Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Heart failure with preserved ejection fraction: targeted pharmacological interventions and rodent disease models
Time:2021-11-15
Number:11991
Author Affiliations

Conditioning Medicine 2021. 4(4): 168-184.
Abstract
Heart failure with preserved ejection fraction (HFpEF) accounts for approximately half of all the reported heart failure cases worldwide, thus representing a huge clinical need in the field of management of cardiovascular diseases. This need has not been met properly, as there is no effective treatment targeting specifically HFpEF, and most clinical trials to date have shown neutral results. The heterogeneity of HFpEF syndrome, which is mainly clinically characterized by left ventricular diastolic dysfunction, as well as by coexistence of multiple comorbidities, such as obesity, diabetes mellitus, or hypertension, has put obstacles in the way of preventing or treating the disease. In this review, we highlight certain important molecular and cellular underlying mechanisms, which are divided into four main categories, depending on the organelles and structures involved. These include microvascular inflammation and endothelial dysfunction, titin-dependent myocardial stiffness, cardiac fibrosis, and mitochondrial dysfunction. Each mechanistic pathway is associated with promising drug targets, which are concisely mentioned. Most of these therapeutic interventions have been in the past or are currently being investigated in clinical studies, data of which are briefly mentioned to provide a comprehensive understanding of their possible clinical benefit for the HFpEF patient. Furthermore, we address the difficulty of developing small animal models, which mimic the human HFpEF phenotype. Despite the lack of “the perfect” model, we provide a synopsis of the most frequently used rodent HFpEF models, as they contribute to the pursuit of discovering innovative therapeutic approaches for HFpEF and “translating” them into clinic.
Keywords: Heart failure with preserved ejection fraction, Endothelial dysfunction, Myocardial stiffness, Mitochondrial dysregulation, Myocardial fibrosis, animal models
Abstract
Heart failure with preserved ejection fraction (HFpEF) accounts for approximately half of all the reported heart failure cases worldwide, thus representing a huge clinical need in the field of management of cardiovascular diseases. This need has not been met properly, as there is no effective treatment targeting specifically HFpEF, and most clinical trials to date have shown neutral results. The heterogeneity of HFpEF syndrome, which is mainly clinically characterized by left ventricular diastolic dysfunction, as well as by coexistence of multiple comorbidities, such as obesity, diabetes mellitus, or hypertension, has put obstacles in the way of preventing or treating the disease. In this review, we highlight certain important molecular and cellular underlying mechanisms, which are divided into four main categories, depending on the organelles and structures involved. These include microvascular inflammation and endothelial dysfunction, titin-dependent myocardial stiffness, cardiac fibrosis, and mitochondrial dysfunction. Each mechanistic pathway is associated with promising drug targets, which are concisely mentioned. Most of these therapeutic interventions have been in the past or are currently being investigated in clinical studies, data of which are briefly mentioned to provide a comprehensive understanding of their possible clinical benefit for the HFpEF patient. Furthermore, we address the difficulty of developing small animal models, which mimic the human HFpEF phenotype. Despite the lack of “the perfect” model, we provide a synopsis of the most frequently used rodent HFpEF models, as they contribute to the pursuit of discovering innovative therapeutic approaches for HFpEF and “translating” them into clinic.
Keywords: Heart failure with preserved ejection fraction, Endothelial dysfunction, Myocardial stiffness, Mitochondrial dysregulation, Myocardial fibrosis, animal models
Introduction
Cardiovascular diseases are the leading cause of death globally, comprising a striking 31% of all deaths (( World Health Organization, n.d. )). Of these, heart failure (HF) continues to increase in prevalence and nearly 8.5 million people in the USA will suffer from HF by 2030, 6 million of whom will be above 65 years old (Borlaug, 2014). HF was initially thought to refer only to patients exhibiting ventricular systolic dysfunction and a reduced ejection fraction (EF). This perception persisted until the late 1980s when it was demonstrated that a significant number of patients who suffered from clinically demonstrated HF had a normal EF and heart failure was attributed to ventricular diastolic dysfunction. Therefore, the earlier term for this syndrome was “diastolic heart failure” (Gladden et al., 2018). Nowadays, this term is replaced by “heart failure with preserved ejection fraction” (HFpEF) and it is widely accepted that HFpEF is a complex clinical syndrome presented hemodynamically as the inability of the heart to pump blood adequately without the requirement for elevated cardiac filling pressures (Ponikowski et al., 2016).
HFpEF accounts for over half of all prevalent HF with high morbidity and mortality rates, the latter of which approaches 30% among patients over 1 year, while a 2–year rate of hospitalization for HF or all–cause death approaches 35% (Owan et al., 2009; Lam et al., 2018a). Effective treatment for HFpEF is a huge unmet medical need of the 21st century, as there is no widely accepted pharmacological therapy that halts the progression of the disease despite numerous clinical trials aiming to prove the opposite. While several successful pharmacological agents have emerged for the treatment of HF with reduced ejection fraction (HFrEF), those such as angiotensin-converting enzyme inhibitors, angiotensin-II receptor blockers and mineralocorticoid receptor antagonists, did not turn out to be significantly beneficial in HFpEF patients (Patel and Shah, 2019). Nowadays, management of HFpEF includes diuretics for decongestion, lifestyle interventions such as regular physical activity, mineralocorticoid antagonists in exceptional cases, and treatment of concomitant comorbidities (Borlaug, 2020).
HFpEF is a heterogenous disorder involving a plethora of organ pathologies and cellular or molecular pathophysiological mechanisms and evolves concurrently with other disease entities. In short, pathomechanisms include ventricular diastolic dysfunction, impaired cardiac reserve, systemic, pulmonary, vascular and renal dysfunction, limited oxygen capacity as well as abnormal skeletal blood flow and metabolism, and they are reviewed extensively elsewhere (Obokata et al., 2020). Most patients suffer from various comorbidities – hypertension, type 2 diabetes mellitus, and obesity – and these conditions are major contributors to the HFpEF pathophysiology (Mishra and Kass, 2021). Atrial fibrillation (AF) is also present in a large proportion of HFpEF patients and may occur before (prevalent), concurrently, or after (incident) the initial diagnosis of the disease and is closely associated with poor prognosis (Zakeri et al., 2013). AF aggravates left ventricular (LV) diastolic and systolic function provoking left atrium dilation and adverse remodeling, as well as limited diastolic and ventricular filling time due to tachyarrhythmias (Lam et al., 2017; Paz et al., 2021).
Additionally, women are more prone to HFpEF than men and the female sex has lately been associated with discrete clinical implications and therapeutic responses. For instance, echocardiographic presence of diastolic dysfunction and risk of all-cause death or HF hospital readmission were independently correlated with female gender (Sotomi et al., 2021). Thus, sex-specific differences pose another crucial parameter to HFpEF multisystemic nature. This multisystemic nature is partly related to the failure of development of effective treatments.
Animal models in which the complexity of this clinical syndrome is sufficiently represented are missing. Most of them focus on unique pathophysiological entities, such as diastolic dysfunction, and develop into at most two different comorbidities, such as hypertension and obesity, failing to recapitulate the vast diversity of findings identified in a HFpEF patient (Roh et al., 2017).
This review aims to summarize potential pharmacotherapies related to four important subcellular pathways, namely microvascular inflammation and endothelial dysfunction, titin-dependent myocardial stiffness, cardiac fibrosis, and mitochondrial dysregulation - metabolic abnormalities involved in the complex HFpEF clinical syndrome. We proceed to discuss available rodent HFpEF models as they remain necessary to comprehend the disease pathophysiology and progress towards novel drug development. Thus, considering exceptional literature reviews surrounding HFpEF, our review combines insights into promising pharmacological interventions with disease models in which those targets could be investigated properly.
Overview of HFpEF cardiac pathophysiological features
The main feature of HFpEF is left ventricular diastolic dysfunction, which is defined as abnormal and prolonged relaxation and increased LV chamber stiffness. LV filling pressures are elevated for a specific LV volume at rest or during exercise when heart rate and blood flow are increased. The heart is unable to complete relaxation during the limited filling time and to demonstrate sufficient end-diastolic and stroke volume (Westermann et al., 2008). LV geometry is typically altered and the most common morphological phenotypes include concentric remodeling or hypertrophy (Gladden et al., 2018). Apart from LV diastolic abnormalities, subtle LV systolic dysfunction, left atrial (LA) enlargement, LA hypertension, impaired contractility and remodeling can be manifested. Other impairments, except for those of the left-sided heart, include abnormal right ventricular-pulmonary coupling, pulmonary hypertension and venous congestion, systemic arterial stiffness, and secondary chronotropic incompetence as a result of autonomic dysfunction (Gladden et al., 2018; Obokata et al., 2020). Another HFpEF pathophysiological component is coronary microvascular dysfunction (CMD), which manifests as myocardial oxidative stress mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), uncoupled nitric oxide (NO) synthases, and as augmented inflammation (Franssen et al., 2016). Moreover, reduced coronary flow reserve, a marker of CMD, was prevalent in three-quarters of HFpEF patients in the large prospective Prevalence of Microvascular Dysfunction in HFpEF (PROMIS – HFpEF) study and was associated with systemic endothelial dysfunction, HF severity, as well as comorbidities, smoking and atrial fibrillation (AF) (Shah et al., 2018). Recently Ozcan et al. (2021) also showed that CMD was a predictor of the coexistence of AF with HFpEF and was positively correlated with increased mortality and HF hospitalization rates in patients with HFpEF or AF. Even though the study lacks mechanistic insights the authors proposed that disruption of myocardial perfusion in the context of CMD exacerbates atrial structural and electrophysiological remodeling (de la Pena et al., 2019; Ozcan et al., 2021). Furthermore, coronary microvascular rarefaction is present as depicted by decreased vessel density and is associated with magnified myocardial fibrosis (Mohammed et al., 2015). Impaired endothelial function restricts vasodilator reserve during exercise, contributing to decreased effort tolerance in addition to limitations in global cardiovascular reserve, including contractile, diastolic, and chronotropic reserves. Depressed reactive hyperemic change in blood flow at rest and exercise also proves that dysfunction is not limited to coronary endothelium but appears in the periphery as well (Borlaug et al., 2010). Arterial stiffness is elevated in HFpEF resulting in afterload surge, decreased arterial compliance and cardiac output, and raised LV filling pressures during exercise (Reddy et al., 2017a). The capacity of the heart to pump blood and deliver oxygen in tissues is severely impaired (Shah et al., 2020).
Therapies targeting microvascular inflammation and endothelial dysfunction
The most promising paradigm for HFpEF is the one proposed by Paulus and Tschöpe back in 2013 who identified a comorbidities-driven systemic proinflammatory state as the root of microvascular inflammation and endothelial dysfunction leading to adverse cardiomyocyte remodeling (Paulus and Tschöpe, 2013). Moreover, comorbidities, including obesity, chronic obstructive pulmonary disease, diabetes, and hypertension often prevalent in HFpEF patients, induce chronic inflammation, evident from elevated plasma inflammation biomarkers. These include tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), soluble ST2, and pentraxin 3, which strongly and positively correlate with mortality (Kalogeropoulos et al., 2010; Matsubara et al., 2011; Shah et al., 2011). Therefore, coronary microvascular endothelial cells express high levels of adhesion molecules, like vascular cell adhesion molecule (VCAM) and E-selectin, facilitating attraction and subsequent transendothelial migration of immune cells (Westermann et al., 2011). Various types of immune cells, including neutrophils or monocytes, infiltrate the failing myocardium initiating a coordinated innate immune response, similar to that of infarcted myocardium, as they secrete proinflammatory cytokines, stimulate attraction and differentiation of neighboring leukocytes, and eventually reactive oxygen species (ROS) production initiating oxidative and nitrosative stress (Tschöpe and Van Linthout, 2014; Andreadou et al., 2019). The presence of such stress is proved by positive dihydroethidium and nitrotyrosine staining in endomyocardial biopsies of HFpEF patients (Westermann et al., 2011; van Heerebeek Loek et al., 2012). It is well established that ROS, like superoxide, limit NO bioavailability because they react with NO, leading to its depletion and formation of reactive nitrogen species (RNS), including peroxynitrite (Daiber et al., 2021). As a result, soluble guanylate cyclase (sGC) activity is reduced in neighboring cardiomyocytes (Paulus and Tschöpe, 2013). Because of decreased sGC activity, cyclic guanosine monophosphate (cGMP) concentration and protein kinase G (PKG) activity are lowered, as it is confirmed immunohistochemically in myocardial tissue samples from HFpEF patients. Lower PKG activity in HFpEF than in HFrEF patients was additionally associated with increased resting tension (Fpassive) of cardiomyocytes, thus deteriorating myocardial diastolic function (van Heerebeek Loek et al., 2012). According to the aforementioned paradigm elevated tension can be due to the ability of RNS to increase protein phosphatase 2a activity, leading to reduced phosphorylation of phospholamban (PLN) (Paulus and Tschöpe, 2013). To elaborate, PLN phosphorylation is necessary for its dissociation from sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) and subsequent reuptake of Ca2+ into sarcoplasmic reticulum, hence decreased PLN phosphorylation increases diastolic Ca2+ in the cytosol (Bibli et al., 2015). Furthermore, low PKG activity results in decreased phosphorylation of the giant cytoskeletal protein titin, making it less compliant and increasing cardiomyocyte stiffness (Paulus and Tschöpe, 2013). Last but not least, it was found that cardiac inflammatory cells, such as monocytes, secrete transforming growth factor β (TGFβ), contributing significantly to differentiation of fibroblasts into myofibroblasts and development of interstitial fibrosis in HFpEF myocardium (Westermann et al., 2011). Mechanisms related to titin and cardiac fibrosis will be addressed more extensively in a following section.
Nitric oxide donors
As NO bioavailability is decreased in HFpEF, a rational therapeutic strategy is enhancement of NO concentration by administration of NO donors, such as organic or inorganic nitrates. These compounds, such as clinically approved nitroglycerine or isosorbide mononitrate, directly activate sGC and are used as vasodilators, even though cGMP – independent effects have been recently observed, including preservation of myocardial eNOS function as a dimer (Bibli et al., 2019). Isosorbide mononitrate has been clinically tested in HFpEF patients using a 6-week dose escalation regimen in Nitrate’s Effect on Activity Tolerance in Heart Failure with Preserved Ejection Fraction (NEAT-HFpEF) clinical trial (NCT02053493). However, compared to the placebo group, patients who received isosorbide mononitrate were less physically active, had decreased blood pressure, and did not present significantly improved quality of life or exercise capacity (Redfield et al., 2015). This adverse outcome may be due to the fact that organic nitrates are more prone to inducing tolerance because they require bioactivation in tissues, and may cause endothelial dysfunction by augmenting nitrosative stress as well as major and rapid alterations in preload and hypotension (Lam et al., 2018b). As a result, the primary focus shifted from organic to inorganic nitrates, which can be reduced to nitrites and NO in vivo in areas of low oxygen concentration and low pH, such as in exercising muscles (Patel and Shah, 2019). Acute intravenous administration of sodium nitrite in a small group of HFpEF patients resulted in a significant decrease in exercise pulmonary capillary wedge pressure versus placebo group, as well as enhanced exercise cardiac output reserve (Borlaug et al., 2015). Nevertheless, administration of nebulized nitrite therapy to patients for four weeks in a randomized, double-blind, placebo-controlled cross-over clinical trial, the Inorganic Nitrite Delivery to Improve Exercise Capacity in Heart Failure With Preserved Ejection Fraction (INDIE-HFpEF), resulted in neutral results, as the primary endpoint of peak oxygen consumption, as well as quality of life and exercise capacity, were not improved compared to placebo (NCT02742129) (Reddy et al., 2017b).
Soluble and particulate guanylate cyclase activation
Another potential drug target is the enzyme sGC, which catalyzes cGMP formation. Enhancement of the enzyme activity can be achieved through sGC activators and stimulators. The former class improves the activity of the enzyme in oxidized (Fe3+) state, as in the case of endothelial dysfunction and oxidative stress. The latter, which has been studied to a greater extent in HFpEF, targets the native non-oxidized (Fe2+) enzyme (Kovács et al., 2016). An example of a sGC stimulator is the compound vericiguat, which has been approved in the USA for HFrEF patients following hospitalization or patients in need of intravenous diuretics (Center for Drug Evaluation and Research, 2021). It was orally administered in HFpEF patients for 12 weeks during the SOLuble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED ejection fraction (SOCRATES-PRESERVED) clinical trial (NCT01951638). Although the primary endpoints of the clinical trial, such as LA volume and N-terminal pro-B-type natriuretic peptide (NT-ProBNP) levels were not different from placebo, vericiguat was well tolerated among patients and patient-reported quality of life scores were significantly improved. Further evidence should be sought in optimally designed clinical trials in which additional clinical endpoints, higher dosing regimen, and longer duration could be applied. Additionally, preclinical studies should be carried out to provide mechanistic insights into both cardiac and non-cardiac effects of sGC stimulation (Pieske et al., 2017).
Particulate guanylate cyclase (pGC) differs from its soluble isoform, sGC and is an additional drug target. It is activated by natriuretic peptides, including atrial natriuretic peptide (ANP) and BNP (Kovács et al., 2016). These peptides are degraded into inactive fragments by vasopeptidase, neprilysin. Inhibition of the enzyme, in combination with renin–angiotensin aldosterone system inhibition, represents a new therapeutic approach, already clinically used in HFrEF. Angiotensin-receptor and neprilysin inhibitors (ARNIs), such as sacubitril/valsartan have been recently tested in clinical trials in HFpEF patients. PARAGON-HF, a phase III clinical trial, studied efficacy and safety of sacubitril/valsartan compared to valsartan on morbidity and mortality in HFpEF patients. Despite the promising results of previous phase II clinical trials, including reduction of NT-proBNP levels at 12 weeks and improvement of LA function and NYHA class at 36 weeks, there were no significant differences in the primary endpoint between groups in the PARAGON-HF trial, which consisted of total HF hospitalizations and cardiovascular death (NCT01920711). However, subsequent subgroup analyses showed a significant clinical benefit in a subgroup of patients with lower LVEF (below 57% median), leading to the conclusion that dual inhibition may be of therapeutic potential in patients with coexisting LV systolic dysfunction (Gronda et al., 2020). Interestingly, HF hospitalizations were also significantly lower in women than men, revealing a sex-related difference in clinical outcome (McMurray et al., 2020). In light of these findings, in a female obese ZSF1 rat model, 12-week treatment with sacubitril/valsartan ameliorated diastolic dysfunction, augmented titin phosphorylation, and decreased LV stiffness and perivascular fibrosis (Schauer et al., 2021). Therefore, potential beneficial effects of this drug combination in female HFpEF cases were confirmed preclinically, even though inclusion of male laboratory animals and direct comparison between both sexes should be sought in future experiments.
Phosphodiesterase enzymes inhibition
The main isoforms of phosphodiesterase (PDE) enzymes degrading cGMP are PDE5 and PDE9, and therefore they represent potential drug targets. In basic research studies, including a canine model of LV concentric hypertrophy or mice subjected to transverse aortic constriction (TAC), sildenafil administration, a clinically used PDE5 inhibitor, improved LV diastolic compliance, decreased LV hypertrophy, and progressive remodeling (Nagayama et al., 2009; Kovács et al., 2016). However, in clinical settings, specifically the RELAX clinical trial, long-term oral sildenafil administration in HFpEF patients with pulmonary hypertension had neutral outcomes, as exercise capacity, quality of life, clinical status, or additional LV diastolic function measures were not enhanced (NCT00763867) (Redfield et al., 2013). Thus, the interest transitioned from PDE5 to PDE9 inhibition. PDE9 is responsible for hydrolysis of cGMP, originating from the NP-pGC pathway instead of cGMP from the NO-sGC pathway, and its expression is cardiomyocyte specific, triggered especially in LV hypertrophy and heart failure (Lee et al., 2015). As this enzyme is not extensively expressed in the systemic vasculature, PDE9 inhibition does not result in significant decreases in blood pressure, in contrast to PDE5. This target had not been addressed in any HFpEF study until recently. Richards et al. (2021) reported that in a TAC mouse model, treatment with CRD-733, a selective PDE9 inhibitor, significantly increased cGMP levels and improved LVEF, LV hypertrophy, LA dilation, and pulmonary edema, revealing important therapeutic potential in HFpEF. In a similar TAC male mouse model, combined with deoxycorticosterone acetate administration, 28-day continuous perfusion with the PDE9 inhibitor, PF-4449613, via subcutaneous implanted osmotic mini-pumps, diminished diastolic dysfunction, as evidenced by a significant decrease in LV chamber stiffness (Methawasin et al., 2020). Moreover, researchers conducted additional experiments in female mice to investigate potential gender-specific outcomes. Female mice, undergoing the same interventions, exhibited similar trends in improvement of diastolic function parameters, including decreased myocardial stiffness and increased LV relaxation velocity (Methawasin et al., 2020). PDE9 inhibition is a promising, under investigated therapeutic approach in HFpEF, independent of NOS activity and NO bioavailability, which are severely impaired but should be further validated as a pharmacological target in basic as well as human studies (McMurray et al., 2020).
Titin-dependent myocardial stiffness and related therapeutic approaches
Diastolic dysfunction is the main component of HFpEF pathophysiology and one main contributor is LV chamber stiffness, which is mainly attributed to alterations in the function of titin (Borbély et al., 2005). Titin is a giant protein of the cardiac sarcomere, extending from Z disc to the M line, which acts as bidirectional molecular spring and regulates passive mechanical and viscoelastic properties of the sarcomere (Patel and Shah, 2019). Two titin isoforms are present in the myocardium. The first one is the shorter and stiffer N2B isoform, and the second one is N2BA, which is longer and more compliant, and both are required for regulation and establishment of moderate passive tension (Virgen-Ortiz et al., 2009; Franssen and González Miqueo, 2016). Myocardial stiffness can be regulated by changes in the ratio of these isoforms as well as posttranslational modifications, including phosphorylation. Changes of the ratio N2B:N2BA (60:40 in normal hearts) induced by alternative splicing evolve during a long period of days to weeks in contrast to posttranslational modifications, which take place sooner (Franssen and González Miqueo, 2016).
Modulation of titin isoforms N2B:N2BA ratio
One potential therapeutic approach is modulation of the titin isoforms ratio in favor of the more compliant isoform, thus decreasing passive tension and increasing left ventricular compliance. RNA binding motif protein-20 (RBM20) is a major factor, which regulates, among other myocardial proteins, the splicing of titin. Homozygosity for mutation in RBM20 causes an increase in the expression of a large and super – compliant NB2A isoform (N2BAsc), as shown in isolated rat hearts, whereas the stiffer N2B isoform was absent. Heterozygosity for the same mutation also resulted in upregulation of the N2BA isoform and downregulation of N2B in rats and humans (Linke and Hamdani, 2014). It was also shown in a genetically modified mouse model that when RBM20 was mutated, compliant titin isoforms were expressed, LV compliance and exercise tolerance were increased, and there were no premature death events. It was further tested if such induced RBM20 dysfunction could provide a benefit in a mouse model of HFpEF. Diastolic dysfunction and stiffness were indeed decreased, depicted as reduced LV end diastolic pressure (LVEDP) and diastolic stiffness coefficients, whereas exercise tolerance and LV relaxation time constant were improved in mice, in which N2BAsc was upregulated in myocardium. These benefits will be significant if they translate to HFpEF patients. Development and screening of small-molecule inhibitors of the RMB20-dependent titin splicing system could represent a novel therapeutic strategy but further research is required (Methawasin et al., 2016).
Intervention in posttranslational phosphorylation of titin
Another potential strategy is switching the phosphorylation status of several titin - specific sites. Titin contains different regions, of which the PEVK spring element, rich in proline, glutamate, valine, lysine, and the N2B unique sequence (N2B-Us) are extensively phosphorylated by various kinases, affecting passive stiffness (Franssen and González Miqueo, 2016). The PEVK region is mainly phosphorylated by protein kinase C, isoform α (PKCα), located widely in myocardial tissue and activated by α1-adrenergic pathways. Phosphorylation of specific serine residues in PEVK by PKCα results in increased passive stiffness and it has been proved in several animal models of HFpEF - canine, rat, mouse - and human failing hearts, that PKCα was upregulated and the PEVK region was hyperphosphorylated (Wolfgang A. and Hamdani, 2014). In contrast, the N2B-Us is phosphorylated by other kinases and mechanical properties and distensibility of titin are differentially affected. These kinases include protein kinase A (PKA), which is activated by β-adrenergic signaling pathways as well as PKG, which is cGMP-dependent and regulated by NO and natriuretic peptides (LeWinter and Granzier, 2013). Phosphorylation of the N2B element reduces passive stiffness and it was demonstrated in HFpEF models and endomyocardial biopsies of HFpEF patients that there was hypophosphorylation of serine residues of this region by PKA and PKG (Wolfgang A. and Hamdani, 2014). Furthermore, when cardiomyocytes, isolated from HFpEF patients or animal HFpEF models, were treated in vitro with PKG or PKA, high passive tension was restored to normal levels indicating the altered phosphorylation state during HFpEF (Borbély et al., 2009; Hamdani et al., 2013). Apart from the aforementioned kinases, Ca2+ and calmodulin – dependent serine/threonine kinase, CaMKII, particularly the delta isoform CaMKIIδ, which is stimulated by increased Ca2+ load in cardiomyocytes, and extracellular signal-regulated kinase 2 (ERK2) phosphorylate N2B-Us reduce myocardial passive tension, similar to PKA and PKG (Perkin et al., 2015). To summarize and considering the beneficial effects of stimulating the cGMP-PKG pathway, the main goal in basic and clinical research studies is the stimulation of PKG and subsequently, phosphorylation of titin elastic regions decreasing therefore, passive stiffness.
Dipeptidyl peptidase-4 (DPP-4) inhibition has been recently proposed as an alternative strategy to limit titin – dependent stiffness and improve LV diastolic function. DPP-4 is a serine protease, which catalyzes the degradation of incretins, such as glucagon-like peptide-1 and glucose-dependent insulinotropic peptide, therefore its inhibitors are used as oral antidiabetic drugs (Hamdani et al., 2014). Furthermore, few cardioprotective peptides, including BNP, are catabolized and inactivated by DPP-4 (Hamdani et al., 2014). Circulating DPP-4 activity has also been found to be significantly elevated in type 2 diabetes mellitus (T2DM) patients and positively correlated with subclinical LV diastolic and systolic dysfunction, as well as in HF patients, when it was associated with adverse cardiovascular outcomes (dos Santos et al., 2013; Ravassa et al., 2013). Such evidence led to application of DPP-4 inhibitors in diastolic dysfunction settings. For instance, 8-week treatment with the DPP-4 inhibitor, sitagliptin, reduced serum DPP-4 activity and myocardial stiffness in obese T2D mice. Interestingly, this effect was mediated by augmentation of cGMP levels, PKG activity and subsequently, phosphorylation of titin N2B segments (Hamdani et al., 2014). A similar molecular mechanism was proposed for linagliptin, which was administered to obese ZSF1 rats for four weeks. As a result, LV relaxation parameters, LV passive stiffness were improved and phosphorylation of titin N2Bus segment was increased (Cuijpers et al., 2021). Findings included also a linagliptin-induced switch from stiffer N2B to the more compliant N2BA isoform in vivo, as well as decreased cleavage of N2BUs and PEVK segments in human cardiomyocytes (Cuijpers et al., 2021). As far as clinical use in HFpEF is concerned, there is an ongoing small clinical trial in HFpEF patients, which aims to evaluate the effects of sitagliptin on LV hypertrophy, diastolic function and hemodynamics (Wintrich et al., 2020). Therefore, further preclinical and clinical studies are needed to explore the potential benefit of those agents.
Cardiac fibrosis – targeting interventions
Among various mechanisms contributing to the complex HFpEF pathophysiology, cardiac interstitial fibrosis is prominent and represents an intriguing drug target. Interstitial fibrosis refers to the presence of collagen-rich extracellular matrix (ECM) among various cells in the interstitial space and is a common histological feature in the majority of endomyocardial biopsies from HFpEF patients (Sweeney et al., 2020). Collagen deposition requires its initial secretion in the interstitial space as procollagen, subsequent cleavage in the N-terminal end by procollagen N-proteinase and C-proteinase and release as mature collagen fibrils (Mishra and Kass, 2021). Besides, PICP (the C-terminal propeptide of procollagen type I) and PIIINP (the N-terminal propeptide of procollagen type III) have been used as biomarkers for myocardial fibrosis in HFpEF patients (Franssen and González Miqueo, 2016). In biopsies from hypertensive HFpEF patients, insoluble, total collagen, and collagen volume fraction, as well as collagen-dependent myocardial stiffness, were significantly increased (Zile et al., 2015). It was also shown that in similar biopsies, the ratio of stiffer collagen type I to the more compliant collagen type III was augmented, whereas matrix metalloproteinase-1 (MMP-1) expression was decreased. Furthermore, there was evidence of a inflammatory cell-driven profibrotic response due to significant expression of the profibrotic factor, TGFβ. These findings were positively correlated with severe diastolic dysfunction parameters (Westermann et al., 2011).
Disrupting transforming growth factor-β signaling
TGFβ stimulates, among various biological effects, procollagen synthesis, collagen deposition in ECM, and fibrogenesis. These actions are mediated by several signaling pathways, the most important of which is the canonical, SMAD-dependent pathway. As TGFβ binds to its transmembrane TGFβRI/II receptors, a heterotetrametric complex is formed and phosphorylation and subsequent activation of SMAD2 and SMAD3 are induced. Both SMAD isoforms, along with SMAD4, are associated to form a complex, which acts as a transcription factor in the nucleus. Several profibrotic genes, such as collagen, smooth muscle actin, and interleukin 11 are greatly expressed and fibrogenesis is induced (Sweeney et al., 2020). Therefore, inhibition of TGFβ activity represents an interesting drug target. In rodent HFpEF models, anti-TGFβ neutralizing antibodies or anti-TGFβRI kinase inhibitors decreased myocardial fibrosis and fibroblast activation and ameliorated diastolic dysfunction (Kuwahara et al., 2002; Sweeney et al., 2020). Similar results were shown when TGFβRI/II or SMAD3 genes were deleted in fibroblasts by genetic manipulation in transgenic HFpEF mouse models (Khalil et al., 2017). However, due to the involvement of TGFβ in additional pathways, such as those promoting survival or cell contractility during stress conditions, TGFβ signaling inhibition often results in significant toxicity, and further research on anti-TGFβ therapies has been halted. For example, TGFβ1 knockout mice are characterized by embryonic lethality and initiation of extensive inflammatory cascades, whereas in human, mutations in the TGFβ1 gene, leading to total loss of functionality, cause colitis or neurological disorders. In addition, mice models in which neutralizing antibodies were administered, exhibited acute and excessive LV dilatation as well as increased mortality despite reduced cardiac fibrosis (Sweeney et al., 2020) .
Mineralocorticoid receptors antagonism
Aldosterone has profibrotic capabilities as it promotes expression of myocardial procollagen N- and C-proteinases enhancer protein (PCPE)-1 and consequently, collagen deposition (Jellis et al., 2010). Binding of aldosterone to mineralocorticoid receptors also results in ROS production via NADPH oxidase facilitating fibrotic processes, initiation of inflammatory cascades, and differentiation of profibrotic M2 type macrophages and T cells (Patel and Shah, 2019; Sweeney et al., 2020). Therefore, mineralocorticoid receptor antagonists, especially potassium-sparing diuretic spironolactone, have been tested in animal HFpEF models and clinical trials. Small preclinical and human studies revealed significant clinical benefit, depicted as decreased myocardial longitudinal strain, collagen deposition, LV hypertrophy, and enhanced LV function (Kosmala et al., 2011; Leader et al., 2019). The most prominent clinical trial to date is the Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist (TOPCAT), an international phase III randomized, double-blind, and placebo-controlled clinical trial of spironolactone therapy in HFpEF patients, with a follow-up of nearly 3.5 years (NCT00094302). The trial did not reach its primary composite endpoint, comprising of death of cardiovascular cause and aborted cardiac arrest, or hospitalization for HF. However, post hoc analyses proved that there were regional variations because in patients recruited in America, spironolactone achieved significant reduction of the primary outcome versus the placebo group, in contrast to patients in Russia and Georgia, where no significant differences were observed. Those differences were attributed partly to disrupted enrollment and drug adherence, as well as different demographic and clinical characteristics, such as younger age and reduced LVEF of Russian and Georgian patients, making them a less relevant target group (Pfeffer et al., 2015). Therefore, the therapeutic potential of these antagonists is under discussion and the ongoing Spironolactone Initiation Registry Randomized Interventional Trial in Heart Failure with Preserved Ejection Fraction (SPIRRIT), a large phase IV pragmatic trial will provide further insights (NCT02901184) (Patel and Shah, 2019).
Galectin-3 inhibition
A novel underexplored drug target is galectin-3 (Gal-3), a β-galactoside binding and matricellular protein that is member of the lectin family (Boer et al., 2013). Gal-3 serum levels were found elevated in patients with HF, independent of their LVEF, and were associated with LV dysfunction and mortality (Ho et al., 2012). In another small cohort of patients, Gal-3 levels were similar between HFrEF and HFpEF patients, but the predictive value of its plasma levels was significantly stronger in the HFpEF group (de Boer et al., 2011). In the preclinical HFpEF models, Gal-3 expression was significantly upregulated. For example, in a hypertensive HF rat model, it was proved that binding of Gal-3 in ECM sites and in cardiac fibroblasts induced fibroblast proliferation, collagen synthesis and deposition, leading to LV dysfunction, excessive fibrosis, and enhanced macrophage infiltration into the myocardium (Sharma et al., 2004). In a TAC mouse model or REN2 rats, genetic deletion of the Gal-3 gene or administration of a Gal-3 inhibitor, N-acetyllactosamine, decreased collagen production, deposition, myocardial fibrogenesis, and attenuated LV dysfunction, reducing LVEDP and improving LV relaxation. This study was crucial because it established Gal-3 as a valid therapeutic target (Yu et al., 2013). As Gal-3 is not extensively explored, intensive research is needed to reveal its relevance to HFpEF and insights into its mechanism of action and development and testing of small molecule inhibitors are required.
Repurposing of pirfenidone from pulmonary to cardiac fibrosis in HFpEF
Pirfenidone, a small-molecule antifibrotic agent, which is currently approved for idiopathic pulmonary fibrosis (Sweeney et al., 2020) has been investigated for possible clinical benefit in cardiac fibrosis and HFpEF, as mechanisms of pulmonary and cardiac fibrosis coincide with each other (Aimo et al., 2020). Although it is not clear how pirfenidone exhibits its antifibrotic action, several mechanisms have been proposed. For example, pirfenidone lowers TGFβ expression, as well as its downstream targets, such as SMAD3. Consequently, pirfenidone inhibits proliferation, migration of cardiac fibroblasts and their differentiation into myofibroblasts (Lewis et al., 2019). It blocks various profibrotic growth factors and mediators, including platelet derived growth factor and basic fibroblast growth factor, and enhances matrix metalloproteinases activity (Aimo et al., 2020). In a mouse model of angiotensin II-induced cardiac hypertrophy, coadministration of pirfenidone with angiotensin II for two weeks reversed the fibrotic and hypertrophic effects of angiotensin II. Pirfenidone decreased heart weight and LV hypertrophy, as well as perivascular and myocardial interstitial fibrosis, and improved LV diastolic function parameters. Furthermore, pirfenidone normalized atrial and brain natriuretic peptides, TGFβ1 receptor and mineralocorticoid receptor expression (Yamazaki et al., 2012). In a TAC mouse model of hypertension and LV hypertrophy, pirfenidone halted profibrotic, hypertrophic, and inflammatory cascades. Short-term administration of pirfenidone after surgery, decreased fibrotic tissue area, collagen type I levels, and TGFβ1 expression. Moreover, LV contractility was enhanced and thickening of the LV chamber wall was reduced to normal levels. Interestingly, pirfenidone attenuated NLRP3 inflammasome expression in vivo, as well as H2O2 and hyaluronan-induced increase in NLRP3 and IL-1β expression in rat myocardial fibroblasts in vitro, exhibiting pleiotropic action (Wang et al., 2013). Considering the above intriguing findings, the efficacy and safety of pirfenidone are being investigated in HFpEF patients in the PIRfenidOne in patients with heart failUre and preserved lEfT venTricular Ejection fraction (PIROUETTE) clinical trial. Hopefully, the PIROUETTE, a randomized double-blind placebo-controlled phase II trial, will provide valuable insights into the antifibrotic effect of long-term treatment with pirfenidone, using specific diagnostic tools for cardiac fibrosis, such as cardiovascular magnetic resonance spectroscopy and recruiting a carefully selected target group (NCT02932566) (Lewis et al., 2019).
Interventions in advanced glycation-end products (AGEs) formation
Myocardial fibrosis can result from extensive cross-linking of collagen fibrils, which is facilitated, partly, by advanced glycation end-products (AGEs) (Nagueh, 2021). AGEs are formed by non-enzymatic glycosylation of proteins or lipids and contribute to oxidation of connective tissue proteins, including collagen and fibronectin, as well as formation of covalent bonds between them in the interstitial space (Jia et al., 2018). Deposition of AGEs is induced in hyperglycemia conditions, and has been proved in both diabetic HFrEF and HFpEF patients and correlated with augmented collagen formation and NO depletion, thus enhancing LV diastolic stiffness (van Heerebeek et al., 2008). Increased plasma AGEs levels of diabetic HF patients proved to be of predictive value for HF hospitalization and mortality (Willemsen et al., 2012). Apart from directly enhancing collagen cross-linking, binding of AGEs to their mutual receptors (RAGEs) in myocardium deteriorates oxidative stress, activates NFκΒ signaling cascade, TGF-β1/SMAD molecular pathways, and provokes cell death and replacement fibrosis (Jia et al., 2018; Paulus and Dal Canto, 2018). Prevention of their formation by administration of aminoguanidine has been tested in rats, which presented severe volume overload by arteriovenous fistula (Herrmann et al., 2003). Aminoguanidine halted AGEs cross-links formation in septum of treated rats and decreased LV longitudinal and septal circumferential stiffness (Herrmann et al., 2003). Another promising compound is alagebrium, which disrupts glucose crosslinks between collagen fibrils and has improved arterial compliance, stiffness, LV mass and distensibility in preclinical diabetic models (Redfield, 2005). Due to its potential in management of diastolic dysfunction and stiffness, it was administered in a small cohort of HFpEF patients resulting in improvement of LV diastolic filling and their quality of life after 16-week treatment (Little et al., 2005). A larger clinical study, recruiting healthy elderly individuals, is ongoing and aims to study the effect of alagebrium on myocardial stiffness, diastolic function and exercise tolerance (NCT01014572) (Zouein et al., 2013). However, according to the initial results, although alagebrium did not exhibit any significant improvement of exercise capacity, 1-year treatment resulted in a modest decrease of LV stiffness (Fujimoto et al., 2013). Therefore, inhibition of AGEs formation and signaling represents an intriguing drug target but it is underexplored in HFpEF setting.
Regenerative stem cell therapy as a therapeutic tool
An innovative therapeutic strategy for HFpEF is regenerative stem cell therapy, using cardiosphere-derived cells (CDCs). CDCs are yielded from cardiospheres, which are multicellular clusters formed by percutaneous endomyocardial biopsy specimens and then grown in primary cultures using specific techniques. These cells possess proliferative and cardiogenic properties and are anti-inflammatory and antifibrotic (Smith et al., 2007). Intracoronary CDCs infusion for four weeks exhibited significant clinical benefit in a HFpEF rat model. CDCs decreased diastolic dysfunction and CDC-treated rats showed dramatically less LV fibrotic changes, depicted as lower collagen type I and III levels. Expression of profibrotic and proinflammatory cytokines was significantly attenuated, as CDCs reduced TNFα, monocyte chemotactic protein-1 (MCP-1), IL-6, and tissue inhibitor of metalloproteinase-1 (TIMP-1), while infiltration of macrophages and leukocytes was also attenuated (Gallet et al., 2016). The randomized double blind placebo-controlled phase IIa clinical trial, Regression of Fibrosis & Reversal of Diastolic Dysfunction in HFPEF Patients Treated With Allogeneic CDCs (Regress-HFpEF), is currently recruiting volunteers. The trial will test whether treatment of HFpEF patients with intracoronary administration of allogeneic CDCs could provide benefit in clinical functional status, exercise tolerance, myocardial interstitial fibrosis, and diastolic function (NCT02941705) (Mishra and Kass, 2021).
Targeting mitochondrial dysregulation and metabolic abnormalities
Abnormal myocardial bioenergetics is typically present in the failing heart. The electron transport chain (ETC) is impaired and there is decreased ATP generation and energy supply (Lam et al., 2018b). Impaired energy homeostasis and reduced oxidative phosphorylation result in changes in substrate utilization and initially increased and subsequently decreased mitochondrial glucose oxidation and reduced fatty acid utilization. Consequently, lactate and proton production are upregulated and the heart manifests signs of intracellular sodium and calcium overload. Mitochondrial abnormalities are also evident in skeletal muscles, as their inability to meet energy demands during rest, and especially exercise, leads to limited working performance and exercise intolerance, a hallmark of HFpEF (Sabbah, 2020).
A study conducted in elderly HFpEF patients demonstrated that mitochondrial content, expressed as the mitochondrial membrane protein porin, and the mitochondrial fusion regulatory protein mitofusin 2 were decreased in Vastus lateralis skeletal muscle biopsy samples. These results, along with lower activity of citrate synthase in the same samples and their correlation with peak oxygen uptake, indicate severe mitochondrial dysfunction in skeletal muscle (Molina et al., 2016). Myocardial bioenergetics dysregulation was evident in HFpEF patients, who presented significantly decreased cardiac creatine phosphate/adenosine triphosphate (PCr/ATP) ratio at rest, as well as decreased peak oxygen uptake, heart rate, cardiac output, and impaired LV active relaxation during intense exercise, indicating poor energy reserves (Phan et al., 2009). Furthermore, in a mouse model of pressure overload-induced cardiac hypertrophy and diastolic dysfunction due to abdominal aortic constriction, severe metabolic defects were present. Mitochondrial oxidative metabolism of fatty acids, lactate, and palmitate was overall suppressed in isolated hearts three weeks after surgery. Mitochondrial complex V activity was also decreased, indicating impairment in energy substrate utilization and ETC function (Zhang et al., 2013). Therefore, mitochondrial function may be an optimal target for HFpEF.
Improving mitochondrial function by cardiolipin association
A promising modulator of mitochondrial bioenergetics in favor of efficient mitochondrial function is elamipretide, a mitochondrion-targeting tetrapeptide, which after penetrating cell membranes associates reversibly with cardiolipin, a phospholipid localized in the inner mitochondrial membrane. Binding of elamipretide to cardiolipin enhances cardiolipin function, as elamipretide protects it against oxidative damage (Birk et al., 2014). This complex contributes positively to maintenance of mitochondrial cristae architecture, optimal ETC function and stability, and mitochondrial biogenesis, thus retaining mitochondrial DNA stability and augmenting ATP synthesis (Karaa et al., 2018; Sabbah, 2020). In a canine model of microembolization-induced HF, chronic monotherapy with subcutaneous elamipretide injections improved LV systolic function, depicted by increased LVEF, restored plasma proinflammatory cytokines TNF-α, IL-6 and C-reactive proteiin (CRP) to normal levels and normalized mitochondrial respiration, ATP synthesis rate, and expression of mitochondrial fusion and fission proteins in LV tissue (Sabbah et al., 2016). The therapeutic potential of elamipretide led to development of a multi-center randomized double-blind placebo-controlled clinical trial (RESTORE-HF) in HFpEF patients to determine the effects of 4-week treatment with subcutaneous elamipretide on LV function (NCT02814097) (Sabbah, 2020). The pending results will provide further knowledge into the mechanism of action of elamipretide and the impact of mitochondrial dysfunction on HFpEF pathophysiology.
Partial adenosine receptors agonists
Adenosine receptors A1 (A1R) activation is an additional therapeutic intervention, as adenosine signaling promotes cardioprotection in multiple ways. For instance, it leads to inhibition of adenyl cyclase, lowering intracellular cAMP levels and halting sympathetic nervous system overexcitation, calcium overload, and extreme contractility. Furthermore, PKC is activated and consequently mitochondrial ATP-dependent K+ channels are modulated, decreasing prolonged mitochondrial permeability transition pore opening and ameliorating mitochondrial function (Sabbah, 2020). Under HF circumstances, partial A1R agonism is more favorable than full, as the latter results in adverse effects, such as atrio-ventricular block, bradycardia, negative inotropy, hypotension, sedation, and antidiuretic effects (Patel and Shah, 2019). Capadenoson is a partial A1R agonist and has been tested as an oral chronic 12-week therapy in dogs with microembolization-induced HF. Indeed, capadenoson administration resulted in LV systolic function improvement, LVEF increase, and reversed adverse LV remodeling, decreasing LV end-systolic volume. In addition, capadenoson decreased plasma norepinephrine levels, NT-proBNP levels, signs of interstitial fibrosis, and restored expression of mitochondrial uncoupling proteins, as well as mitochondrial citrate synthase activity (Sabbah et al., 2013). However, it was not well tolerated in clinical trials due to the presence of vertigo and dizziness, which were attributed to a high degree of agonism; it also exhibited poor solubility. Thus, a more soluble and stable prodrug, neladenoson bialanate hydrochloride, was developed. The prodrug is converted in vivo into neladenoson, a less potent partial agonist (Meibom et al., 2017). A chronic 20-week long therapy with oral neladenoson bialanate was evaluated in a Trial to Study Neladenoson Bialanate Over 20 Weeks in Patients With Chronic Heart Failure With Preserved Ejection Fraction (PANACHE), a randomized, placebo-controlled, parallel-group, double blind dose-finding phase II trial, which evaluated the potential effect of neladenoson on exercise capacity, LV function biomarkers, physical activity, and quality of life in HFpEF patients (NCT03098979). Nevertheless, initial results were disappointing, because it was reported that there were no clinically relevant increase in 6-minute walk test distance from baseline and no significant dose-response relationship (Shah et al., 2019). Further investigation is required if the potential clinical benefits of partial A1R agonism are to be explored in the future.
Inhibitors of beta-oxidation of long-chain fatty acids
Perhexiline is licensed as an antianginal agent in Australia and Asia and is starting to gain interest as a potential therapeutic strategy for HFpEF. The main mechanism of action is inhibition of carnitine palmitoyltransferase-1 (CPT-1). CPT-1 regulates mitochondrial uptake of long-chain fatty acids and their access to sites of beta-oxidation in the mitochondrial matrix. Thus, its inhibition shifts the main energy metabolic pathway from fatty acid to glucose oxidation (Lam et al., 2018b). Perhexiline improved diastolic function, without impact on systolic function, in isolated rat hearts during ischemia (Kennedy et al., 2000). A small, randomized, double-blind, placebo-controlled trial will test whether perhexiline improves exercise capacity and quality of life in HFpEF patients (NCT00839228). The pending results will provide insights into its possible role in enhancing cardiac energetics and diastolic function (Singh et al., 2014).
Ranolazine, commercially available as antianginal medication, is currently evaluated for its efficacy in HFpEF. Ranolazine exerts its antianginal pharmacological action via metabolic as well as membrane current potential-modifying mechanisms and has also exhibited infarct-limiting effects via activation of cardioprotective salvage pathways (Efentakis et al., 2016). In particular, ranolazine inhibits late inward sodium currents by blocking late INa channels. Consequently, it reduces sodium-triggered intracellular calcium overload and ameliorates LV diastolic tension and relaxation, as well as myocardial oxygen consumption (Steggall et al., 2017). Beta oxidation of fatty acids is also inhibited, and as a compensatory mechanism, glucose oxidation and pyruvate dehydrogenase activity are increased, promoting linkage of the glycolysis pathway to the citric acid cycle to carry out cellular respiration, hence the potential use of ranolazine in HF is being tested. (Mishra and Kass, 2021). In HF canine models, treatment with ranolazine demonstrated restoration of normal cardiomyocyte relaxation, decrease in resting tension, and in LVEDP (Sabbah et al., 2002; Undrovinas et al., 2006). Treatment with ranolazine was evaluated initially in HFpEF patients in the prospective, randomized, double-blind, placebo-controlled small proof-of-concept study, RAnoLazIne for the Treatment of Diastolic Heart Failure (RALI-DHF) clinical study (NCT01163734). Intravenous infusion of ranolazine, followed by 13-day oral administration, improved hemodynamic parameters, including LVEDP, but did not exhibit significant benefit in LV relaxation parameters (Maier et al., 2013). An additional clinical trial, The Effects of Ranolazine on Exercise Capacity in Patients with Heart Failure with Preserved Ejection Fraction (RAZE) trial, sought to provide further evidence of potential clinical benefit of ranolazine in HFpEF patients, evaluating its effect, primarily, on exercise capacity and peak oxygen consumption (NCT01505179). The first results showed that ranolazine significantly increased exercise capacity but did not change LV filling pressures and cardiac performance, revealing a potential role on partly ameliorating specific symptoms of HFpEF patients, including breathlessness and the ability to exercise (Miranda-Silva et al., 2021a).
A schematic representation of the four main pathomechanisms contributing to HFpEF development is illustrated in Figure 1.
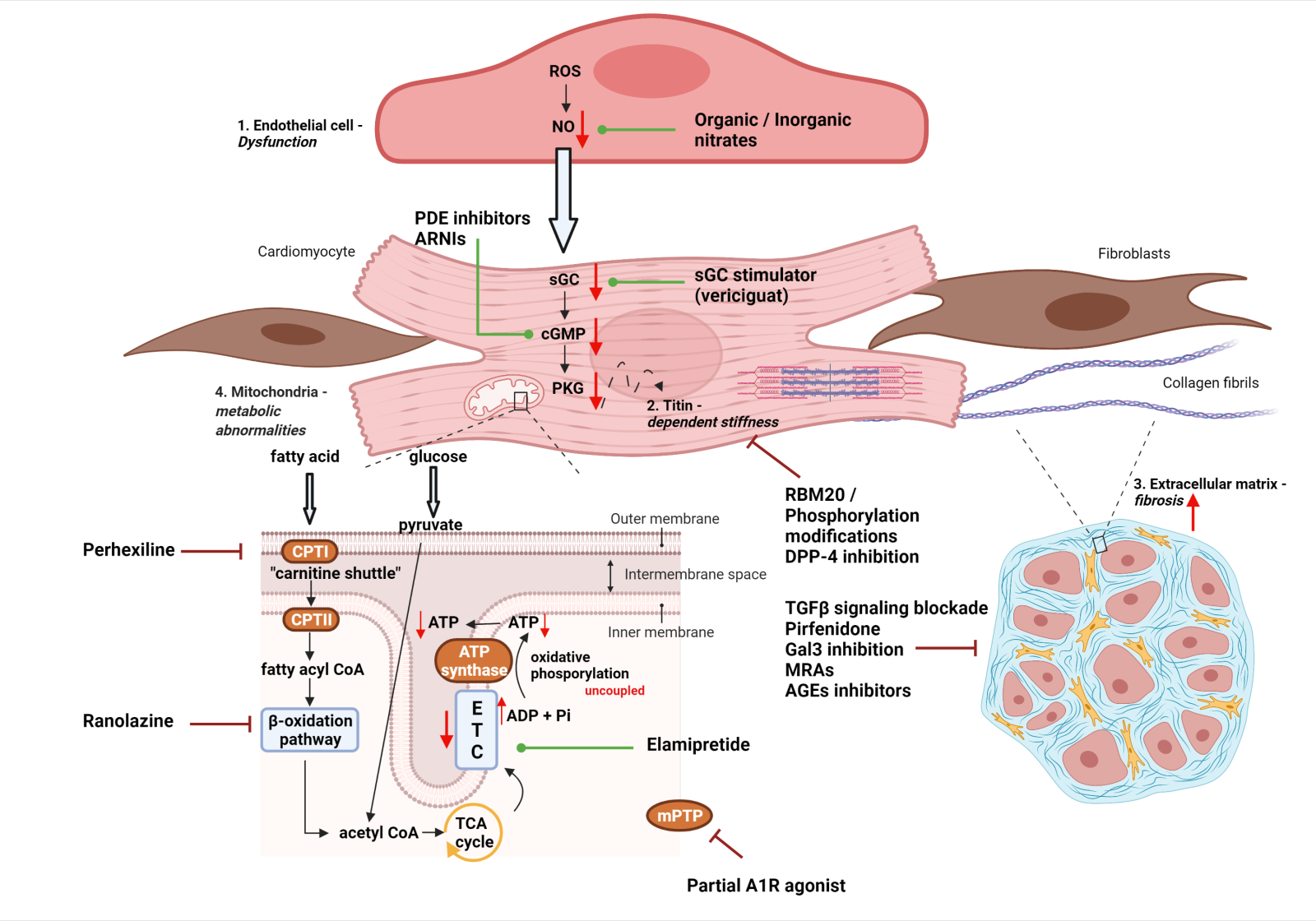
In a new window | Download PPT
Figure 1: Schematic representation of four main pathomechanisms (See respective modules) contributing to HFpEF development: 1. Endothelial dysfunction and microvascular inflammation 2. Titin – dependent myocardial stiffness 3. Cardiac fibrosis 4. Mitochondrial dysregulation. Potential pharmacological interventions are also highlighted in each mechanistic pathway. Red arrows (down/up) symbolize downregulation or upregulation, green lines leading to dots refer to positive pharmacological modulation, whereas dark red inhibitor lines refer to inhibitory interventions. Abbreviations: ADP, adenosine diphosphate; AGEs, advanced glycation end-products; ARNIs, angiotensin receptor and neprilysin inhibitors; ATP, adenosine tripshosphate; cGMP, cyclic guanosine monophosphate; CPT, carnitine palmitoyltransferase; DPP-4, dipeptidyl peptidase-4; ETC, electron transport chain; Gal3, galectin -3; MRAs, mineralocorticoid receptor antagonists; NO, nitric oxide; PDE, phosphodiesterase; PKG, protein kinase G; RBM20, RNA-binding motif protein 20; ROS, reactive oxygen species; sGC, soluble guanylate cyclase; TCA, tricarboxylic acid; TGFβ, transforming growth factor – β. (Created with BioRender.com)
Experimental rodent models of heart failure with preserved ejection fraction
Our review focuses, specifically, on small animal models as they are widely used in HFpEF preclinical studies. Rodents are preferred over large animals due to their ease of handling and breeding, short-term gestation and cheap laboratory housing. Use of large animals, despite their resemblance to human physiology, is limited because of demanding housing facilities, higher cost and ethical considerations expressed by animal welfare committees (Andreadou et al., 2020). Rodents are also more convenient in terms of executing genetic manipulations and developing various genetic strains in short period of time. These genetically modified models can provide insights into the function of specific molecular targets or signaling pathways (Riehle and Bauersachs, 2019). Therefore, they can be utilized to explore discrete HFpEF pathomechanisms, individual contributions of risk factors and potential therapeutic strategies, without presence of confounding factors, observed in larger animal species (Valero-Muñoz et al., 2017). Moreover, small animals can undergo multiple surgical and pharmacological interventions simultaneously, in a high throughput-like manner at a lower cost because they require short follow-up periods due to their short lifespan and fewer resources (e.g. lower dosage schemes respective to lower body mass) (Milani-Nejad and Janssen, 2014). Hence, we firmly believe that small animal models could provide a relatively fast-paced and readily manipulatable platform for testing candidate drug molecules and deciphering their mechanism of action in HFpEF.
Challenges in development of HFpEF animal models
It has proved challenging to carry over HFpEF heterogeneity to disease models. Most of the experimental models focus on distinct pathophysiological features, such as diastolic dysfunction, and fail to recapitulate the whole spectrum of HFpEF phenotypes. An approach toward this direction is development of comorbidities, including diabetes mellitus or hypertension, in otherwise healthy animal models to result in the intended phenotype. However, animal models eventually exhibit no more than two comorbidities, whereas HFpEF patients have a complex medical history (Roh et al., 2017). HFpEF is also typically referred to as a “disease of the elderly”, whereas most animal models used relatively young animals. Furthermore, heart failure in humans is a progressive disease, spanning over a long period. In animal models though, heart failure occurs all at once, as a consequence of a surgical or pharmacological intervention (Conceição et al., 2016).
Despite the above considerations, several animal models have been developed. They are far more money and time-consuming, and they are more demanding to develop than HFrEF ones and present only a fraction of all HFpEF clinical entities. However, they are considered to be an asset in our way of validating the aforementioned drug targets and answering biological questions.
Hypertensive models
Hypertension usually coexists and contributes to the progression of HFpEF. About 55-86% HFpEF patients are hypertensive. Thus, hypertensive rodent models, typically characterized by increased LV afterload and concentric hypertrophy, are a significant research tool (Horgan et al., 2014; Conceição et al., 2016).
Dahl salt-sensitive (Dahl/SS) rats represent a popular model, which is bred from Sprague-Dawley rats and is hypersensitive to salt intake (Conceição et al., 2016). Dahl/SS rats are fed with a high-salt diet, including 8% sodium chloride, starting at 7 weeks old (Rapp and Dene, 1985). Elevation in systolic blood pressure (>175 mmHg) is evident by 9 weeks and developed hypertension leads to HF and cardiac hypertrophy. HF symptoms such as tachypnea, dyspnea, and exercise intolerance show up by the 12th week and clinical signs of severe diastolic dysfunction and LV chamber stiffness are present. Nevertheless, LV systolic function and LVEF are not decreased until week 19, when HFpEF shifts to a HFrEF phenotype (Doi et al., 2000; Qu et al., 2000). In most studies, investigational drugs are administered from week 13 and experiments are terminated at weeks 19-21. A disadvantage of this model is limited clinical relevance to HFpEF patients, because in those rats, in contrast to patients, HFpEF comes from a single dietary intervention and subsequent water and sodium retention (Horgan et al., 2014). In this preclinical setting, aforementioned cardiosphere-derived cells attenuated inflammatory infiltration, fibrotic markers, and normalized diastolic function (Gallet et al., 2016).
The deoxycorticosterone acetate (DOCA) salt rat model is an example of pharmacologically induced hypertension. DOCA is administered at a high dose via intraperitoneal injection or subcutaneous pellet implantation one week after unilateral nephrectomy, which is usually performed at 6 weeks of age. Sodium chloride at 1% is added to drinking water, expediting transition into severe hypertension (Sun and Zhang, 2005). After 4-5 weeks of treatment, hypertension, cardiac and renal hypertrophy, as well as myocardial and perivascular fibrosis are evident (Grobe et al., 2006; Gomes et al., 2013). Myocardial inflammation, oxidative stress, increased stiffness, and diastolic dysfunction have been also reported (Conceição et al., 2016). Variations combine DOCA administration with TAC to induce further pressure overload by surgical means. For instance, genetic modulation of RBM20, upregulating compliant titin isoforms, ameliorated myocardial stiffness in a TAC/DOCA mice model (Methawasin et al., 2016). However, requirements for surgery of unilateral nephrectomy, large DOCA amounts, and strict control of dietary salt make the model pretty arduous (Gomes et al., 2013).
TAC in rodents is an established model of LV pressure overload. It involves surgical intervention, during which blood flow across the aortic arch is restricted by banding, as sutures or surgical clips are placed onto the ascending aorta. Considerable elevation in LV after load, nd impaired diastolic filling are provoked, causing LV concentric hypertrophy and HF (Riehle and Bauersachs, 2019). Aortic banding is performed during the 3rd or 4th week of age in rats, hypertrophy is evident by week 6 and diastolic function abnormalities are fully present at the 12th week until the 18th week, when the HFpEF phenotype is transitioning to LV dilatation, systolic dysfunction, and HFrEF (Litwin et al., 1995). N-acetyllactosamine, a Gal-3 inhibitor, reversed adverse LV remodeling in a 4-week long TAC-induced mouse model (Yu et al., 2013). However, this model is not translatable, as essential hypertension and aortic stenosis develop gradually in humans contrary to the acute onset of TAC - induced hypertension in rodents (Horgan et al., 2014).
Angiotensin II subcutaneous infusion is applied in order to develop rodent HFpEF models, because the subsequent β-adrenergic stimulation results in cardiac hypertrophy, hypertension, inflammation, and diastolic dysfunction (Horgan et al., 2014). Fourteen day treatment with AngII in male 9-week old C57/BL6 wild-type mice led to development of concentric hypertrophy, impaired cardiac energy metabolism, raised interstitial fibrosis, and natriuretic peptide levels, whereas systolic function was preserved (Mori et al., 2012). It should be noted that frequently used BALB/c mice present, additionally, strain-specific dilated LV chamber and severe systolic dysfunction with reduced ejection fraction as a consequence of a similar intervention. This model has been used to reveal mechanistic insights into the anti-fibrotic role of pirfenidone and the capacity of sildenafil to decrease immune cells infiltration, apoptosis, and adverse LV remodeling (Westermann et al., 2012; Yamazaki et al., 2012).
Models featuring diabetes mellitus and/or obesity
Obesity and T2DM are common causes of impaired myocardial diastolic function, due to their capacity to induce oxidative stress, mitochondrial dysregulation, abnormal myocardial energetics and cellular lipotoxicity (Andreadou et al., 2021). For instance, diastolic dysfunction is already evident in young diabetic patients, who display abnormal diastolic filling (Schannwell et al., 2002). Thus, it comes as no surprise that T2DM is present in one-third of HFpEF patients and obesity is prevalent in 41-46% of HFpEF patients as well (Lam et al., 2011). Obesity and T2DM rodent models are also deployed in HFpEF preclinical studies because they exhibit characteristics observed in HFpEF patients and can be utilized to investigate pathophysiological features and therapeutic interventions for the disease (Conceição et al., 2016).
Common genetically modified mice DM models include db/db (diabetic) and ob/ob (obese) models. These models are characterized by either hypothalamic leptin receptor or functional leptin deficiency, respectively, and consequently increased appetite and body weight, and decreased energy expenditure, leading to obesity by 4 weeks, with hyperinsulinemia and diabetes by 15 weeks. Diastolic dysfunction develops progressively, as myocardial oxygen consumption rises, limiting cardiac efficiency (Noll et al., 2020). Db/db mice bear a point mutation in the diabetes gene (db), responsible for encoding the leptin receptor (Chen et al., 1996). Nine week old db/db mice exhibit increased LV mass and wall thickness but decreased LV end-diastolic volume by 13 weeks, when diastolic dysfunction and impaired ventricular compliance are evident (Horgan et al., 2014). Ob/ob leptin – deficient mice also have manifested neutral lipid accumulation into cardiomyocytes, which correlate with diastolic dysfunction (Christoffersen et al., 2003). Disadvantages of these models include compounding effects of defective leptin signaling. For instance, tyrosine kinase signaling pathways are not affected in db/db mice cardiomyocytes in contrast to the failing human heart, and ob/ob mice exhibit altered responses to myocardial injury because of their repressed innate and acquired immune responses (Noll et al., 2020).
The Zucker fatty rat model is another model, with a mutation called “fatty or fa” in the Lepr gene, which lowers affinity of leptin for its respective receptor. These rats manifest hyperphagia, obesity, and insulin resistance but are not diabetic. However, there is a variation of this model, the Zucker diabetic fatty rat model (ZDF), which occurred by inbreeding, using obese male and diabetic Zucker rats (Velez et al., 2014). Both gradually develop LV hypertrophy, elevated LV mass, major diastolic and moderate systolic dysfunction, and preserved LVEF. Cardiac fibrosis and microvascular dysfunction have also been reported (Valero-Muñoz et al., 2017). Long-term PDE5 inhibition by vardenafil administration restored cGMP/PKG signaling and alleviated hypertrophy, fibrosis, and diastolic dysfunction in ZDF rats (Mátyás et al., 2017).
Models with multiple cardiometabolic syndrome features
The Dahl/SS/obese rat model developed from crossbreeding between Dahl/SS and Zucker rat strains. Diastolic dysfunction appears by 15 weeks of age in female Dahl/SS/obese rats. They present signs of marked LV concentric hypertrophy, perivascular and interstitial fibrosis and, at the cellular level, oxidative stress and inflammation, depicted as upregulation of NADPH oxidase, TNF-α, and IL-6 expression (Murase et al., 2012).
Crossing the lean female ZDF rats with the lean male spontaneously hypertensive HF ones, resulted in development of the diabetic Zucker fatty spontaneously hypertensive heart failure F1 hybrid (ZSF1) model (Valero-Muñoz et al., 2017). By 20 weeks of age, ZSF1 rats present a wide spectrum of high metabolic risk features, including hypertension, obesity, T2DM, insulin resistance, hyperinsulinemia, hypercholesterolemia, and hypertriglyceridemia (Hamdani et al., 2003). HFpEF is established between 10 and 20 weeks, accompanied by concentric LV hypertrophy and aggravating diastolic function (Valero-Muñoz et al., 2017). HFpEF in a ZSF1 background shares common features with human HFpEF, because LV systolic function and LVEF are preserved, with exercise intolerance along with decreased effort tolerance and peak oxygen capacity being evident too (Horgan et al., 2014; Conceição et al., 2016). Important features shared between rats and human patients are evident, such as myocardial stiffness due to hypophosphorylation of N2B titin isoforms, and replacement fibrosis (Hamdani et al., 2003). Indeed, titin phosphorylation and collagen expression levels were normalized, whereas LVEDP and heart weight were decreased in ZSF1 rats, which were administered 12-week long treatment with sacubitril/valsartan (Schauer et al., 2021). ZSF1 is one of the most translatable HFpEF models as it resembles the HFpEF human disease state, recapitulating multiple comorbidities and histological features. It is not suitable, though, for preclinical studies, which seek to address each HFpEF phenotype separately, owing to undesirable confounding factors.
Last but not least, a new mouse “two-hit” HFpEF model was developed recently, which combines metabolic syndrome manifestations – obesity and glucose intolerance – with hypertension and nitrosative stress (Schiattarella et al., 2019). To elaborate, C57BL/6N mice were fed with a high – fat diet and treated with the NO synthases inhibitor, Nω-nitro-L-arginine methyl ester (L-NAME) for 5 to 15 weeks. Those mice exhibited elevated filling pressure, effort intolerance, signs of cardiac hypertrophy, fibrosis, inflammation, and pulmonary congestion (Amgalan and Kitsis, 2019; Schiattarella et al., 2019). These features, along with preserved LVEF even at one-year time course, render this model rather promising and close to the human phenotype. A downside of the model is onset of hypertension due to L-NAME intervention rather than fluid, salt retention, or other human – related conditions (Mishra and Kass, 2021).
A schematic diagram of the above-mentioned HFpEF animal models is presented in Figure 2.
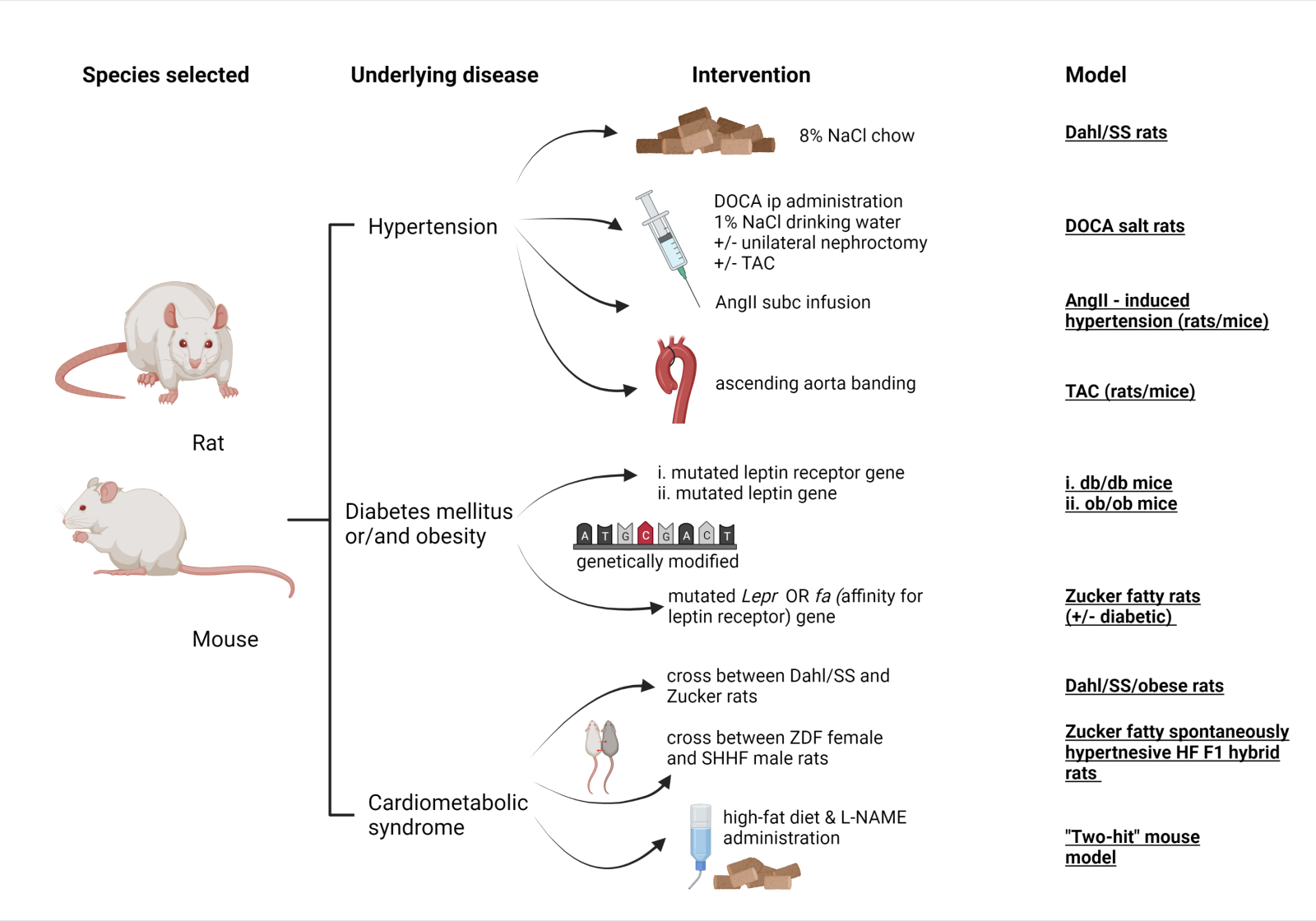
In a new window | Download PPT
Figure 2: Schematic diagram of the referred HFpEF rodent (mice, rats) models, including species (1st column), main comorbidity (2nd column), stressor for induction of the disease (3rd column) and commonly used name of the model (4th column). Abbreviations: AngII, angiotensin – II; DOCA, deoxycorticosterone acetate; L-NAME, L-NG-Nitro arginine methyl ester; SHHF, spontaneously hypertensive heart failure; SS, salt – sensitive; subc, subcutaneous; TAC, transverse aortic constriction; ZDF, Zucker diabetic fatty (Created with BioRender.com)
Conclusions and future perspectives
HFpEF remains an unsolved public health problem, due to high event rates, comparable to other cardiovascular diseases or cancer entities. It places a significant burden on the healthcare system and patients’ quality of life. Even though preclinical studies show encouraging results in early drug development, the majority of large, phase II/III trials have not met their primary composite endpoints and there is currently no successful targeted treatment. It is evident that HFpEF is a multisystem disease and intensive preclinical research is needed before onset of new clinical trials. Preclinical research must focus on elucidating key pathogenic processes taking place at a cellular or subcellular level, as these will open the door for the discovery of novel interventions, in addition to those referred in our review.
We reported herein four distinctive HFpEF pathomechanisms and focused on possible drug interventions. Nevertheless, we acknowledge the presence of additional molecular and cellular mechanisms, which can provide novel drug targets. For instance, a principal component of diastolic dysfunction which is oddly underexplored in HFpEF, in contrast to HFrEF, is myocardial excitation-contraction coupling and calcium handling (Gladden et al., 2014). There is evidence of decreased sarcoplasmic reticulum Ca2+ ATPase pump (SERCA2a) and Na+/Ca2+ exchanger activity in preclinical models of diastolic dysfunction, in accordance with increased diastolic Ca2+ concentrations and abnormal relaxation. These findings, along with promising therapeutic strategies to enhance SERCA2a function (e.g. gene therapy or SERCA2a activators) and ameliorate calcium dysregulation are beyond the scope of this review and we refer the reader to exceptional reviews (Lin et al., 2020; Miranda-Silva et al., 2021).
Nonetheless, thorough preclinical research is needed to address all those unanswered questions regarding the multifactorial nature of HFpEF and establishment of novel drug targets. Shortage of relevant preclinical animal models represents one of the main reasons why HFpEF treatment is still not successful, thus halting the progress of innovative preclinical research.
The existing animal models do not relate adequately to human phenotypes and are complicated to develop, creating a great barrier. For instance, we consider the progressive transition to HFrEF as a major limitation of these models, because preserved EF, along with evident diastolic dysfunction, result in only a transient state throughout the development of the diseased phenotype. We posit that most rodent models recapitulate HFpEF pathophysiological characteristics due to external stressors, instead of a gradual comorbidity – driven dysregulation. Therefore, abrupt insults, such as myocardial infarction, are frequent and consequently rapid development of LV remodeling, fibrosis, hypertrophy, or wall thinning lead to systolic dysfunction and moderate to reduced EF (Noll et al., 2020). This phenomenon contrasts with the human HFpEF phenotype, because abrupt decline of EF and systolic function rarely occurs in HFpEF patients, but occurs over a multi-year time course as evident also from recent long-term longitudinal studies (Lupón et al., 2019; Mishra and Kass, 2021). Frequent monitoring of diastolic and systolic function of small animals via echocardiographic measurements may prove efficient in preventing the aforementioned transition, even though development of more appropriate HFpEF preclinical models remains crucial for enhancing our understanding of the HFpEF fundaments (Noll et al., 2020). Hopefully, such optimization of preclinical HFpEF models will increase their external validity, thus mimicking the human condition and bridging the translational gap between the species being studied and the species needing to be treated.
Another crucial aspect to be addressed is the proper design of clinical trials, so that target groups of HFpEF patients are correctly enrolled using appropriate biomarkers and diagnostic approaches, including imaging, biopsies, and blood analyses, and these should be carefully monitored throughout the whole study. Phenotyping of patients according to the most prominent underlying pathomechanism should also become a priority for a phenotype-specific and disease-modifying therapeutic approach to be developed. At last, there are still much to be elucidated about the challenging HFpEF syndrome and establishment of inter – laboratory collaboration or an international consortium of multidisciplinary HFpEF experts should be taken into consideration so as to boost fruitful discussion and exchange of knowledge about underexplored aspects of HFpEF.
Conflicts of interest
The authors declare that they have no conflicts of interest.
References
Theano Dermintzoglou1
1Laboratory of Pharmacology, School of Pharmacy, National and Kapodistrian University of Athens, 15771, Athens, Greece
Ioanna Andreadou1
1Laboratory of Pharmacology, School of Pharmacy, National and Kapodistrian University of Athens, 15771, Athens, Greece
Corresponding author:
Andreadou Ioanna
Email: janandread@pharm.uoa.gr
In a new window | Download PPT
Figure 1: Schematic representation of four main pathomechanisms (See respective modules) contributing to HFpEF development: 1. Endothelial dysfunction and microvascular inflammation 2. Titin – dependent myocardial stiffness 3. Cardiac fibrosis 4. Mitochondrial dysregulation. Potential pharmacological interventions are also highlighted in each mechanistic pathway. Red arrows (down/up) symbolize downregulation or upregulation, green lines leading to dots refer to positive pharmacological modulation, whereas dark red inhibitor lines refer to inhibitory interventions. Abbreviations: ADP, adenosine diphosphate; AGEs, advanced glycation end-products; ARNIs, angiotensin receptor and neprilysin inhibitors; ATP, adenosine tripshosphate; cGMP, cyclic guanosine monophosphate; CPT, carnitine palmitoyltransferase; DPP-4, dipeptidyl peptidase-4; ETC, electron transport chain; Gal3, galectin -3; MRAs, mineralocorticoid receptor antagonists; NO, nitric oxide; PDE, phosphodiesterase; PKG, protein kinase G; RBM20, RNA-binding motif protein 20; ROS, reactive oxygen species; sGC, soluble guanylate cyclase; TCA, tricarboxylic acid; TGFβ, transforming growth factor – β. (Created with BioRender.com)
In a new window | Download PPT
Figure 2: Schematic diagram of the referred HFpEF rodent (mice, rats) models, including species (1st column), main comorbidity (2nd column), stressor for induction of the disease (3rd column) and commonly used name of the model (4th column). Abbreviations: AngII, angiotensin – II; DOCA, deoxycorticosterone acetate; L-NAME, L-NG-Nitro arginine methyl ester; SHHF, spontaneously hypertensive heart failure; SS, salt – sensitive; subc, subcutaneous; TAC, transverse aortic constriction; ZDF, Zucker diabetic fatty (Created with BioRender.com)
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11991 | 48 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA