein kinase-1).
The PI3K/Akt pathway in ischemic PostC regulates a great part of the processes named above; therefore, this pathway should contribute to PostC-induced neuroprotection against cerebral ischemia. In 2008, Gao et al. demonstrated that rapid PostC increases both phosphorylation (by Western blot) and Akt activity (by in vitro kinase assay) (Gao et al., 2008a). Moreover, Akt inhibition by injection of the PI3K inhibitor, LY294002, partially blocks the protective effect of rapid PostC (Pignataro et al., 2008; Gao et al., 2008b). These results are further supported by a recent in vitro experiment showing that Akt inhibition abolished the protective effect of OGD and DHPG, PostC in hippocampal slice cultures (Scartabelli et al., 2008). Furthermore, similar results were shown in other PostC models, such as ischemic remote PostC, pharmacological, as sevoflurane PostC, and delayed PostC (Prasad et al., 2011; Zhan et al., 2012).
4.2 mTOR pathway
As an important regulation factor, Akt has many downstream targets, but which downstream target relates to the protective effects of ischemic PostC is unclear. The mammalian target of the rapamycin (mTOR) signaling pathway plays a central role in metabolism, cell growth, differentiation, development, and cell survival (Jewell and Guan, 2015). A few studies have shown that brain injury induced by ischemia involves the mTOR pathway (Mao et al., 2013; Wu et al., 2012; Wang et al., 2012), but the effects of the mTOR pathway in cerebral ischemia-reperfusion injury remain controversial. There is evidence that several neuroprotectants upregulate mTOR activity, including the antioxidants melatonin and estradiol (Koh et al., 2008) and the anti-inflammatory agent silibinin (Wang et al., 2012). mTOR activity is inhibited by hypoxia, adenosine triphosphate depletion, and DNA damage, but activated by nutrients (amino acids) and growth factors. These inhibitory or stimulatory factors also modulate Akt, probably through the activation of the mTOR cell signaling pathway. In addition, rapamycin treatment increased cleaved caspase protein levels and promoted neuronal death after hypoxia-ischemia (Chen et al., 2012).
In 2013, Xie et al. found that lentiviral-mediated over-expression of Akt3 resulted in stronger protection than cAkt1 over-expression. Western blot analyses further showed that Akt3 promoted significantly higher levels of phosphorylated Akt and phosphorylated mTOR than cAkt1. The mTOR inhibitor, rapamycin, blocked the protective effects of both Akt1 and Akt3. In conclusion, Akt3 offers stronger protection than Akt1 by maintaining Akt levels and promoting mTOR activity (Xie et al., 2013a).
Next, Xie et al. sought to understand the relationship between the mTOR pathway and protection of ischemic PostC. They first investigated if ischemic PostC protected against brain injury induced by focal ischemia in T cell-deficient nude rats, and examined its effects on Akt and the mTOR pathway. They found that PostC did not attenuate infarction in nude rats compared with wild-type rats, but PostC still improved neurological function in nude rats 1-30 days after ischemic-reperfusion injury, and reduced the extent of brain damage 30 days after stroke. The mTOR inhibitor, rapamycin, abolished the long-term protective effects of PostC (Xie et al., 2013b). Next, in 2014, Xie and colleagues reported that stroke resulted in reduced levels of phosphorylated proteins in the mTOR pathway, including S6K1, S6, and 4EBP1, and that ischemic PostC increased these proteins. mTOR inhibition, both by rapamycin and by mTOR short hairpin RNA, worsened ischemic outcomes in vitro and in vivo, and abolished the protective effects of ischemic PostC on neuronal death in vitro and on brain injury size in vivo (Xie et al., 2014).
Based on this research, Xie and colleagues found that other molecular factors, including PRAS40 overexpression and p53 inhibition, had protective effects against ischemic-reperfusion injury; they also found that these were determined by the mTOR pathway (Xiong et al., 2014; Li et al., 2015c), which strongly supported the neuroprotective role of the mTOR pathway against I/R injury.
4.3 MAPK and Others
Ischemic injury and neuronal survival are modulated by the MAPK pathways, including extracellular signal-regulated kinase 1/2 (ERK1/2), P38, and c-Jun N-terminal kinase (JNK) pathways. Sawe et al. found that ERK1/2 phosphorylation (P-ERK1/2) levels increased from 1 to 24 hours after stroke, and rapid PostC reduced their levels in the penumbra (Sawe et al., 2008), which implied a detrimental role of P-ERK1/2 after ischemia.
Ischemic PostC can also alter BDNF expression. BDNF is a member of the neurotrophin family, which is related with neuronal development and differentiation (Singh and Su, 2013, Alboni et al., 2011). The cAMP response element-binding protein (CREB), a transcription factor of BDNF (Alboni et al., 2011), the extracellular signal-regulated kinases 1/2 (ERK1/2), and upstream phosphorylating BDNF enzymes are upregulated by PostC to enhance BDNF production in rats after focal ischemia (Zhang et al., 2017).
Tim-3, a member of the T cell immunoglobulin and mucin domain family, is activated by its ligand, galectin-9, and it can be expressed on microglia and neurons in the brain to regulate the inflammatory response (Anderson et al., 2015), Recently, Wei et al. found that ischemic PostC robustly inhibits the expression of Tim-3 and galectin-9 at 1, 5, and 24 hours after reperfusion in the dMCAo rat model (Wei et al., 2015).
Sirtuin 1 (SIRT 1) is a member of the sirtuin enzyme family in mammals (Hernandez-Jimenez et al., 2013). SIRT 1 has been implicated as a key neuroprotective molecule against ischemic-reperfusion injury (Miao et al., 2016; Wan et al., 2016). Kaur also reported that the neuroprotective effects of ischemic PostC were related to the activation of the SIRT1 enzyme (Kaur et al., 2015).
5. Future direction and conclusion
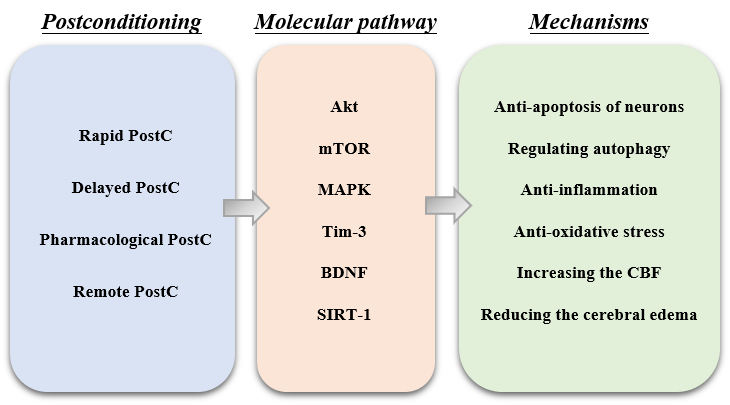
PostC has been established as a promising neuro-protective strategy against ischemic stroke. The multiple mechanisms of PostC are robust and sufficient enough to conquer most of the pathological processes of cerebral I/R injury (Fig. 1). In animal models in vivo, PostC can effectively reduce neural cell death, decrease cerebral edema and infarct size, improve cerebral circulation, and relieve the inflammation, apoptosis and oxidative stress of the neurons.
Compared to single target neuroprotectants, PostC has many more advantages. Remote PostC, pharmacological PostC and chemical PostC are much easier and safer for clinical application. However the risks, efficacy, therapeutic time windows, and above all the underlying protective mechanisms of PostC must be assessed and cleared before clinical translation. Therefore, future studies should be aimed at investigating the detailed neuroprotective mechanisms of PostC and optimizing the operational protocol, in order to accelerate its clinical translation.
Acknowledgements
We thank Felicia F. Beppu and Christine Plant at the Department of Neurosurgery, Stanford University School of Medicine for their help in manuscript preparation and editing. This study was supported by NIH grant R01NS06413606 (HZ).
References
Rong Xie
1Department of Neurosurgery, Huashan Hospital, Fudan University, Shanghai, China
Jinquan Li
1Department of Neurosurgery, Huashan Hospital, Fudan University, Shanghai, China
Heng Zhao
2Department of Neurosurgery, Stanford University, Stanford, California, USA
Rong Xie and Jinquan Li contributed equally to this article.
Corresponding author:
Rong Xie
Email: xierong3034@126.com
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 9584 | 41 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA