Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Conditioning Against the Pathology of Parkinson’s disease
Time:2018-04-29
Number:13254
Author Affiliations

Conditioning Medicine, 2018. 1(3):143-162.
Abstract
Parkinson’s disease is delayed in clinical onset, asymmetric in initial appearance, and slow in progression. One explanation for these characteristics may be a boost in natural defenses after early exposure to mild cellular stress. As the patient ages and resilience recedes, however, stress levels may become sufficiently high that toxic cellular responses can no longer be curbed, culminating in inverted U-shaped stress-response curves as a function of disease duration. If dopaminergic systems are indeed capable of responding to mild stress with effective natural defenses, experimental models of Parkinson’s disease should adhere to the principles of preconditioning, whereby stress exposure fortifies cells and tempers the toxic sequelae of subsequent stressors. Here, I review evidence favoring the efficacy of preconditioning in dopaminergic systems. Recent animal work also raises the possibility that cross-hemispheric preconditioning may arrest the spread of asymmetric Parkinson’s pathology to the opposite side of the brain. Indeed, compensatory homeostatic systems have long been hypothesized to maintain neurological function until a threshold of cell loss is exceeded, and are often displayed as inverted U-shaped curves. However, some stress responses assume an exponential or sigmoidal profile as a function of disease severity, suggesting end-stage deceleration of disease processes. Thus, surviving dopaminergic neurons may become progressively harder to kill, with the dorsal nigral tier dying more slowly due to superior baseline defenses, inducible conditioning capacity, or delayed dorsomedial nigral spread of disease. In addition, compensatory processes may be useful as biomarkers to distinguish “responder patients” from “nonresponders” before clinical trials. However, another possibility is that defenses are already maximally conditioned in most patients and that no further boost is possible. A third alternative is that genuinely diseased human cells cannot be conditioned, in contrast to cells in preclinical models, none of which faithfully simulate age-related human conditions. Disease-related “conditioning deficiencies” would then help explain how Parkinson’s pathology takes root, progressively shrinks defenses, and eventually kills the patient.
Keywords: preconditioning, hormesis, dopamine, compensatory, basal ganglia, Lewy, synuclein
Abstract
Parkinson’s disease is delayed in clinical onset, asymmetric in initial appearance, and slow in progression. One explanation for these characteristics may be a boost in natural defenses after early exposure to mild cellular stress. As the patient ages and resilience recedes, however, stress levels may become sufficiently high that toxic cellular responses can no longer be curbed, culminating in inverted U-shaped stress-response curves as a function of disease duration. If dopaminergic systems are indeed capable of responding to mild stress with effective natural defenses, experimental models of Parkinson’s disease should adhere to the principles of preconditioning, whereby stress exposure fortifies cells and tempers the toxic sequelae of subsequent stressors. Here, I review evidence favoring the efficacy of preconditioning in dopaminergic systems. Recent animal work also raises the possibility that cross-hemispheric preconditioning may arrest the spread of asymmetric Parkinson’s pathology to the opposite side of the brain. Indeed, compensatory homeostatic systems have long been hypothesized to maintain neurological function until a threshold of cell loss is exceeded, and are often displayed as inverted U-shaped curves. However, some stress responses assume an exponential or sigmoidal profile as a function of disease severity, suggesting end-stage deceleration of disease processes. Thus, surviving dopaminergic neurons may become progressively harder to kill, with the dorsal nigral tier dying more slowly due to superior baseline defenses, inducible conditioning capacity, or delayed dorsomedial nigral spread of disease. In addition, compensatory processes may be useful as biomarkers to distinguish “responder patients” from “nonresponders” before clinical trials. However, another possibility is that defenses are already maximally conditioned in most patients and that no further boost is possible. A third alternative is that genuinely diseased human cells cannot be conditioned, in contrast to cells in preclinical models, none of which faithfully simulate age-related human conditions. Disease-related “conditioning deficiencies” would then help explain how Parkinson’s pathology takes root, progressively shrinks defenses, and eventually kills the patient.
Keywords: preconditioning, hormesis, dopamine, compensatory, basal ganglia, Lewy, synuclein
Introduction
Parkinson’s disease is a chronic neurodegenerative condition characterized by slow, progressive loss of dopaminergic neurons in the substantia nigra pars compacta, following retrograde degeneration of their efferent projections to the dorsal striatum (Kish et al., 1988; Rinne, 1993; Cheng et al., 2010). There has been a major revision of the traditional view that the loss of neurons in Parkinson’s disease is restricted to the nigrostriatal pathway, and it is now widely accepted that this is a systemic, proteinopathic disorder with hallmark inclusions known as Lewy bodies deposited in many parts of the brain, spinal cord, and peripheral organs, including the olfactory bulb and gastrointestinal tract (Braak et al., 2002; Braak et al., 2003b; Braak et al., 2003a; Hawkes et al., 2007; Beach et al., 2009b; Beach et al., 2009a; Jucker and Walker, 2013; Fasano et al., 2015; Adler and Beach, 2016; Del Tredici and Braak, 2016). Aside from well-established loss of somata in the substantia nigra, there appears to be additional cell loss in the anterior olfactory nucleus (Pearce et al., 1995), locus coeruleus (German et al., 1992; Zarow et al., 2003), pedunculopontine tegmental nucleus and raphe nuclei (Hirsch et al., 1987; Jellinger, 1988; Halliday et al., 1990a; Halliday et al., 1990b), rostral ventrolateral medulla and dorsomotor nucleus of the vagus (Halliday et al., 1990b), hypothalamic orexin+ neurons (Fronczek et al., 2007; Thannickal et al., 2007), and cholinergic basal forebrain (Candy et al., 1983; Nakano and Hirano, 1984; Hall et al., 2014), among other sites. The widely distributed extranigral cell loss and the gradual expansion of Lewy pathology across the neuraxis likely contribute to nonmotor parkinsonian deficits that were not widely discussed until recently—including anosmia, depression, sleep disturbances, and, at end stages, dementia (Pellicano et al., 2007; Verbaan et al., 2007; Simuni and Sethi, 2008). Nevertheless, the majority of experimental and clinical studies of Parkinson’s disease concentrate on dopaminergic cell death, as loss of the nigrostriatal pathway is clearly associated with some of the movement deficits originally reported by James Parkinson in 1817 (Goetz, 2011; Goedert et al., 2013).
The present review focuses on the concept that endogenous mechanisms are mobilized by disease-related stressors to spur tissue repair and recovery, galvanize defenses, and buttress cells against future challenges in the context of Parkinson’s disease. This topic is significant because functional outcomes are often better associated with neuroplasticity and compensatory mechanisms than classic, histological hallmarks of neurodegenerative disorders (Iacono et al., 2009; Lo, 2010). Furthermore, the insoluble Lewy pathology in Parkinson’s disease may reflect an adaptive attempt to sequester damaged proteins and shunt toxic oligomeric species away from wreaking havoc in the watery cytosol (Tanaka et al., 2004; Au and Calne, 2005; Beyer et al., 2009). Compensatory mechanisms may predominate during prodromal stages of Parkinson’s disease, before massive dopaminergic cell loss unfolds. Therefore, the likelihood of identifying clinically effective neuroprotectants is higher in earlier disease stages, although robust biomarkers will be needed to identify these largely asymptomatic subjects (Beach, 2017). Indeed, the major distinction between functional responders and nonresponders in a clinical trial may be the compensatory mechanisms (Lo, 2010), and even “nonspecific” biomarkers of compensatory stress responses might serve as bellwethers of the efficacy of therapeutic interventions before the trial commences. An alternative possibility, however, is that natural defenses are already maximally deployed in most patients, and that they may have decelerated disease onset and progression as far as possible until age-related damage became unsurmountable. Therefore, it may not be possible to boost this optimized system with anything other than lifestyle interventions. This alternative is supported by a large body of work showing that the amplitude of beneficial stress responses is usually restricted to 30-60% of control values (discussed further in Section III). A third grim alternative is that genuinely diseased human brains exhibit from the beginning intractable deficiencies in compensation, repair, and recovery, unlike non-diseased, albeit injured cells in the classic models of dopaminergic cell loss. The third scenario would give the proteinopathic seeds unfettered license to plant themselves and propagate throughout the neuraxis. Although there are many other variables not considered here, the second and third scenarios would impede the translation of preclinically efficacious neuroprotectants into fruitful clinical interventions.
What underlies the slow rate of progression of most neurodegenerative disorders?
The proteinopathy that characterizes all neurodegenerative disorders may commence in the form of insoluble protein deposits at surprisingly young ages, extending as far as 25-50 years prior to the manifestation of cardinal symptoms (Claassen et al., 2010; Savica et al., 2010; Braak et al., 2011). In contrast, dopaminergic cell loss per se is thought to extend ~5-6 years before the movement disorder is diagnosed by a clinician and to continue for many additional years, based partly on extrapolation of 18F-Fluorodopa positron emission tomography (PET) of dopaminergic terminal loss in the striatum and histological patterns of nigral cell death in postmortem tissue (Fearnley and Lees, 1991; Morrish et al., 1998; Hilker et al., 2005; Savica et al., 2010). This type of progressive cell loss is fundamentally distinct from the relative abrupt loss of massive cell numbers in stroke or traumatic brain injury. Here I will list three possible explanations for the slow progression of proteinopathy and dopaminergic cell loss in Parkinson’s disease:
A) Biphasic (nonlinear, non-monotonic) or U-shaped stress responses: Natural compensatory defenses may be upregulated by mild stress in early disease phases and may delay the onset and slow disease progression as a form of “conditioning.” At later disease stages, endogenous defenses may be depleted or overwhelmed by inexorable accrual of cellular damage, age-related decay in resilience, and engagement of senescence programs. These opposing effects would result in biphasic stress-response patterns plotted as inverted U-shaped curves as a function of disease duration or severity.
B) Exponential or sigmoidal (nonlinear, monotonic) stress responses: Early in the course of the disease, there may be relatively rapid loss of cells, followed by a deceleration as the more resistant, surviving cells become progressively harder to kill and their recalcitrance slows disease progression. Rather than assuming a U-shaped profile, these stress responses would be plotted as negative exponential or sigmoidal curves with rapid early response rates yielding to saturating kinetics at end stages of the disease. Thus, disease progression would decline as it approaches a theoretical asymptote or maximum (e.g., zero remaining dopaminergic cells) while death draws near.
C) Linear, monotonic stress responses: There might be linear, albeit sluggish (i.e., low-sloped) decreases in endogenous defenses and cell numbers, combined with linear increases in insoluble protein deposits as the disease completes its natural course, with no change in the rate of progression over time after disease onset and until death.
In the vast clinical literature on Parkinson’s disease, all three types of progression have been plotted as a function of years from clinical diagnosis or extrapolated into the prodromal phase of the illness. The most frequently cited example of compensatory changes in early-to-mid Parkinson’s disease is a potential increase in dopaminergic tone in the remaining axon terminals of the mildly denervated striatum (Zigmond, 1997; Nandhagopal et al., 2011). According to this perspective, a clinical syndrome is only manifested once a critical threshold of cell loss is exceeded in the substantia nigra, and the compensatory mechanisms in the striatum are no longer sufficient to maintain normal dopamine equilibrium. Although currently debated, the mechanisms underlying the delay in movement deficits (relative to the onset of dopaminergic cell loss) have been attributed to increases in dopamine release and metabolic turnover, upregulation of dopamine synthesis by the enzymes aromatic L-amino-acid decarboxylase and tyrosine hydroxylase, less removal of synaptic dopamine via downregulation of the membrane-bound dopamine transporter, non-classical volume transmission of dopamine, increased expression of striatal dopamine receptors, and/or favorable shifts in the activation patterns of movement-controlling neural circuitry, including hyperactivity in the motor cortex, subthalamic nucleus, and internal globus pallidus (Zigmond et al., 1990; Zigmond, 1997; Bezard et al., 2001; Bezard et al., 2003; Brotchie and Fitzer-Attas, 2009; Obeso and Schapira, 2009; Appel-Cresswell et al., 2010; Nandhagopal et al., 2011; Navntoft and Dreyer, 2016; Blesa et al., 2017). Some authors have proposed that maintenance of motor function in the face of early cell loss may not be dopamine-mediated after all, as dopaminergic changes (e.g., heightened efflux, synthesis, and turnover) may not unfold sufficiently early, and passive dopamine stabilization mechanisms may prevail in early disease stages without any true “plasticity” of release or clearance (Garris et al., 1997; Bezard et al., 2001; Bezard et al., 2003; Navntoft and Dreyer, 2016; Blesa et al., 2017).
Based on a large body of longitudinal and cross-sectional clinical work, Jankovic favored the first two types of disease progression—nonlinear stress responses defined above in A and B—depending on the specific outcome measure (Hilker et al., 2005; Jankovic, 2005). Jankovic’s illustration of disease progression displays early compensatory mechanisms with a characteristic, inverted U-shaped profile, but also accelerated early loss of nigral cell bodies, striatal terminals, and non-dopaminergic neurons, followed by deceleration at late disease stages in negative exponential curves (Figure 1A). In non-dopaminergic circuitry, Wu and Hallett also proposed an inverted U-shaped curve for compensatory cerebellar function in preclinical and early clinical stages of Parkinson’s disease (Figure 1B) (Wu and Hallett, 2013). Hyperactivation of extrastriatal circuitry has long been proposed to compensate against early nigrostriatal pathway loss and to explain why frank loss of motor function does not surface until a threshold of loss of 18Fluorodopa uptake in the putamen of the dorsal striatum is exceeded (Bezard et al., 2003; Moore, 2003; Palmer et al., 2010). For example, Whone and colleagues reported upregulation of 18Fluorodopa uptake in the internal globus pallidus in early Parkinson’s disease, followed by a decrease in the same measure in more advanced disease stages, conforming to a U-shaped profile (Whone et al., 2003). Similar compensatory increases in 18Fluorodopa uptake in early stages of Parkinson’s disease are evident in the anterior cingulate gyrus, pineal gland, and midbrain raphe, some or all of which may oppose the effects of mild striatal denervation (Rakshi et al., 1999; Moore et al., 2008).
A large number of studies have assumed monotonic progression of Parkinson’s disease—usually linear or exponential rates of decline—particularly in variables such as nigral neuron counts as a function of disease duration (Bhattaram et al., 2009; Maetzler et al., 2009; Cheng et al., 2010; Darbin et al., 2013; Reinoso et al., 2015; Venuto et al., 2016). For example, longitudinal multi-tracer imaging of striatal dopamine terminals and Hoehn and Yahr clinical stages suggest rapid early disease processes followed by deceleration at late stages (Muller et al., 2000; Greffard et al., 2006; Nandhagopal et al., 2009). Holford et al. modelled longitudinal Unified Parkinson's Disease Rating Scale (UPDRS) scores and discovered that both exponential and Gompertz (i.e., sigmoidal) asymptotic curves exhibited better goodness-of-fit with disease progression than linear models, with the Gompertz model being superior (Holford et al., 2006). Vu and colleagues similarly favored the Gompertz asymptotic model as a better predictor of cardinal motor symptoms and nonmotor depressive symptoms, but reported that performance on the mini-mental status exam declined in a linear fashion as a function of disease course (Vu et al., 2012). However, other longitudinal studies suggest that the scores on the mini-mental status exam are not a linear function of Parkinson’s disease duration (Aarsland et al., 2011).
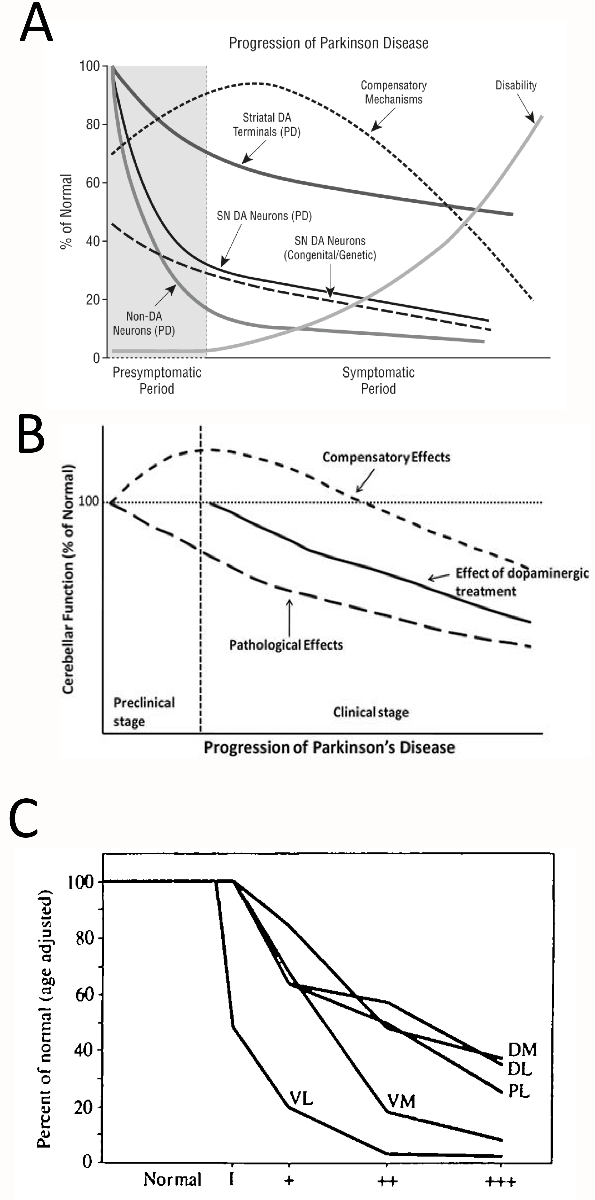
In a new window | Download PPT
Figure 1: Nonlinear stress responses as a function of the duration (severity) of Parkinson’s disease. (A) Jankovic noted the existence of inverted U-shaped and negative exponential stress response curves during the progression of Parkinson’s disease. Copied with permission from Jankovic’s editorial (Jankovic, 2005). (B) Wu and Hallet hypothesized a U-shaped compensatory phase in cerebellar function early in the course of Parkinson’s disease. Copied with permission from Wu and Hallet’s review (Wu and Hallett, 2013). (C) Fearnley and Lees reported that overall nigral cell loss assumes a negative exponential profile but that different subregions of the substantia nigra lose cells at varying rates. Phase 1 on the X axis in panel C reflects cases with incidental Lewy body disease (assumed to be prodromal Parkinson’s disease), and the plus signs reflect increasing severity of Parkinson’s disease. Dorsal tier: medial part (DM), lateral part (DL) and pars lateralis (PL). Ventral tier: medial part (VM) and lateral part (Vl-intermediate and VL). Copied legend and graph with permission from Fearnley and Lees’ research report (Fearnley and Lees, 1991). For a graph of the alignment of motor deficits and nigral cell loss as exponential, saturating curves, please also consult Figure 4 in Greffard and colleagues’ work (Greffard et al., 2006).
In an influential cross-sectional study on postmortem nigral tissue, Fearnley and Lees plotted loss of dopaminergic cells as an exponential function of symptom duration, and reported the earliest and fastest loss of somata in the ventrolateral tier of the substantia nigra pars compacta (Figure 1C) (Fearnley and Lees, 1991). The authors wrote, “neuronal loss appears to be confined to the lateral ventral tier early in the disease, but by the time symptoms appear it has spread to the other regions and in particular the medial ventral tier,” consistent with the view that a clinical syndrome only emerges after loss of compensatory defenses within resilient subpopulations of cells. Damier and colleagues subsequently reported that loss of nigral somata commences in the calbindin-poor ventrolateral nigrosome of the caudal substantia nigra and advances progressively rostrally, medially, and dorsally across the nigrosomes over time, while sparing the calbindin-rich nigral matrix (Damier et al., 1999). According to longitudinal tracer imaging, Nandhagopal et al. reported that all striatal subregions exhibit similar rates of dopaminergic terminal decline, although the degree of denervation varies across subregions and the posterior putamen is the earliest to lose dopaminergic tone (Nandhagopal et al., 2009). Nevertheless, dopamine axon terminal loss for each striatal subregion assumes a negative exponential profile as a function of disease duration, even if there are no regional variations in the response rate (Nandhagopal et al., 2009)
The diminished loss of striatal 18Fluorodopa uptake compared to other dopaminergic tracers (dopamine transporter/vesicular monoamine transporter) has been viewed as indirect evidence for potential compensatory upregulation of aromatic L-amino acid decarboxylase per remaining axon terminal, (Cheng et al., 2010; Kaasinen and Vahlberg, 2017). Aromatic L-amino acid decarboxylase catalyzes the conversion of L-DOPA to dopamine. Furthermore, the contrast between near-complete loss of dopaminergic fibers in histopathological studies and only moderate loss of tracer binding in imaging studies (averaging only ~50% and still detectable after 20 years of disease) also supports the existence of compensatory changes in remaining striatal fibers (Kaasinen and Vahlberg, 2017). Kaasinen and Vahlberg’s recent meta-analysis further suggests that cross-sectional tracer binding studies tend to report linear progression of disease, whereas longitudinal studies favor negative exponential progression (Kaasinen and Vahlberg, 2017). However, there may be exceptions to this rule; for example, cross-sectional studies of dopaminergic fiber density in the putamen as a function of time from diagnosis are not consistent with linear decay, and neither are cross-sectional studies of the pattern of nigral cell loss (Fearnley and Lees, 1991; Kordower et al., 2013). Kaasinen wrote that negative exponential decay in longitudinal studies reflects “the tail-end of a longer, nonlinear process that follows a sigmoid shape,” consistent with the abovementioned superior goodness-of-fit of the sigmoidal Gompertz curve in Holford’s work (Holford et al., 2006; Kaasinen and Vahlberg, 2017). In Gompertz functions, the rate of change is slowest at the start and end of a timed process; in this context, it is important to recognize that the movement disorder cannot be diagnosed until Braak stages III-IV (after nigral cell loss surfaces), which represents roughly mid-stages of the illness on a six-point scale of Lewy pathology (Braak et al., 2003b; Halliday et al., 2006). This implies that data plotted as a function of years from clinical diagnosis will miss the earliest, milder disease stages, when compensatory reactions have greater likelihood of prevailing. Therefore, one key difference between U-shaped, linear, exponential, and sigmoidal stress-response curves may be the range of stimulus doses (or durations) that are assessed. Edward Calabrese’s body of work suggests that linear and sigmoidal responses may be extrapolated when outcomes are not measured over a sufficiently wide range of stimulus intensities (Calabrese and Baldwin, 2001; Calabrese, 2004). A separate concern about graphing disease progression based on cross-sectional histological studies is that “supernormal agers” may harbor greater densities of dopaminergic cells than less-fit individuals dying early (Kubis et al., 2000), which could lead to spurious U-shaped profiles of dopaminergic cell counts or fiber density as a function of disease duration in postmortem studies.
In summary, there is evidence of divergent rates of progression of Parkinson’s disease, depending upon the measurement—curves of dopaminergic cell loss may be monotonic and sigmoidal while activation of extrastriatal circuitry in the same patient may follow a biphasic, U-shaped pattern. Given the heterogeneous natures of human populations and their disease states, the three patterns described above are probably not the only possibilities.
The ubiquity of nonlinear stress-response curves
As argued above, mild stressors that only slightly perturb homeostasis may elicit compensatory responses with upregulation of endogenous defenses, but when applied for sufficiently long durations and high intensities, stressful stimuli turn destructive to the same defense systems, and may even be lethal to the cell or organism. This non-monotonic stress response is expressed as an inverted U or J-shaped dose- response curve in the fields of toxicology, pharmacology, agricultural science, radiation, exercise, and dietary fasting (Calabrese and Baldwin, 2003; Mattson, 2008; Calabrese, 2010, 2014). The principle of stimulation at low doses but inhibition at high doses is defined as “hormesis,” a word derived from the Greek hormaein—to excite, stimulate, or set in motion (Calabrese and Baldwin, 2002; Calabrese and Mattson, 2017). Hormesis is consistent with the adage, “the dose makes the poison,” by the 15th-century physician Philippus Theophrastus von Hohenheim, widely known as Paracelsus, or the father of toxicology. The German pharmacologist Hugo Schulz observed biphasic effects of disinfectants on yeast metabolism in the 1800s (Calabrese and Baldwin, 2002; Calabrese and Mattson, 2017). Ehrlich and Southam subsequently defined this nonlinear property as “hormesis” in their own studies of the biphasic effects of red cedar tree extracts on fungal metabolism (Southam and Ehrlich, 1943; Calabrese and Baldwin, 2000; Calabrese, 2004, 2014). Potential mechanisms underlying hormesis have been reported in many recent studies, including the work of Marini and colleagues, who proposed that neuronal hormesis can be elicited by activation of ionotropic glutamate receptors (Marini et al., 2008a; Marini et al., 2008b). According to this view, low concentrations of otherwise excitotoxic glutamate transform neurons into a “conditioned” phenotype that survives high energy demands in an RNA/protein synthesis-dependent manner, perhaps by boosting expression of brain-derived neurotrophic factor.
Similar to the early observers of hormesis, Hans Selye, the father of stress research, described a “general adaptation syndrome” following an initial alarm reaction after stress exposure (Selye, 1950, 1975). Beneficial, mild stressors that stimulate endogenous defenses were defined by Selye as “eustress.” Following chronic, unremitting stress (“distress”), Selye reported a period of exhaustion characterized by immune suppression and eventually death, consistent with a non-monotonic, multiphasic response as a function of stress intensity (i.e., dose). Selye also wrote that “heredity, species differences, previous exposure to stress, and the nutritional state of the organism” serve as “conditioning factors” that underlie “polymorphic” responses to stress (Selye, 1950).
Since the period of Selye’s foresights on conditioning factors, the term “conditioning” has been used extensively in the ischemia literature. When sufficiently mild ischemic attacks are followed by a recovery interval during which compensatory adaptations are engaged, the organism is usually much better prepared to face, tolerate, and survive a severe ischemic challenge. This form of “preconditioning” of the myocardium was described in seminal publications by Murry, Reimer, and colleagues in the 1980s (Murry et al., 1986; Reimer et al., 1986), although, there are earlier reports of mechanical stress-induced tolerance against whole-body trauma and anoxia-induced resistance against subsequent anoxic challenges, and the terms “preconditioning” and “tolerance” were also employed in Janoff’s 1964 publication on lysosome shock (Noell and Chinn, 1948; Dahl and Balfour, 1964; Janoff, 1964; Simon, 2013). However, Murry’s 1986 study was more influential and inspired a slew of work confirming the parallel existence of ischemic preconditioning in the brain (Kitagawa et al., 1990; Perez-Pinzon et al., 1993; Simon et al., 1993; Gidday et al., 1994; Chen et al., 1996; Chen and Simon, 1997; Kitagawa et al., 1997; Roth et al., 1998; Miller et al., 2001; Kitagawa, 2012).
It is important to recognize that preconditioning responses to increasing intensities of ischemic stimuli adhere to the quantitative properties of the U-shaped hormetic curve (Calabrese et al., 2007; Calabrese, 2008, 2016a, b; Mollereau et al., 2016). Calabrese has calculated that the typical maximal amplitude of the hormetic curve is 30-60% of control values (Calabrese and Blain, 2005; Calabrese et al., 2007; Calabrese, 2010; Calabrese and Blain, 2011; Calabrese, 2014; Calabrese and Mattson, 2017). Second, the duration of stress exposure is defined as the stimulus “dose” in the ischemic preconditioning literature and in Selye’s original work on stress responses. Similarly, longitudinal studies of Parkinson’s patients assume that disease duration is in proportion to disease severity.
Despite the pervasiveness of inverted U-shaped dose-response curves, stress responses as a function of disease duration are not all plotted in this manner (see Figure 1), although one can make the arguments that disease duration may not be in direct proportion to disease severity (stimulus dose) and that compensatory responses in prodromal phases are readily missed, as argued above. In order to render plots of dopaminergic cell numbers as a function of disease duration U-shaped, nigral cells would have to undergo neurogenesis at early or end stages of the disease, a highly controversial proposition (Gould, 2007; Arias-Carrion et al., 2009). An alternative means of observing a U-shaped profile in dopaminergic cell numbers would be if phenotypically quiescent, melanized nigral cells that do not normally express tyrosine hydroxylase (estimated at ~18% by Kordower and colleagues (Kordower et al., 2013)) would abnormally gain a dopaminergic phenotype either early or late in Parkinson’s disease. An old body of experimental work demonstrates compensatory increases in tyrosine hydroxylase activity in response to stress exposure and compensatory sprouting of dopaminergic fibers in the striatum after injury (Zigmond et al., 1984; Bezard et al., 2000; Song and Haber, 2000). However, tyrosine hydroxylase mRNA levels appear to be reduced—not increased—in remaining nigral neurons in Parkinson’s disease (Javoy-Agid et al., 1990), and melanized nigral cells in primates exhibit a loss of tyrosine hydroxylase expression in response to aging (McCormack et al., 2004). Furthermore, the work of Hirsch et al. suggests that 17% of melanized nigral neurons do not express tyrosine hydroxylase in Parkinson’s patients (calculations from their Table 1) (Hirsch et al., 1988), which seems no different from Kordower’s estimate of 18% in subjects without Parkinson’s disease (Kordower et al., 2013). Thus, when postmitotic neuron numbers are plotted as a function of disease duration or stressor dose, the curves will be monotonic if there is no chance of cell proliferation or phenotypic recovery, with deceleration of cell loss over time as the fixed asymptote is approached. More simply put, one cannot plot fewer than zero cells remaining in the brain. Figure 1 also suggests that stress response curves as a function of disease duration may vary according to the clinical outcome measure, be it corticocerebellar/nigropallidal activation (perhaps U-shaped) or loss of nigral cell numbers (perhaps exponential or sigmoidal). Calabrese and Baldwin built a database showing that U-shaped curves predominate over sigmoidal curves, and also graphed multiphasic curves beginning with an inverted U-shape but ending as a negative exponential with increasing stressor dose (Calabrese and Baldwin, 2001). Di Veroli and colleagues similarly argued that pharmacological responses are not adequately modeled by the classic Hill equation, which only predicts monotonic, sigmoidal curves (Di Veroli et al., 2015). Thus, if reliable biomarkers for prodromal Parkinson’s disease could be identified, some but not all of the longitudinal responses to disease duration may eventually be plotted as multiphasic, with early inverted U-shaped curves yielding to a negative exponential profile as the disease forges into advanced stages (e.g., see Figure 3, panels f, j, t, and u in Calabrese’s report (Calabrese and Baldwin, 2001)).
What might underlie a deceleration of nigral cell loss as the asymptote is approached, instead of linear loss of dopaminergic nigral cells until all have disappeared from the brain or the patient dies? Most patients in Kordower’s report still have some dopaminergic somata remaining in the nigra at the time of death (see Figure 8 in Kordower et al., 2013). Our recent mechanistic in vitro work demonstrates that primary cortical astrocytes surviving high concentrations of proteinopathic stressors become progressively harder to kill, either because successive stress exposures enrich the remaining heterogeneous population in resistant survivors with inherently superior endogenous defenses, such as basally higher glutathione levels, or because stress exposure actively stimulates upregulation of defenses, such as a boost in glutathione production capacity (Titler et al., 2013; Gleixner et al., 2016). If these phenomena are applicable to human dopaminergic systems, they might underlie the gradual slowing of nigral cell loss. Indeed, Fearnley and Lees’ data in Figure 1C reveal heterogeneity in the defense capacity of nigral neurons, with ventrolateral cells dying at the fastest rate (Fearnley and Lees, 1991). The caudal ventrolateral nigra projects into the dorsal, lateral, and caudal aspects of the striatum, whereas the rostral dorsomedial nigra projects rostrally, medially, and ventrally (Fallon and Moore, 1978). For this reason, the dorsal posterior (post-commissural) putamen exhibits the greatest loss of dopaminergic signal according to clinical imaging studies (Nandhagopal et al., 2009). These topographic distinctions may reflect a number of mechanisms, perhaps including: 1) regionally distinct patterns of activation/upregulation of defense systems in response to injury, such as heterogeneity in stress-inducible heat shock proteins or phase II antioxidant enzymes; 2) regionally distinct differences in basal defenses, such as inherent variability in cytosolic calcium oscillations and calcium-buffering capacity by calbindin (Surmeier et al., 2017); or 3) varying degrees of exposure to a transmissible pathogenic insult that slowly spreads from ventrolateral to dorsomedial nigral cells over time. The current state of Parkinson’s disease research does not distinguish among these and other possibilities.
Can stress-induced conditioning ameliorate oxidative and proteinopathic pathology in experimental Parkinson’s disease?
An enormous body of preclinical and clinical work suggests that dopaminergic cell death in Parkinson’s disease is related to defective redox equilibrium, protein quality control, and mitochondrial integrity (Ganguly et al., 2017). If it is true that the response to the pathology of Parkinson’s disease elicits a functional compensatory reaction, it should be possible to demonstrate preconditioning with low doses of oxidative, proteinopathic, or mitochondrial stress in various experimental models of this condition. Aside from numerous reports of the salutary effects of exercise conditioning in patients with movement disorders, there are relatively few studies of stress-induced conditioning in Parkinson’s disease or models thereof, compared to the vast ischemic preconditioning literature, which boasts 10,815 studies in PubMed with the keywords and Boolean operators “preconditioning AND (ischemia OR ischemic)” (accessed November 24th 2017). Furthermore, most conditioning studies in the field of Parkinson’s disease have been completed in dopaminergic cell lines, including our own in vitro studies, which suffer from a number of obvious drawbacks, such as mutagenesis and oncogenic transformation. Tumor cell lines are specifically selected for their propensities to survive insults and may therefore be more likely to display hormesis than postmitotic dopaminergic neurons in vitro or in vivo. Second, even in primary cells, in vitro studies only capture the behavior of cells hardy enough to survive the culturing process, which might predispose the culture system to display hormetic effects. Third, studies of conditioning in Parkinson’s disease models—including our own in vivo rodent studies—involve relatively acute cell death, which is difficult to translate to the slowly progressive neurodegeneration observed in humans. Fourth, preconditioning is generally thought to wane within a few weeks after a transient exposure, whereas prolonged stress exposure is typically associated with morbidity and mortality; most researchers therefore do not expect conditioning to be a major player in the slow progression of a chronic neurodegenerative disease. The abovementioned caveats or perspectives have contributed to the lack of expansion of the field relative to ischemic preconditioning. Bearing these provisos in mind, below we briefly review the literature summoned on PubMed with the keywords “Parkinson’s disease AND preconditioning” (accessed until November 24th 2017). The term “preconditioning” was more discerning than “conditioning,” given the historical use of the former term in the ischemia literature and use of the latter for Pavlovian and pain behaviors.
Although the studies are relatively few, the beneficial impact of exposure to mild stress on the parkinsonian phenotype and its ability to allay dopaminergic cell loss has been reported in experimental models since the late 1990s. Duan and Mattson tackled this question in an early report showing that the mild stress of either dietary restriction or 2-deoxyglucose administration (a glucose analogue that cannot be metabolized) preconditioned mice against loss of dopaminergic neurons and appearance of motor deficits following exposure to the neurotoxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Duan and Mattson, 1999; Mattson et al., 2004), which rapidly induces a parkinsonian syndrome in humans (Langston et al., 1983). Since these observations, the beneficial effects of dietary restriction have been reproduced in other studies modeling Parkinson’s disease, including in primates (for some examples, see Maswood et al., 2004; Holmer et al., 2005; Griffioen et al., 2013; Mattson, 2014; Bayliss et al., 2016; Coppens et al., 2017).
A number of cell culture studies support the view that transformed dopaminergic cell lines can be preconditioned quite effectively against Parkinson’s-related oxidative toxicants. Tai and Truong reported that activation of ATP-sensitive potassium channels or exposure to pro-oxidants protected dopaminergic cells against subsequent exposure to the mitochondrial poison rotenone, and these effects were abolished by protein synthesis inhibitors (Tai and Truong, 2002). Years later, Tai and Truong also reported that green tea polyphenols mitigated dichlorodiphenyl-trichloroethane (DDT)-induced dopaminergic cell loss in a manner dependent upon the concentration and number of exposures (Tai and Truong, 2010). Similarly, the polyphenol resveratrol can precondition cells against dopaminergic neurotoxins, such as the complex I inhibitor 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of MPTP, perhaps through sirtuin 1 and the antioxidant methionine sulfoxide reductase A (Wu et al., 2012). Resveratrol is also efficacious in vivo as a therapeutic intervention against the dopaminergic oxidative toxicant 6- hydroxydopamine (6-OHDA), an established animal model of Parkinson’s disease, in which resveratrol seems to blunt the inflammatory response (Jin et al., 2008). These findings are consistent with the view that dietary polyphenols exert hormetic effects (Mattson and Cheng, 2006; Mattson et al., 2007). For example, low concentrations of the phytochemicals in Panax notoginseng stimulate dopaminergic cell growth at modest rates in vitro (~30%, as is typical for hormetic curves) and prevent the toxicity of 6- OHDA in zebrafish in vivo, whereas higher concentrations are toxic (Zhang et al., 2017).
In the same year as Tai and Truong’s original paper on in vitro dopaminergic preconditioning, Andoh and colleagues reported that sublethal serum deprivation for two hours preconditioned dopaminergic cells against MPP+, and this was associated with upregulation of nitric oxide synthesis and expression of the antioxidant enzyme superoxide dismutase and the prosurvival protein Bcl2 (Andoh et al., 2002). One year later, Quigney and colleagues demonstrated that exposure of dopaminergic cells to mild heat stress induced thermotolerance and preconditioned against necrotic death elicited by MPP+ (Quigney et al., 2003). The preconditioning effect of heat shock was associated with upregulation of the chaperones heat shock protein 25 (Hsp25) and Hsp70. Similarly, Fan and colleagues subsequently confirmed that exposure to mild heat shock or forced overexpression of HDJ-1 (the homologue of human Hsp40) in dopaminergic cells reduced MPP+ toxicity and oxidative stress and maintained the mitochondrial membrane potential (Fan et al., 2005).
The older studies of preconditioning in experimental Parkinson’s disease typically employed pro-oxidants and mitochondrial toxicants because markers of oxidative stress and complex 1 dysfunction have long been observed in postmortem tissue from Parkinson’s victims and proposed as etiological factors (Dexter et al., 1989; Schapira et al., 1989; Alam et al., 1997; Floor and Wetzel, 1998; Hauser and Hastings, 2013; Blesa et al., 2015). For example, low concentrations of hydrogen peroxide were shown to mitigate the pro-apoptotic effects of subsequent exposures to hydrogen peroxide and the pro-oxidant dopamine (Chen et al., 2005; Xiao-Qing et al., 2005). We demonstrated that pretreatment of dopaminergic cells with 6-OHDA (5-10 µM) prevented cell loss in response to subsequent exposures to higher concentrations of 6-OHDA (50 µM), an effect abolished by co-application of the antioxidant N-acetyl cysteine or the protein synthesis inhibitor cycloheximide (Leak et al., 2006). In our model, low concentrations of 6-OHDA upregulated expression of Bcl2 and rapidly activated the phosphokinases ERK1/2, Akt, and JNK. Inhibition of activation of all three kinases abolished 6-OHDA preconditioning-mediated protection. Subsequent studies revealed that subtoxic concentrations of methamphetamine also protected dopaminergic cells against 6-OHDA toxicity, whereas exposure to higher concentrations exerted toxicity, conforming to a biphasic concentration-response profile (El Ayadi and Zigmond, 2011). Similarly, royal jelly fatty acid derivatives such as 4-hydroperoxy-2-decenoic acid ethyl ester increased the production of reactive oxygen species and prevented 6-OHDA-induced dopaminergic cell loss, perhaps by activating Nrf2, a master gatekeeper of redox equilibrium (Inoue et al., 2017). Collectively, these studies established that exposure of dopaminergic cells to subtoxic levels of reactive oxygen species is an effective preconditioning stimulus. The beneficial effects of 6-OHDA preconditioning in dopaminergic cells can be further enhanced by the phytochemical triterpenoid oleanolic acid (Ndlovu et al., 2014). Oleanolic acid also blunted L-DOPA-induced abnormal involuntary movements (dyskinesia) in 6-OHDA-treated animals (Ndlovu et al., 2016).
Fewer studies have reported the effects of proteinopathic stress-induced preconditioning in models of Parkinson’s disease, perhaps because the view that Parkinson’s disease originates from protein misfolding is more recent than the concept that nigrostriatal loss is precipitated by oxidative toxicity (Zhou et al., 2008; Hastings, 2009; Jucker, 2010; Jucker and Walker, 2013; Segura-Aguilar et al., 2014). Nevertheless, the protective impact of heat shock in the dopaminergic cell models described above reflects an adaptive response to misfolded proteins, which might stimulate proteasomal and autophagic clearance of damaged proteins, as well as a beneficial endoplasmic reticulum stress response at low concentrations. Mollereau, Hetz, and colleagues recently proposed that mild perturbations in cellular proteostasis—including endoplasmic reticulum stress—show promise as preconditioning stimuli in Parkinson’s disease (Hetz and Mollereau, 2014; Mollereau et al., 2016). For example, the endoplasmic reticulum stress inducers thapsigargin and tunicamycin prevent the cytotoxicity of 6-OHDA in vitro (Hara et al., 2011). Thapsigargin induces heme oxygenase 1 expression by activating the antioxidant response element and mitigating the production of reactive oxygen species by 6-OHDA. Subsequent reports by Fouillet et al. suggested that mild endoplasmic reticulum stress protects neurons by crosstalk with autophagy pathways in vivo, an effect attributed by Matus and colleagues to hormesis (Fouillet et al., 2012; Matus et al., 2012). In the Fouillet studies, tunicamycin pretreatment reduced rotational behavior and dopaminergic cell loss in 6-OHDA-lesioned mice. The unfolded protein response transcription factor X-box binding protein 1 (XBP1) may be critical for the protection of dopaminergic neurons against 6-OHDA toxicity in adult animals, and deletion of this protein in developing animals induces an adaptive endoplasmic reticulum stress response that prevents dopaminergic neurotoxicity (Valdes et al., 2014).
Preconditioning dopaminergic cells with proteotoxic stress has also been achieved by direct exposure to proteasome inhibitors. We observed that weeks- to months-long exposure to low levels of the proteasome inhibitor MG132 resulted in structural and functional protection of dopaminergic cells against subsequent exposures to high concentrations of either 6-OHDA or MG132 (Leak et al., 2008). Cells were protected in this model despite the chronic nature of the stimulus because the MG132 concentration was sufficiently low. MG132 exposure reduced the chymotrypsin-like activity of the ubiquitin-proteasome system without causing cell loss and increased protein expression as well as enzymatic activity of the antioxidant CuZn superoxide dismutase. Although glutathione plays a major role in many models of preconditioning (Maher, 2005), the glutathione synthesis inhibitor buthionine sulfoximine failed to reduce MG132-induced protection against 6-OHDA, whereas siRNA-mediated knockdown of CuZn superoxide dismutase with two independent sequences abolished protection readily. These findings suggest that exposure to proteinopathic stress elicits “cross-tolerance” against oxidative stress by upregulation of antioxidant defense systems, and confirm the existence of divergence in the mechanisms underlying preconditioning (i.e., not unchangingly dependent on glutathione). The results also confirm a link between proteinopathic and oxidative stress, as presumed to be the case in Parkinson’s disease (Ganguly et al., 2017).
Aside from the studies on dopaminergic cells, proteotoxic stress has also been employed as a preconditioning tool in microglia in the context of Parkinson’s pathology. First, exposure to the Lewy body protein α-synuclein primes microglia toward higher cytokine secretion and caspase activation after stimulation of toll-like receptors 2/1 (TLR2/1) with Pam3, but reduces caspase 3 activation after stimulation with TLR7 ligands, and shifts microglial polarization status without an effect on phagocytic capacity (Roodveldt et al., 2013). Second, microglial activation and cytokine release in response to rotenone treatment can be mitigated by preconditioning with rifampicin from Streptomyces mediterranei, perhaps by upregulation of autophagic clearance systems, mitigation of oxidative stress, and preservation of the mitochondrial membrane potential, which culminate in neuroprotection in co-cultures of microglia with neuronal cells (Liang et al., 2017).
Other studies have also linked inflammatory stress to conditioning in the context of Parkinson’s pathology. For example, Ding and Li reported that 24-hour-long exposure of organotypic midbrain slices to low concentrations of the archetypal pro-inflammatory endotoxin lipopolysaccharide protected dopaminergic neurons against subsequent 72-hour-long exposure to higher concentrations of the same agent (Ding and Li, 2008). The authors proposed that reductions in the cytokine tumor necrosis factor-α and microglial activation contributed to endotoxin tolerance. Recent in vivo studies confirm that low-dose exposure to lipopolysaccharides induces a state of tolerance against subsequent brain injections of higher doses and alleviates cytokine production, microglial activation, and dopaminergic cell loss (Liu et al., 2017). These studies are consistent with the recent proposal that the response to inflammation in Parkinson’s disease is U-shaped (Calabrese et al., 2017), and with long-standing observations that inflammatory stress can elicit either tolerance or sensitization, depending upon the dose (Schneider, 2011; Quintin et al., 2012; Ifrim et al., 2014; Schmidt et al., 2014).
A series of in vivo studies by Cannon and colleagues demonstrated that infusions of low doses of the serine protease thrombin above the medial forebrain bundle preconditioned against the toxicity of subsequent infusions of 6-OHDA into the medial forebrain bundle (Cannon et al., 2005; Cannon et al., 2006). Although prevention of striatal dopamine depletion was not achieved, thrombin preconditioning mitigated neurological deficits, histological loss of dopaminergic terminals, and ventricular enlargement in 6-OHDA-infused animals. Notably, the protective effects of thrombin preconditioning were prevented by infusions of an antagonist against protease-activated receptors (PARs) and appeared to be mediated by PAR-1 but not PAR-4 activation (Cannon et al., 2006). Subsequent studies on the temporal kinetics of thrombin preconditioning revealed that co-application of thrombin or a PAR1 receptor agonist with 6-OHDA exacerbated behavioral deficits (Cannon et al., 2007). In contrast, delaying thrombin administration until 1 or 7 days after 6-OHDA assuaged the toxic effects of 6-OHDA on neurological function and depletion of dopamine and its metabolites. These experiments provided the first evidence of post-conditioning in an animal model of Parkinson’s disease and are consistent with the view that a recovery interval between sequential insults is critical in determining the direction of the stress response and that synergistic effects may otherwise be elicited.
The findings of Cannon and colleagues are important in light of increases in PAR1, prothrombin, and thrombin in astrocytes and/or the endothelial lining of the substantia nigra in Parkinson’s disease (Ishida et al., 2006; Sokolova and Reiser, 2008). Furthermore, thrombin exposure has been shown to raise expression of glial cell-derived growth factor and glutathione peroxidase in human astrocytes and to increase their proliferation (Ishida et al., 2006). More recent work in the 6-OHDA animal model suggests that apomorphine-induced rotations and performance on the Rotarod test and the elevated body swing test may also be ameliorated by preconditioning rats with hyperoxia for 24 hours before 6-OHDA application (Hamidi et al., 2012). Collectively, the thrombin and hyperoxia studies in the 6-OHDA model of Parkinson’s disease suggest that cross-tolerance against a different insult can be achieved in dopaminergic neurons in vivo.
The studies described above do not capture the entire complement of natural defenses expressed in Parkinson’s disease models. Additional descriptions of endogenous defenses in neurodegenerative disorders and the underlying mechanisms can be found in recent reviews (Stetler et al., 2014; Golpich et al., 2015; Mollereau et al., 2016). Other than discussions of oxidative stress, the present review also does not address the role of mitochondria in dopaminergic preconditioning; many other groups have already discussed the influence of mitohormesis and the general importance of mitochondrial viability in Parkinson’s disease (Calabrese et al., 2001; Greenamyre and Hastings, 2004; Tapia, 2006; Correia et al., 2011; Van Laar and Berman, 2013; Ristow and Schmeisser, 2014; Carvalho et al., 2015). Recent studies also suggest that mitohormesis might be effective against conditions that are specifically rooted in mitochondrial dysfunction, such as neurodegenerative disorders (Jain et al., 2016).
Fewer studies have examined the intersection of conditioning responses with mutant genes associated with familial Parkinson’s disease. For example, only 22 studies are summoned in PubMed with the Boolean operators “(preconditioning OR conditioning OR pre-conditioning) AND Parkinson's AND (synuclein OR LRRK2 OR PINK OR Parkin OR DJ-1 OR DJ1)”. Some of those studies are highlighted here. First, three months of exercise conditioning improves cognitive and motor function and reduces the levels of aggregated α-synuclein in transgenic mice overexpressing a mutant form of α-synuclein (Zhou et al., 2017). Second, α-synuclein mutations also hamper the natural increases in neuroplasticity that occur within the limbic system after fear conditioning (Schell et al., 2012). Third, deficiencies in DJ-1 (PARK7) or Parkin (PARK2) render animals resistant to ischemic preconditioning and increase cardiovascular or stroke-related injury (Huang et al., 2011; Lu et al., 2012; Dongworth et al., 2014; Mukherjee et al., 2015). Fourth, low doses of polyphenols and the oxidative toxicant paraquat delay mortality and improve motor output in flies exhibiting loss of the Parkinson’s-related protein parkin (Bonilla-Ramirez et al., 2013). Finally, Mukherjee and colleagues have argued that Parkin, PINK1, DJ-1, LRRK2, and α-synuclein maintain mitochondrial function and mitophagy, reduce free-radical toxicity, and mitigate apoptotic cell death (Mukherjee et al., 2015). Nevertheless, much work remains to be done on the intersection of conditioning pathways with Parkinson’s-related genes.
Does the slow spread of Parkinson’s pathology to the contralateral side reflect cross-hemispheric conditioning?
The nigrostriatal dopamine depletion in Parkinson’s disease and the resulting motor deficits commence on one side of the body and spread to the contralateral side over the course of years (Djaldetti et al., 2006; Wang et al., 2015). The reason underlying this asymmetric pathology is not known, but may reflect slow spread of a pathogenic insult from one hemisphere to the other side (Hobson, 2012). Exposure of one hemisphere to an ischemic event has been shown to precondition the contralateral side against experimental stroke, and this stress response is associated with bilateral upregulation of superoxide dismutase (Schumacher et al., 2014). These findings raise the possibility that the unilateral onset of Parkinson’s pathology upregulates natural defenses in the contralateral hemisphere, perhaps via diffusible factors or other means of cross-hemispheric communication. In order to test this hypothesis, we recently completed a series of behavioral and histological studies following sequential infusions of 6-OHDA into the right and left mouse caudoputamen (i.e., dorsal striatum) spaced three days apart (Weilnau et al., 2017). The 72-hour interval between the two brain infusions was chosen as sufficient time for adaptive changes in gene expression and is considered an optimal interval between ischemic preconditioning and subsequent challenges (Kirino et al., 1991; Chen and Simon, 1997; Zhang et al., 2008; Lusardi et al., 2010). Therefore, we infused 6-OHDA (4 µg) or an equivalent volume of vehicle in the right striatum, followed three days later by an infusion of vehicle or the same dose of 6-OHDA in the left striatum. We observed that the group receiving two sequential hits of 6-OHDA (6-OHDA R/6-OHDA L; group d in Figure 2) exhibited similar asymmetries in spontaneous turning behavior and dopaminergic axon terminal and cell body loss as the group that only received vehicle as the second hit (6-OHDA R/vehicle L; group c), as if the left hemisphere exposed to the second 6-OHDA hit was less damaged than the right hemisphere exposed to the first 6-OHDA hit. Furthermore, the behavioral and histological patterns in the dual-hit group d (6-OHDA R/6-OHDA L) appeared the reverse of group b animals receiving 6-OHDA in the left hemisphere without any previous preconditioning (vehicle R/6-OHDA L; Figure 2). Thus, dual-hit mice in group d (6-OHDA R/6-OHDA L) preferred to turn to the right side, consistent with the established view that unilaterally lesioned rodents prefer to turn contralateral to the hemisphere with greater dopaminergic tone (Ungerstedt, 1968; Ungerstedt and Arbuthnott, 1970; Fornaguera et al., 1993; Schwarting and Huston, 1996).
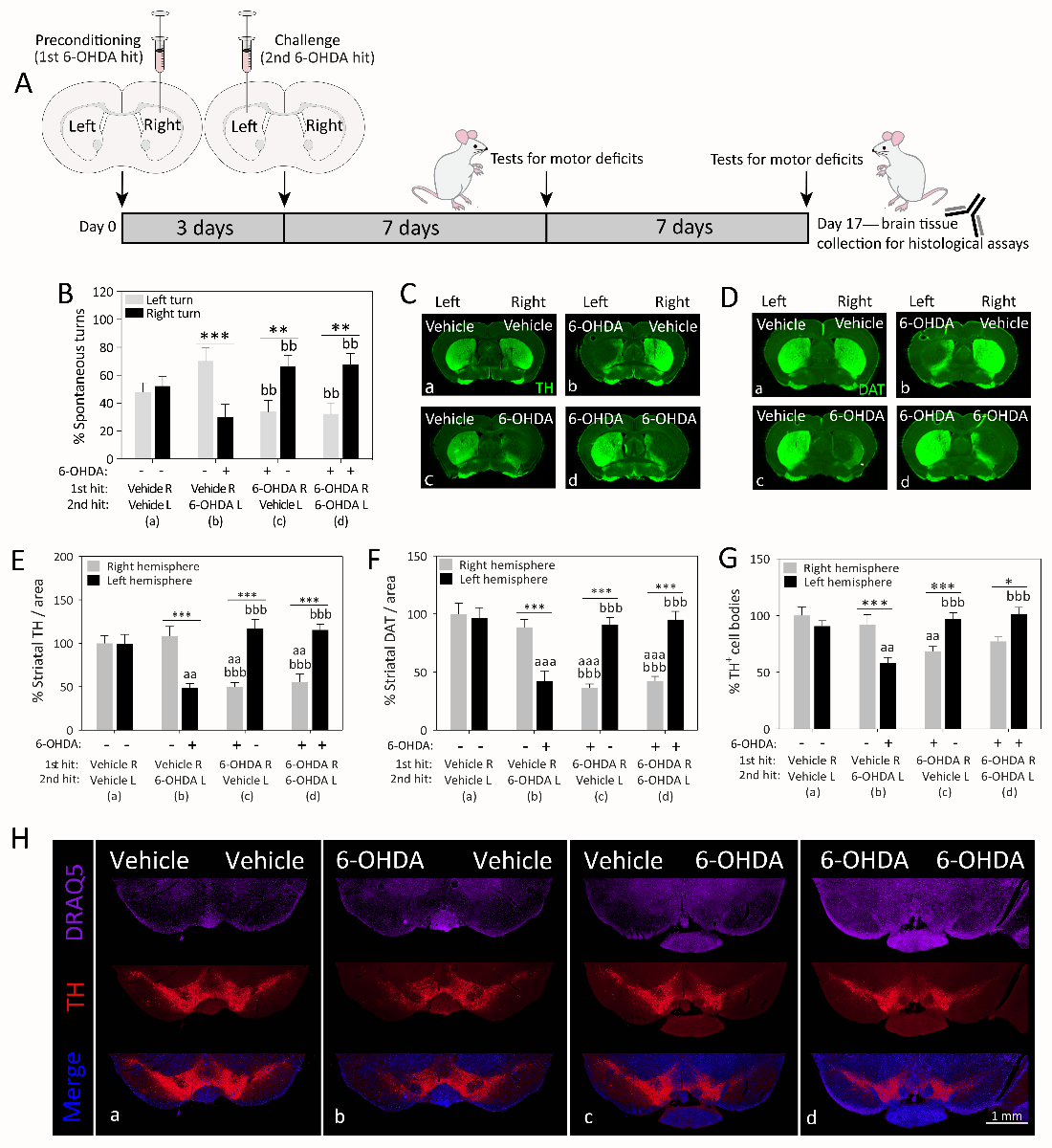
In a new window | Download PPT
Figure 2: Cross-hemispheric preconditioning mitigates motor deficits and nigrostriatal somata and terminal loss in an animal model of Parkinson’s disease. (A) Schematized illustration of the experimental design. Mice were infused in the right hemisphere with 4 μg 6-hydroxydopamine (6-OHDA) or an equal volume of saline with 0.02% ascorbic acid (preconditioning 1st hit). Three days later, animals were infused in the left hemisphere with 4 μg 6-OHDA or an equal volume of saline with 0.02% ascorbic acid (challenging 2nd hit). One and two weeks later, animals were placed in a transparent cylindrical enclosure and spontaneous behavior was videotaped. (B) A blinded observer measured the number of spontaneous turns to the left or right side one week after the second hit. (C-F) Two weeks following the second hit, coronal brain sections through the forebrain were immunostained for two markers of the dopaminergic phenotype—the rate-limiting enzyme for dopamine biosynthesis, tyrosine hydroxylase (TH) and the dopamine transporter (DAT). A blinded observer traced the anatomical boundaries of the dorsal striatum and measured the immunofluorescent signal within the region of interest using an ultrasensitive, low- resolution imager. (G-H) Two weeks following the second hit, coronal brain sections through the midbrain were immunostained for the dopaminergic marker TH and viewed by higher-resolution fluorescence microscopy. The numbers of TH+ cells were counted in the substantia nigra by a blinded observer. Panel H contains representative stitched photomontages of the ventral midbrain in all four groups. A second blinded observer confirmed key behavioral and histological data (not shown). Tissue from all groups was stained simultaneously with the same stock solutions and photographed in parallel at equivalent camera settings. Data are presented as the mean and SEM from 5-6 mice per group. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 left versus right hemisphere; a p ≤ 0.05, aa p ≤ 0.01, aaa p ≤ 0.001 versus group a; b p ≤ 0.05, bb p ≤ 0.01, bbb p ≤ 0.001 versus group b; two-way ANOVA followed by Bonferroni post hoc correction. Legend and figure copied with permission from main text or supplemental files of our recent report (Weilnau et al., 2017).
According to repeated measurements by blinded investigators, stressing the right hemisphere with 6-OHDA in the dual-hit group d completely prevented contralateral loss of dopaminergic cells in response to the second 6-OHDA hit, as demonstrated by two independent markers of dopaminergic fibers in the striatum (tyrosine hydroxylase and dopamine transporter), as well as counts of tyrosine hydroxylase+ somata in the substantia nigra pars compacta (Weilnau et al., 2017). Our prior work with the retrograde tracer FluoroGold confirmed true loss of nigral neurons and not merely downregulation of the dopaminergic phenotype when the same dose of 6-OHDA (4 µg) was applied to the striata of the same mouse strain (Nouraei et al., 2016). In sum, cross-hemispheric preconditioning ablated the toxic influence of 6-OHDA on the entire contralateral nigrostriatal pathway.
In the cross-hemispheric preconditioning model, measurements of asymmetry in forelimb contacts with a vertical wall and forepaw landing on the floor showed smaller-sized effects and some recovery of function (Weilnau et al., 2017), but were nevertheless significantly correlated with dopaminergic markers, as was spontaneous turning asymmetry (previously unpublished graphs in Figure 3). These correlations suggest that structural protection of the nigrostriatal pathway by cross-hemispheric preconditioning is associated with functional preservation of motor output. Furthermore, the behavioral tests consistently reveal a preference for turning, forelimb use, and floor landing behavior ipsiversive to the side of greater dopamine loss, confirming that the hemisphere with higher dopaminergic tone in the dual-hit group d was indeed the left hemisphere (exposed to the second 6-OHDA hit).
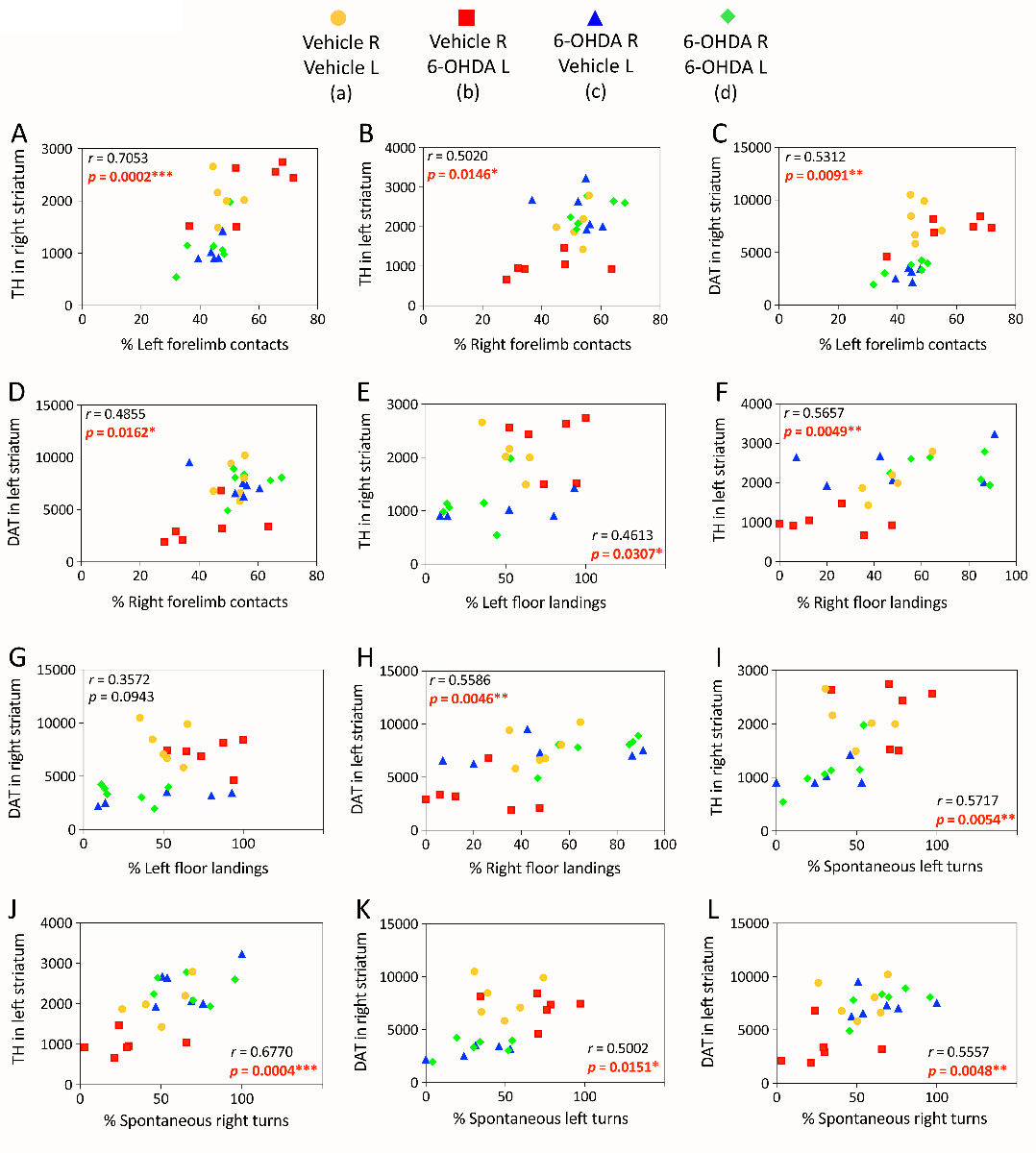
In a new window | Download PPT
Figure 3: Linear correlations between structural and functional outcomes of cross-hemispheric preconditioning in experimental Parkinson’s disease. Mice were preconditioned with 6-OHDA, as described in the legend of Figure 2, and behavioral and histological outcomes were measured, as described in our previous report (Weilnau et al., 2017). Raw striatal TH or DAT measurements (arbitrary fluorescence units) were employed as indicators of the structural integrity of nigrostriatal axon terminals and plotted as a function of the percentage of left versus right forelimb contacts with a vertical wall (A-D), left versus right forelimb landings on the floor (E-H), and left versus right spontaneous turns (I-L), as measured 7 or 14 days after the second hit (data from Weilnau et al., 2017). Two-tailed Pearson correlation analyses were applied to all the data. The Pearson correlation coefficient r and the corresponding p value are shown. Statistically significant results are presented in red font. Each animal is represented as one dot and each group is represented by a unique color and symbol. Note that some of the values are so similar that the dots overlap. n = 5-6 mice per group.
Previous studies indicated that dopamine receptor changes might underlie compensatory effects against 6-OHDA (Hornykiewicz and Kish, 1987; Zigmond et al., 1990; Brotchie and Fitzer-Attas, 2009), but our cross-hemispheric preconditioning effect was not associated with changes in expression of the D1 or D2 receptors. Furthermore, the protection could not be explained by activation of astrocytes or upregulation of the growth-associated protein 43, which is expressed in growth cones during axonal sprouting (Harding et al., 1999). However, unilateral 6-OHDA infusions in the striatum elicited significant bilateral upregulation of CuZn superoxide dismutase and phosphorylation of ERK2 three days later, in agreement with our previous mechanistic work showing that siRNA-induced knockdown of CuZn superoxide dismutase expression or pharmacological inhibitors of ERK1/2 abolished preconditioning of dopaminergic cells against 6-OHDA in vitro (Leak et al., 2006; Leak et al., 2008).
Functional recovery in the 6-OHDA animal model has been reported by others when the degree of dopamine depletion fails to exceed ~83% (Fornaguera et al., 1994; Schwarting and Huston, 1996; Yuan et al., 2005). Thus, we do not expect continued, progressive loss of function at longer time points than assessed in our cross-hemispheric preconditioning study (14 days after second hit). On the other hand, Parkinson’s disease in humans progresses ineluctably with age and eventually evolves into a more bilaterally symmetrical pattern (Nandhagopal et al., 2009; Marinus and van Hilten, 2015), suggesting that 1) mice might also lose cross-hemispheric preconditioning capacity with age, and 2) our model does not recapitulate the longitudinal pattern of the progressive human condition. For these reasons, we interpreted our results to support the potential of cross-hemispheric preconditioning only in the early stages of Parkinson’s disease (Weilnau et al., 2017). Furthermore, it is important to emphasize that our studies are correlative and do not establish any causal links, as we have not yet tested whether inhibition or loss of ERK1/2 and CuZn superoxide dismutase abolishes cross-hemispheric preconditioning. Given the limitations of acute nigrostriatal loss in the 6-OHDA model, studies are underway to determine if slowly progressive proteinopathic stress elicits similar cross-hemispheric preconditioning responses in a model of transmission of Lewy pathology from olfactory induction sites recently developed by us (Mason et al., 2016) and other groups (Rey et al., 2016; Rey et al., 2017).
In sum, cross-hemispheric preconditioning in the 6-OHDA model is consistent with the phenomenon of “cross-education,” whereby exercising only one half of the body also results in strength improvements on the non-exercised side (Lee and Carroll, 2007). Furthermore, previous studies show that unilateral 6- OHDA application elicits compensatory adjustments in the non-affected limb (Miklyaeva et al., 1994; Miklyaeva et al., 1997; Woodlee et al., 2008). Similarly, patients with Parkinson’s disease learn to rely on the non-affected side of their bodies (Boonstra et al., 2014; Roemmich et al., 2014) and exhibit a compensatory increase in glucose metabolism in the less-affected hemisphere (Dethy et al., 1998).
Does conditioning delay the onset and slow the progression of Parkinson’s disease or not?
An enormous body of work from the 1950s onwards and out of the scope of this review suggests that increased physical activity mitigates motor deficits in human subjects with Parkinson’s disease, consistent with animal studies (Bilowit, 1956; Clark et al., 1957; Zigmond et al., 2009; Gerecke et al., 2010; Hubble et al., 2017; Mak et al., 2017; Schenkman et al., 2017; Storzer et al., 2017). In addition, dietary protein restriction in Parkinson’s patients has been shown to alleviate motor fluctuations and lack of responsiveness to L-DOPA therapy, but these effects may be mediated by liberating the neutral amino acid transporter for greater L-DOPA delivery across the blood-brain barrier (Pincus and Barry, 1987a; Pincus and Barry, 1987b). Low carbohydrate or fat intake is also associated with a reduced risk of developing Parkinson’s disease, but some of the results are contradictory (Johnson et al., 1999; Abbott et al., 2003; Agim and Cannon, 2015). Potential conditioning effects of dietary restriction and exercise are readily explained in the context of hominid evolution: our ancestors had far less access to calories but nevertheless required sharp cognitive function and vigorous physical activity to solve the problem of scarce resources while hungry (Mattson, 2015).
Few publications other than the exercise literature noted above discuss adaptive conditioning of Parkinson’s patients. In one meta-analysis, a lack of association between welding or manganese exposure and an increased risk of Parkinson’s disease was interpreted as evidence of possible hormetic effects in humans (Mortimer et al., 2012). The term preconditioning has also been employed in the context of repetitive transcranial magnetic stimulation (rTMS) or transcranial direct current stimulation (tDCS), which are proposed to act as hormetic stimuli and exert favorable effects upon cognition and performance (Giordano et al., 2017). In Parkinson’s patients, stimulation of the primary motor cortex with rTMS increased simple index finger and hand tapping movements and horizontal pointing (Gruner et al., 2010), whereas rTMS applied over the primary motor cortex also improved hypokinetic gait when preceded by anodal tDCS preconditioning (von Papen et al., 2014). These studies typically assess acute conditioning effects on motor output rather than the long-term progression of Parkinson’s disease. However, one study reported that benefits of rTMS in Parkinson’s patients endured for one month after treatment (Lomarev et al., 2006).
Despite the paucity of studies of conditioning in Parkinson’s patients, there are many examples of compensatory reactions in the disorder, as conveyed above. Furthermore, with pro-dopaminergic interventions, there is symptomatic early improvement in UPDRS scores, and the profile of these scores plotted as a function of disease duration conforms remarkably well to a J-shaped curve (Guimaraes et al., 2005; Reinoso et al., 2015). However, these pharmacological responses cannot be viewed as expressions of natural defense mechanisms. Exposure to pro-dopaminergic agents is a prerequisite for the placebo response in humans, and these effects are also sometimes described as “conditioning” (Haour, 2005; Benedetti et al., 2016; Frisaldi et al., 2017), although they are not stress-induced. In contrast, the reduction in Parkinson’s disease risk with tobacco consumption may exemplify genuine stress-induced conditioning (van der Mark et al., 2014).
Measurements of stress responses across disease stages are complicated by regional differences in stress reactivity, tolerance, and vulnerability. For example, the U-shaped changes in 18Fluorodopa uptake in the nigropallidal pathway reported by Whone and colleagues are paralleled by simultaneous decreases of the same measure in the nigrostriatal pathway and motor cortex (Whone et al., 2003). While relatively resistant brain regions exhibit an increase in natural defenses, such as an increase in glutathione levels in the cerebral cortex in Parkinson’s disease, more vulnerable regions such as the substantia nigra show a simultaneous decline in glutathione levels (Perry et al., 1982; Sofic et al., 1992; Sian et al., 1994; Mythri et al., 2011). Calbindin and calcium channel expression differences in ventral versus dorsal tiers and in nigrosomes versus matrix may further contribute to selective vulnerabilities (Surmeier et al., 2011).
Tim Schallert has argued that injured tissue might be particularly sensitive to overstimulation, suggesting that the hormetic “sweet spot” will be tricky to achieve in the clinic, even with physical exercise, which is considered relatively innocuous (Schallert et al., 1997). Furthermore, the biphasic nature of U-shaped stress responses dictates that sufficiently severe stress above a threshold should sensitize cells to even greater injury in response to subsequent challenges, rather than mitigating subsequent injury. Indeed, synergistic effects of multiple exposures to severe stress form the basis of the “two hit” or “dual hit” hypothesis of neurodegeneration (Ling et al., 2004; Zhu et al., 2004; Carvey et al., 2006; Sulzer, 2007; Zhu et al., 2007; Boger et al., 2010; Unnithan et al., 2012; Leak, 2014; Unnithan et al., 2014; Heinemann et al., 2016; Mori, 2017). Therefore, it is conceivable that sensitization to cumulative, relentless stressors underlies the defeat of natural defense systems in Parkinson’s disease, due to slow accretion of protein deposits, oxidative damage, and cell loss, until only the most resistant, stress-refractory dopaminergic cells remain alive. Self-amplifying, synergistic processes might also explain the acceleration of nigral cell loss in mid-disease stages (after Braak stage III, see above and Figure 1). Eventual enrichment of the shrinking nigral cell population in highly resistant neurons of the dorsal nigrosomes might subsequently decelerate the rate of cell loss. As argued above, these features may underlie the exponential or sigmoidal shape of the dopaminergic cell loss curve. Stress-response curves in Parkinson’s disease might also assume a number of multiphasic shapes because of: 1) regional differences in vulnerability (e.g., neocortex versus allocortex, and ventral versus dorsal nigral tier); 2) the duration of the illness that is feasible to assess (i.e., early enough to capture compensatory changes versus late enough to reveal the asymptote); and 3) the specific nature of the response (e.g., dopamine cell counts in the nigra versus extracellular dopaminergic tone in the striatum). Age-related failures in stress compensation may also contribute to variability in disease progression according to the age at diagnosis (Collier et al., 2007).
Conclusions
Armed largely with correlative and observational clinical work and preclinical studies on transformed cell lines or acute, toxicant-induced animal models, it is difficult to conclude whether or not Parkinson’s progression in humans is indeed impeded by the kindling of conditioning defenses. This question might be better addressed by measuring viability in response to multiple hits in patient-derived stem cells, blood cells, or fibroblasts collected prior to the appearance of motor deficits, provided specific biomarkers of prodromal disease such as Lewy pathology in the submandibular gland are employed (Beach et al., 2016; Adler et al., 2017). However, researchers may discover that the conditioning achieved readily in experimental models cannot be actualized in authentically diseased human cells. Indeed, it may be the very absence (or diminution) of conditioning capacity that allows the disease to germinate in the first place and spread its shoots throughout the neuraxis. On the other hand, the benefits of conditioning might still be leveraged during transplantation of non-diseased cells into patients with Parkinson’s disease (Pan-Montojo and Funk, 2012). Transplantation of dopaminergic tissue into the brains of Parkinson’s patients was originally attempted with adrenal medulla tissue many decades ago (Kelly et al., 1989). More recently, human embryonic cells (Olanow et al., 2001; Brundin and Kordower, 2012) and induced pluripotent stem cells have been employed (Takahashi, 2017), although there is evidence that the transplanted dopaminergic cells are transformed into diseased cells over time (Kordower et al., 2008; Li et al., 2008; Li et al., 2010). In the stroke literature, it is well established that stem cells can be conditioned for improved survival after transplantation into the brain by mild hypoxia exposure (Bernstock et al., 2017). Notably, exposure of mesencephalic progenitors to low partial pressure of oxygen increases their branching, branch length, and mRNA expression of markers of maturation such as Nurr1, Pitx3, and the dopamine transporter (Liu et al., 2009). Therefore, a hypoxia-conditioned transplant preparation of allogeneic, otherwise healthy cells may have superior chances of surviving 1) the shock of the transplant procedure itself and 2) the initial shock of the hostile environment within severely diseased brains. In the context of transplants, it is important to remember that the brains of patients with Parkinson’s disease teem with free radicals, denatured proteins, dysfunctional mitochondria, and, even in the early stages, suffer from defective complex I activity and depressed glutathione (Schapira et al., 1990; Dexter et al., 1994; Sian et al., 1994; Gu et al., 1998; Zeevalk et al., 2008). The latter two characteristics probably set in motion stress responses early in the disease but may also wear down natural defenses over the course of years, consistent with the biphasic nature of hormesis and Hans Selye’s classic studies of acute eustress versus chronic distress.
Acknowledgements
I must apologize to all those whose research I had no space to discuss. I remain immensely grateful to Justin Weilnau for his work on preconditioning in my research laboratory. I have no conflicts to disclose. Supported by NINDS grant R15NS093539.
References
Aarsland D, Muniz G, Matthews F (2011) Nonlinear decline of mini-mental state examination in Parkinson's disease. Mov Disord 26:334-337.
Abbott RD, Ross GW, White LR, Sanderson WT, Burchfiel CM, Kashon M, Sharp DS, Masaki KH, Curb JD, Petrovitch H (2003) Environmental, life-style, and physical precursors of clinical Parkinson's disease: recent findings from the Honolulu-Asia Aging Study. J Neurol 250 Suppl 3:III30-39.
Adler CH, Beach TG (2016) Neuropathological basis of nonmotor manifestations of Parkinson's disease.
Mov Disord 31:1114-1119.
Adler CH, Dugger BN, Hentz JG, Hinni ML, Lott DG, Driver-Dunckley E, Mehta S, Serrano G, Sue LI, Duffy A, Intorcia A, Filon J, Pullen J, Walker DG, Beach TG (2017) Peripheral synucleinopathy in early Parkinson's disease: Submandibular gland needle biopsy findings. Mov Disord 32:722-723.
Agim ZS, Cannon JR (2015) Dietary factors in the etiology of Parkinson's disease. Biomed Res Int 2015:672838.
Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B (1997) Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. Journal of neurochemistry 69:1196-1203.
Andoh T, Chock PB, Chiueh CC (2002) Preconditioning-mediated neuroprotection: role of nitric oxide, cGMP, and new protein expression. Ann N Y Acad Sci 962:1-7.
Appel-Cresswell S, de la Fuente-Fernandez R, Galley S, McKeown MJ (2010) Imaging of compensatory mechanisms in Parkinson's disease. Curr Opin Neurol 23:407-412.
Arias-Carrion O, Yamada E, Freundlieb N, Djufri M, Maurer L, Hermanns G, Ipach B, Chiu WH, Steiner C, Oertel WH, Hoglinger GU (2009) Neurogenesis in substantia nigra of parkinsonian brains? J Neural Transm Suppl:279-285.
Au WL, Calne DB (2005) A reassessment of the Lewy body. Acta Neurol Taiwan 14:40-47.
Bayliss JA, Lemus MB, Stark R, Santos VV, Thompson A, Rees DJ, Galic S, Elsworth JD, Kemp BE, Davies JS, Andrews ZB (2016) Ghrelin-AMPK Signaling Mediates the Neuroprotective Effects of Calorie Restriction in Parkinson's Disease. J Neurosci 36:3049-3063.
Beach TG (2017) A Review of Biomarkers for Neurodegenerative Disease: Will They Swing Us Across the Valley? Neurol Ther 6:5-13.
Beach TG, White CL, 3rd, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, Sue LI, Sasse J, Bachalakuri J, Henry- Watson J, Akiyama H, Adler CH, Arizona Parkinson's Disease C (2009a) Olfactory bulb alpha- synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169-174.
Beach TG, Adler CH, Serrano G, Sue LI, Walker DG, Dugger BN, Shill HA, Driver-Dunckley E, Caviness JN, Intorcia A, Filon J, Scott S, Garcia A, Hoffman B, Belden CM, Davis KJ, Sabbagh MN, Arizona Parkinson's Disease C (2016) Prevalence of Submandibular Gland Synucleinopathy in Parkinson's Disease, Dementia with Lewy Bodies and other Lewy Body Disorders. J Parkinsons Dis 6:153-163.
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL, 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG, Arizona Parkinson's Disease C (2009b) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613-634.
Benedetti F, Frisaldi E, Carlino E, Giudetti L, Pampallona A, Zibetti M, Lanotte M, Lopiano L (2016) Teaching neurons to respond to placebos. J Physiol 594:5647-5660.
Bernstock JD, Peruzzotti-Jametti L, Ye D, Gessler FA, Maric D, Vicario N, Lee YJ, Pluchino S, Hallenbeck JM (2017) Neural stem cell transplantation in ischemic stroke: A role for preconditioning and cellular engineering. J Cereb Blood Flow Metab 37:2314-2319.
Beyer K, Domingo-Sabat M, Ariza A (2009) Molecular pathology of Lewy body diseases. Int J Mol Sci 10:724-745.
Bezard E, Gross CE, Brotchie JM (2003) Presymptomatic compensation in Parkinson's disease is not dopamine-mediated. Trends Neurosci 26:215-221.
Bezard E, Dovero S, Imbert C, Boraud T, Gross CE (2000) Spontaneous long-term compensatory dopaminergic sprouting in MPTP-treated mice. Synapse 38:363-368.
Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie JM, Gross CE (2001) Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson's disease. J Neurosci 21:6853-6861.
Bhattaram VA, Siddiqui O, Kapcala LP, Gobburu JV (2009) Endpoints and analyses to discern disease- modifying drug effects in early Parkinson's disease. AAPS J 11:456-464.
Bilowit DS (1956) Establishing physical objectives in the rehabilitation of patients with Parkinson's disease; gymnasium activities. Phys Ther Rev 36:176-178.
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson's disease. Front Neuroanat 9:91.
Blesa J, Trigo-Damas I, Dileone M, Del Rey NL, Hernandez LF, Obeso JA (2017) Compensatory mechanisms in Parkinson's disease: Circuits adaptations and role in disease modification. Exp Neurol 298:148- 161.
Boger HA, Granholm AC, McGinty JF, Middaugh LD (2010) A dual-hit animal model for age-related parkinsonism. Progress in neurobiology 90:217-229.
Bonilla-Ramirez L, Jimenez-Del-Rio M, Velez-Pardo C (2013) Low doses of paraquat and polyphenols prolong life span and locomotor activity in knock-down parkin Drosophila melanogaster exposed to oxidative stress stimuli: implication in autosomal recessive juvenile parkinsonism. Gene 512:355-363.
Boonstra TA, Schouten AC, van Vugt JP, Bloem BR, van der Kooij H (2014) Parkinson's disease patients compensate for balance control asymmetry. J Neurophysiol 112:3227-3239.
Braak H, Rub U, Gai WP, Del Tredici K (2003a) Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517-536.
Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960-969.
Braak H, Del Tredici K, Bratzke H, Hamm-Clement J, Sandmann-Keil D, Rub U (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol 249 Suppl 3:III/1-5.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003b) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197-211.
Brotchie J, Fitzer-Attas C (2009) Mechanisms compensating for dopamine loss in early Parkinson disease.
Neurology 72:S32-38.
Brundin P, Kordower JH (2012) Neuropathology in transplants in Parkinson's disease: implications for disease pathogenesis and the future of cell therapy. Prog Brain Res 200:221-241.
Calabrese EJ (2004) Hormesis: a revolution in toxicology, risk assessment and medicine. EMBO Rep 5 Spec No:S37-40.
Calabrese EJ (2008) Converging concepts: adaptive response, preconditioning, and the Yerkes-Dodson Law are manifestations of hormesis. Ageing research reviews 7:8-20.
Calabrese EJ (2010) Hormesis is central to toxicology, pharmacology and risk assessment. Human & experimental toxicology 29:249-261.
Calabrese EJ (2014) Hormesis: a fundamental concept in biology. Microb Cell 1:145-149.
Calabrese EJ (2016a) Preconditioning is hormesis part I: Documentation, dose-response features and mechanistic foundations. Pharmacol Res 110:242-264.
Calabrese EJ (2016b) Preconditioning is hormesis part II: How the conditioning dose mediates protection: Dose optimization within temporal and mechanistic frameworks. Pharmacol Res 110:265-275.
Calabrese EJ, Baldwin LA (2000) Chemical hormesis: its historical foundations as a biological hypothesis.
Hum Exp Toxicol 19:2-31.
Calabrese EJ, Baldwin LA (2001) Hormesis: U-shaped dose responses and their centrality in toxicology.
Trends Pharmacol Sci 22:285-291.
Calabrese EJ, Baldwin LA (2002) Defining hormesis. Hum Exp Toxicol 21:91-97.
Calabrese EJ, Baldwin LA (2003) Hormesis: the dose-response revolution. Annu Rev Pharmacol Toxicol 43:175-197.
Calabrese EJ, Blain R (2005) The occurrence of hormetic dose responses in the toxicological literature, the hormesis database: an overview. Toxicol Appl Pharmacol 202:289-301.
Calabrese EJ, Blain RB (2011) The hormesis database: the occurrence of hormetic dose responses in the toxicological literature. Regulatory toxicology and pharmacology : RTP 61:73-81.
Calabrese EJ, Mattson MP (2017) How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech Dis 3:13.
Calabrese EJ et al. (2007) Biological stress response terminology: Integrating the concepts of adaptive response and preconditioning stress within a hormetic dose-response framework. Toxicology and applied pharmacology 222:122-128.
Calabrese V, Scapagnini G, Giuffrida Stella AM, Bates TE, Clark JB (2001) Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res 26:739-764.
Calabrese V, Santoro A, Monti D, Crupi R, Paola RD, Latteri S, Cuzzocrea S, Zappia M, Giordano J, Calabrese EJ, Franceschi C (2017) Aging and Parkinson's Disease: Inflammaging, Neuroinflammation and Biological Remodelling as Key Factors in Pathogenesis. Free Radic Biol Med.
Candy JM, Perry RH, Perry EK, Irving D, Blessed G, Fairbairn AF, Tomlinson BE (1983) Pathological changes in the nucleus of Meynert in Alzheimer's and Parkinson's diseases. J Neurol Sci 59:277-289.
Cannon JR, Keep RF, Hua Y, Richardson RJ, Schallert T, Xi G (2005) Thrombin preconditioning provides protection in a 6-hydroxydopamine Parkinson's disease model. Neurosci Lett 373:189-194.
Cannon JR, Keep RF, Schallert T, Hua Y, Richardson RJ, Xi G (2006) Protease-activated receptor-1 mediates protection elicited by thrombin preconditioning in a rat 6-hydroxydopamine model of Parkinson's disease. Brain Res 1116:177-186.
Cannon JR, Hua Y, Richardson RJ, Xi G, Keep RF, Schallert T (2007) The effect of thrombin on a 6- hydroxydopamine model of Parkinson's disease depends on timing. Behav Brain Res 183:161-168. Carvalho C, Correia SC, Cardoso S, Placido AI, Candeias E, Duarte AI, Moreira PI (2015) The role of mitochondrial disturbances in Alzheimer, Parkinson and Huntington diseases. Expert Rev Neurother 15:867-884.
Carvey PM, Punati A, Newman MB (2006) Progressive dopamine neuron loss in Parkinson's disease: the multiple hit hypothesis. Cell Transplant 15:239-250.
Chen J, Simon R (1997) Ischemic tolerance in the brain. Neurology 48:306-311.
Chen J, Graham SH, Zhu RL, Simon RP (1996) Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab 16:566-577.
Chen ZH, Yoshida Y, Saito Y, Niki E (2005) Adaptation to hydrogen peroxide enhances PC12 cell tolerance against oxidative damage. Neurosci Lett 383:256-259.
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715-725.
Claassen DO, Josephs KA, Ahlskog JE, Silber MH, Tippmann-Peikert M, Boeve BF (2010) REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 75:494- 499.
Clark EC, Mulder DW, Erickson DJ, Clements BG, Maccarty CS (1957) Parkinson's disease: therapeutic exercises in its management. Postgrad Med 21:301-308.
Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, Bakay RA, Sladek JR, Jr., Kordower JH (2007) Aging- related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol Dis 26:56-65.
Coppens J, Bentea E, Bayliss JA, Demuyser T, Walrave L, Albertini G, Van Liefferinge J, Deneyer L, Aourz N, Van Eeckhaut A, Portelli J, Andrews ZB, Massie A, De Bundel D, Smolders I (2017) Caloric Restriction Protects against Lactacystin-Induced Degeneration of Dopamine Neurons Independent of the Ghrelin Receptor. Int J Mol Sci 18.
Correia SC, Cardoso S, Santos RX, Carvalho C, Santos MS, Perry G, Smith MA, Moreira PI (2011) New insights into the mechanisms of mitochondrial preconditioning-triggered neuroprotection. Curr Pharm Des 17:3381-3389.
Dahl NA, Balfour WM (1964) Prolonged Anoxic Survival Due to Anoxia Pre-Exposure: Brain Atp, Lactate, and Pyruvate. Am J Physiol 207:452-456.
Damier P, Hirsch EC, Agid Y, Graybiel AM (1999) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain 122 ( Pt 8):1437-1448.
Darbin O, Adams E, Martino A, Naritoku L, Dees D, Naritoku D (2013) Non-linear dynamics in parkinsonism.
Front Neurol 4:211.
Del Tredici K, Braak H (2016) Review: Sporadic Parkinson's disease: development and distribution of alpha- synuclein pathology. Neuropathol Appl Neurobiol 42:33-50.
Dethy S, Van Blercom N, Damhaut P, Wikler D, Hildebrand J, Goldman S (1998) Asymmetry of basal ganglia glucose metabolism and dopa responsiveness in parkinsonism. Mov Disord 13:275-280.
Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD (1989) Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem 52:381-389.
Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira AH, et al. (1994) Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol 35:38-44.
Di Veroli GY, Fornari C, Goldlust I, Mills G, Koh SB, Bramhall JL, Richards FM, Jodrell DI (2015) An automated fitting procedure and software for dose-response curves with multiphasic features. Sci Rep 5:14701.
Ding Y, Li L (2008) Lipopolysaccharide preconditioning induces protection against lipopolysaccharide- induced neurotoxicity in organotypic midbrain slice culture. Neurosci Bull 24:209-218.
Djaldetti R, Ziv I, Melamed E (2006) The mystery of motor asymmetry in Parkinson's disease. Lancet Neurol 5:796-802.
Dongworth RK, Mukherjee UA, Hall AR, Astin R, Ong SB, Yao Z, Dyson A, Szabadkai G, Davidson SM, Yellon DM, Hausenloy DJ (2014) DJ-1 protects against cell death following acute cardiac ischemia- reperfusion injury. Cell Death Dis 5:e1082.
Duan W, Mattson MP (1999) Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson's disease. J Neurosci Res 57:195-206.
El Ayadi A, Zigmond MJ (2011) Low concentrations of methamphetamine can protect dopaminergic cells against a larger oxidative stress injury: mechanistic study. PLoS One 6:e24722.
Fallon JH, Moore RY (1978) Catecholamine innervation of the basal forebrain. IV. Topography of the dopamine projection to the basal forebrain and neostriatum. J Comp Neurol 180:545-580.
Fan GH, Qi C, Chen SD (2005) Heat shock proteins reduce toxicity of 1-methyl-4-phenylpyridinium ion in SK-N-SH cells. J Neurosci Res 82:551-562.
Fasano A, Visanji NP, Liu LW, Lang AE, Pfeiffer RF (2015) Gastrointestinal dysfunction in Parkinson's disease. Lancet Neurol 14:625-639.
Fearnley JM, Lees AJ (1991) Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 114 ( Pt 5):2283-2301.
Floor E, Wetzel MG (1998) Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem 70:268-275.
Fornaguera J, Schwarting RK, Boix F, Huston JP (1993) Behavioral indices of moderate nigro-striatal 6- hydroxydopamine lesion: a preclinical Parkinson's model. Synapse 13:179-185.
Fornaguera J, Carey RJ, Huston JP, Schwarting RK (1994) Behavioral asymmetries and recovery in rats with different degrees of unilateral striatal dopamine depletion. Brain Res 664:178-188.
Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, Belaidi E, Ovize M, Touret M, Nataf S, Mollereau B (2012) ER stress inhibits neuronal death by promoting autophagy. Autophagy 8:915- 926.
Frisaldi E, Carlino E, Zibetti M, Barbiani D, Dematteis F, Lanotte M, Lopiano L, Benedetti F (2017) The placebo effect on bradykinesia in Parkinson's disease with and without prior drug conditioning. Mov Disord 32:1474-1478.
Fronczek R, Overeem S, Lee SY, Hegeman IM, van Pelt J, van Duinen SG, Lammers GJ, Swaab DF (2007) Hypocretin (orexin) loss in Parkinson's disease. Brain 130:1577-1585.
Ganguly G, Chakrabarti S, Chatterjee U, Saso L (2017) Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease. Drug Des Devel Ther 11:797-810.
Garris PA, Walker QD, Wightman RM (1997) Dopamine release and uptake rates both decrease in the partially denervated striatum in proportion to the loss of dopamine terminals. Brain Res 753:225- 234.
Gerecke KM, Jiao Y, Pani A, Pagala V, Smeyne RJ (2010) Exercise protects against MPTP-induced neurotoxicity in mice. Brain Res Brain Res Protoc 1341:72-83.
German DC, Manaye KF, White CL, 3rd, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM (1992) Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 32:667-676.
Gidday JM, Fitzgibbons JC, Shah AR, Park TS (1994) Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett 168:221-224.
Giordano J, Bikson M, Kappenman ES, Clark VP, Coslett HB, Hamblin MR, Hamilton R, Jankord R, Kozumbo WJ, McKinley RA, Nitsche MA, Reilly JP, Richardson J, Wurzman R, Calabrese E (2017) Mechanisms and Effects of Transcranial Direct Current Stimulation. Dose Response 15:1559325816685467.
Gleixner AM, Posimo JM, Pant DB, Henderson MP, Leak RK (2016) Astrocytes Surviving Severe Stress Can Still Protect Neighboring Neurons from Proteotoxic Injury. Mol Neurobiol 53:4939-4960.
Goedert M, Spillantini MG, Del Tredici K, Braak H (2013) 100 years of Lewy pathology. Nat Rev Neurol 9:13-24.
Goetz CG (2011) The history of Parkinson's disease: early clinical descriptions and neurological therapies.
Cold Spring Harb Perspect Med 1:a008862.
Golpich M, Rahmani B, Mohamed Ibrahim N, Dargahi L, Mohamed Z, Raymond AA, Ahmadiani A (2015) Preconditioning as a potential strategy for the prevention of Parkinson's disease. Mol Neurobiol 51:313-330.
Gould E (2007) How widespread is adult neurogenesis in mammals? Nat Rev Neurosci 8:481-488.
Greenamyre JT, Hastings TG (2004) Biomedicine. Parkinson's--divergent causes, convergent mechanisms.
Science 304:1120-1122.
Greffard S, Verny M, Bonnet AM, Beinis JY, Gallinari C, Meaume S, Piette F, Hauw JJ, Duyckaerts C (2006) Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body- associated neuronal loss in the substantia nigra. Arch Neurol 63:584-588.
Griffioen KJ, Rothman SM, Ladenheim B, Wan R, Vranis N, Hutchison E, Okun E, Cadet JL, Mattson MP (2013) Dietary energy intake modifies brainstem autonomic dysfunction caused by mutant alpha- synuclein. Neurobiol Aging 34:928-935.
Gruner U, Eggers C, Ameli M, Sarfeld AS, Fink GR, Nowak DA (2010) 1 Hz rTMS preconditioned by tDCS over the primary motor cortex in Parkinson's disease: effects on bradykinesia of arm and hand. J Neural Transm (Vienna) 117:207-216.
Gu M, Owen AD, Toffa SE, Cooper JM, Dexter DT, Jenner P, Marsden CD, Schapira AH (1998) Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J Neurol Sci 158:24-29.
Guimaraes P, Kieburtz K, Goetz CG, Elm JJ, Palesch YY, Huang P, Ravina B, Tanner CM, Tilley BC (2005) Non- linearity of Parkinson's disease progression: implications for sample size calculations in clinical trials. Clin Trials 2:509-518.
Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, Thompson L, Halliday G, Kirik D (2014) Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson's disease. Brain 137:2493-2508.
Halliday GM, Del Tredici K, Braak H (2006) Critical appraisal of brain pathology staging related to presymptomatic and symptomatic cases of sporadic Parkinson's disease. J Neural Transm Suppl:99-103.
Halliday GM, Blumbergs PC, Cotton RG, Blessing WW, Geffen LB (1990a) Loss of brainstem serotonin- and substance P-containing neurons in Parkinson's disease. Brain Res 510:104-107.
Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, Blessing WW, Geffen LB (1990b) Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol 27:373-385.
Hamidi GA, Faraji A, Zarmehri HA, Haghdoost-Yazdi H (2012) Prolonged hyperoxia preconditioning attenuates behavioral symptoms of 6-hydroxydopamine-induced Parkinsonism. Neurological research 34:636-642.
Haour F (2005) Mechanisms of the placebo effect and of conditioning. Neuroimmunomodulation 12:195- 200.
Hara H, Kamiya T, Adachi T (2011) Endoplasmic reticulum stress inducers provide protection against 6- hydroxydopamine-induced cytotoxicity. Neurochem Int 58:35-43.
Harding DI, Greensmith L, Mason M, Anderson PN, Vrbova G (1999) Overexpression of GAP-43 induces prolonged sprouting and causes death of adult motoneurons. Eur J Neurosci 11:2237-2242.
Hastings TG (2009) The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson's disease. J Bioenerg Biomembr 41:469-472.
Hauser DN, Hastings TG (2013) Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis 51:35-42.
Hawkes CH, Del Tredici K, Braak H (2007) Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 33:599-614.
Heinemann SD, Posimo JM, Mason DM, Hutchison DF, Leak RK (2016) Synergistic stress exacerbation in hippocampal neurons: Evidence favoring the dual-hit hypothesis of neurodegeneration. Hippocampus 26:980-994.
Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15:233-249.
Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, Rudolf J, Herholz K, Heiss WD (2005) Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch Neurol 62:378-382.
Hirsch E, Graybiel AM, Agid YA (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature 334:345-348.
Hirsch EC, Graybiel AM, Duyckaerts C, Javoy-Agid F (1987) Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A 84:5976-5980.
Hobson DE (2012) Asymmetry in parkinsonism, spreading pathogens and the nose. Parkinsonism Relat Disord 18:1-9.
Holford NH, Chan PL, Nutt JG, Kieburtz K, Shoulson I, Parkinson Study G (2006) Disease progression and pharmacodynamics in Parkinson disease - evidence for functional protection with levodopa and other treatments. J Pharmacokinet Pharmacodyn 33:281-311.
Holmer HK, Keyghobadi M, Moore C, Menashe RA, Meshul CK (2005) Dietary restriction affects striatal glutamate in the MPTP-induced mouse model of nigrostriatal degeneration. Synapse 57:100-112. Hornykiewicz O, Kish SJ (1987) Biochemical pathophysiology of Parkinson's disease. Adv Neurol 45:19-34.
Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA (2011) Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 6:e20975.
Hubble RP, Naughton G, Silburn PA, Cole MH (2017) Trunk exercises improve gait symmetry in Parkinson disease: A blind phase II randomised-controlled trial. Am J Phys Med Rehabil.
Iacono D, Markesbery WR, Gross M, Pletnikova O, Rudow G, Zandi P, Troncoso JC (2009) The Nun study: clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology 73:665-673.
Ifrim DC, Quintin J, Joosten LA, Jacobs C, Jansen T, Jacobs L, Gow NA, Williams DL, van der Meer JW, Netea MG (2014) Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol 21:534-545.
Inoue Y, Hara H, Mitsugi Y, Yamaguchi E, Kamiya T, Itoh A, Adachi T (2017) 4-Hydroperoxy-2-decenoic acid ethyl ester protects against 6-hydroxydopamine-induced cell death via activation of Nrf2-ARE and eIF2alpha-ATF4 pathways. Neurochem Int.
Ishida Y, Nagai A, Kobayashi S, Kim SU (2006) Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol 65:66-77.
Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, Zhang F, Goessling W, Zapol WM, Mootha VK (2016) Hypoxia as a therapy for mitochondrial disease. Science 352:54-61.
Jankovic J (2005) Progression of Parkinson disease: are we making progress in charting the course? Arch Neurol 62:351-352.
Janoff A (1964) Alterations in Lysosomes (Intracellular Enzymes) during Shock; Effects of Preconditioning (Tolerance) and Protective Drugs. Int Anesthesiol Clin 2:251-269.
Javoy-Agid F, Hirsch EC, Dumas S, Duyckaerts C, Mallet J, Agid Y (1990) Decreased tyrosine hydroxylase messenger RNA in the surviving dopamine neurons of the substantia nigra in Parkinson's disease: an in situ hybridization study. Neuroscience 38:245-253.
Jellinger K (1988) The pedunculopontine nucleus in Parkinson's disease, progressive supranuclear palsy and Alzheimer's disease. J Neurol Neurosurg Psychiatry 51:540-543.
Jin F, Wu Q, Lu YF, Gong QH, Shi JS (2008) Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson's disease in rats. Eur J Pharmacol 600:78-82.
Johnson CC, Gorell JM, Rybicki BA, Sanders K, Peterson EL (1999) Adult nutrient intake as a risk factor for Parkinson's disease. Int J Epidemiol 28:1102-1109.
Jucker M (2010) The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med 16:1210-1214.
Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501:45-51.
Kaasinen V, Vahlberg T (2017) Striatal dopamine in Parkinson disease: A meta-analysis of imaging studies.
Ann Neurol.
Kelly PJ, Ahlskog JE, van Heerden JA, Carmichael SW, Stoddard SL, Bell GN (1989) Adrenal medullary autograft transplantation into the striatum of patients with Parkinson's disease. Mayo Clin Proc 64:282-290.
Kirino T, Tsujita Y, Tamura A (1991) Induced tolerance to ischemia in gerbil hippocampal neurons. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 11:299-307.
Kish SJ, Shannak K, Hornykiewicz O (1988) Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N Engl J Med 318:876-880.
Kitagawa K (2012) Ischemic tolerance in the brain: endogenous adaptive machinery against ischemic stress. Journal of neuroscience research 90:1043-1054.
Kitagawa K, Matsumoto M, Mabuchi T, Yagita Y, Mandai K, Matsushita K, Hori M, Yanagihara T (1997) Ischemic tolerance in hippocampal CA1 neurons studied using contralateral controls. Neuroscience 81:989-998.
Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. (1990) 'Ischemic tolerance' phenomenon found in the brain. Brain Res 528:21- 24.
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 14:504-506.
Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136:2419-2431.
Kubis N, Faucheux BA, Ransmayr G, Damier P, Duyckaerts C, Henin D, Forette B, Le Charpentier Y, Hauw JJ, Agid Y, Hirsch EC (2000) Preservation of midbrain catecholaminergic neurons in very old human subjects. Brain 123 ( Pt 2):366-373.
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979-980.
Leak RK (2014) Adaptation and sensitization to proteotoxic stress. Dose-response : a publication of International Hormesis Society 12:24-56.
Leak RK, Liou AK, Zigmond MJ (2006) Effect of sublethal 6-hydroxydopamine on the response to subsequent oxidative stress in dopaminergic cells: evidence for preconditioning. J Neurochem 99:1151-1163.
Leak RK, Zigmond MJ, Liou AK (2008) Adaptation to chronic MG132 reduces oxidative toxicity by a CuZnSOD-dependent mechanism. J Neurochem 106:860-874.
Lee M, Carroll TJ (2007) Cross education: possible mechanisms for the contralateral effects of unilateral resistance training. Sports Med 37:1-14.
Li JY, Englund E, Widner H, Rehncrona S, Bjorklund A, Lindvall O, Brundin P (2010) Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson's disease. Mov Disord 25:1091-1096.
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 14:501-503.
Liang Y, Zhou T, Chen Y, Lin D, Jing X, Peng S, Zheng D, Zeng Z, Lei M, Wu X, Huang K, Yang L, Xiao S, Liu J, Tao E (2017) Rifampicin inhibits rotenone-induced microglial inflammation via enhancement of autophagy. Neurotoxicology 63:137-145.
Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM (2004) Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Experimental neurology 190:373-383.
Liu S, Tian Z, Yin F, Zhao Q, Fan M (2009) Generation of dopaminergic neurons from human fetal mesencephalic progenitors after co-culture with striatal-conditioned media and exposure to lowered oxygen. Brain Res Bull 80:62-68.
Liu Y, Xie X, Xia LP, Lv H, Lou F, Ren Y, He ZY, Luo XG (2017) Peripheral immune tolerance alleviates the intracranial lipopolysaccharide injection-induced neuroinflammation and protects the dopaminergic neurons from neuroinflammation-related neurotoxicity. J Neuroinflammation 14:223.
Lo EH (2010) Degeneration and repair in central nervous system disease. Nat Med 16:1205-1209.
Lomarev MP, Kanchana S, Bara-Jimenez W, Iyer M, Wassermann EM, Hallett M (2006) Placebo-controlled study of rTMS for the treatment of Parkinson's disease. Mov Disord 21:325-331.
Lu HS, Chen HP, Wang S, Yu HH, Huang XS, Huang QR, He M (2012) Hypoxic preconditioning up-regulates DJ-1 protein expression in rat heart-derived H9c2 cells through the activation of extracellular- regulated kinase 1/2 pathway. Mol Cell Biochem 370:231-240.
Lusardi TA, Farr CD, Faulkner CL, Pignataro G, Yang T, Lan J, Simon RP, Saugstad JA (2010) Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J Cereb Blood Flow Metab 30:744-756.
Maetzler W, Liepelt I, Berg D (2009) Progression of Parkinson's disease in the clinical phase: potential markers. Lancet Neurol 8:1158-1171.
Maher P (2005) The effects of stress and aging on glutathione metabolism. Ageing Res Rev 4:288-314.
Mak MK, Wong-Yu IS, Shen X, Chung CL (2017) Long-term effects of exercise and physical therapy in people with Parkinson disease. Nat Rev Neurol 13:689-703.
Marini AM, Jiang H, Pan H, Wu X, Lipsky RH (2008a) Hormesis: a promising strategy to sustain endogenous neuronal survival pathways against neurodegenerative disorders. Ageing Res Rev 7:21-33.
Marini AM, Popolo M, Pan H, Blondeau N, Lipsky RH (2008b) Brain adaptation to stressful stimuli: a new perspective on potential therapeutic approaches based on BDNF and NMDA receptors. CNS & neurological disorders drug targets 7:382-390.
Marinus J, van Hilten JJ (2015) The significance of motor (a)symmetry in Parkinson's disease. Mov Disord 30:379-385.
Mason DM, Nouraei N, Pant DB, Miner KM, Hutchison DF, Luk KC, Stolz JF, Leak RK (2016) Transmission of alpha-synucleinopathy from olfactory structures deep into the temporal lobe. Mol Neurodegener 11:49.
Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, Grondin R, Roth GS, Mattison J, Lane MA, Carson RE, Cohen RM, Mouton PR, Quigley C, Mattson MP, Ingram DK (2004) Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc Natl Acad Sci U S A 101:18171-18176.
Mattson MP (2008) Awareness of hormesis will enhance future research in basic and applied neuroscience. Crit Rev Toxicol 38:633-639.
Mattson MP (2014) Interventions that improve body and brain bioenergetics for Parkinson's disease risk reduction and therapy. J Parkinsons Dis 4:1-13.
Mattson MP (2015) Lifelong brain health is a lifelong challenge: from evolutionary principles to empirical evidence. Ageing Res Rev 20:37-45.
Mattson MP, Cheng A (2006) Neurohormetic phytochemicals: Low-dose toxins that induce adaptive neuronal stress responses. Trends Neurosci 29:632-639.
Mattson MP, Son TG, Camandola S (2007) Viewpoint: mechanisms of action and therapeutic potential of neurohormetic phytochemicals. Dose Response 5:174-186.
Mattson MP, Duan W, Wan R, Guo Z (2004) Prophylactic activation of neuroprotective stress response pathways by dietary and behavioral manipulations. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics 1:111-116.
Matus S, Castillo K, Hetz C (2012) Hormesis: protecting neurons against cellular stress in Parkinson disease.
Autophagy 8:997-1001.
McCormack AL, Di Monte DA, Delfani K, Irwin I, DeLanney LE, Langston WJ, Janson AM (2004) Aging of the nigrostriatal system in the squirrel monkey. J Comp Neurol 471:387-395.
Miklyaeva EI, Castaneda E, Whishaw IQ (1994) Skilled reaching deficits in unilateral dopamine-depleted rats: impairments in movement and posture and compensatory adjustments. J Neurosci 14:7148- 7158.
Miklyaeva EI, Woodward NC, Nikiforov EG, Tompkins GJ, Klassen F, Ioffe ME, Whishaw IQ (1997) The ground reaction forces of postural adjustments during skilled reaching in unilateral dopamine- depleted hemiparkinson rats. Behav Brain Res 88:143-152.
Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM (2001) Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport 12:1663-1669.
Mollereau B et al. (2016) Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations. Brain Res 1648:603-616.
Moore RY (2003) Organization of midbrain dopamine systems and the pathophysiology of Parkinson's disease. Parkinsonism Relat Disord 9 Suppl 2:S65-71.
Moore RY, Whone AL, Brooks DJ (2008) Extrastriatal monoamine neuron function in Parkinson's disease: an 18F-dopa PET study. Neurobiol Dis 29:381-390.
Mori I (2017) Viremic attack explains the dual-hit theory of Parkinson's disease. Med Hypotheses 101:33- 36.
Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ (1998) Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with [18F]dopa PET. J Neurol Neurosurg Psychiatry 64:314-319.
Mortimer JA, Borenstein AR, Nelson LM (2012) Associations of welding and manganese exposure with Parkinson disease: review and meta-analysis. Neurology 79:1174-1180.
Mukherjee UA, Ong SB, Ong SG, Hausenloy DJ (2015) Parkinson's disease proteins: Novel mitochondrial targets for cardioprotection. Pharmacol Ther 156:34-43.
Muller J, Wenning GK, Jellinger K, McKee A, Poewe W, Litvan I (2000) Progression of Hoehn and Yahr stages in Parkinsonian disorders: a clinicopathologic study. Neurology 55:888-891.
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124-1136.
Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, Srinivas Bharath MM, Shankar SK (2011) Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson's disease brains. Neurochemical research 36:1452-1463.
Nakano I, Hirano A (1984) Parkinson's disease: neuron loss in the nucleus basalis without concomitant Alzheimer's disease. Ann Neurol 15:415-418.
Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, McKenzie J, McCormick S, Ruth TJ, Sossi V, de la Fuente-Fernandez R, Stoessl AJ (2011) Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson's disease. Brain 134:3290-3298.
Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, McKenzie J, McCormick S, Samii A, Troiano A, Ruth TJ, Sossi V, de la Fuente-Fernandez R, Calne DB, Stoessl AJ (2009) Longitudinal progression of sporadic Parkinson's disease: a multi-tracer positron emission tomography study. Brain 132:2970-2979.
Navntoft CA, Dreyer JK (2016) How compensation breaks down in Parkinson's disease: Insights from modeling of denervated striatum. Mov Disord 31:280-289.
Ndlovu BC, Daniels WM, Mabandla MV (2014) Oleanolic Acid enhances the beneficial effects of preconditioning on PC12 cells. Parkinsons Dis 2014:929854.
Ndlovu BC, Daniels WM, Mabandla MV (2016) Amelioration of L-Dopa-Associated Dyskinesias with Triterpenoic Acid in a Parkinsonian Rat Model. Neurotox Res 29:126-134.
Noell WK, Chinn HI (1948) The cerebral survival time of rabbits in anoxia; effects of previous oxygenation.
Q Res Rep 60.
Nouraei N, Zarger L, Weilnau JN, Han J, Mason DM, Leak RK (2016) Investigation of the therapeutic potential of N-acetyl cysteine and the tools used to define nigrostriatal degeneration in vivo. Toxicol Appl Pharmacol 296:19-30.
Obeso JA, Schapira AH (2009) Compensatory mechanisms in Parkinson's disease. Mov Disord 24:153-154. Olanow CW, Freeman T, Kordower J (2001) Transplantation of embryonic dopamine neurons for severe
Parkinson's disease. N Engl J Med 345:146; author reply 147.
Palmer SJ, Li J, Wang ZJ, McKeown MJ (2010) Joint amplitude and connectivity compensatory mechanisms in Parkinson's disease. Neuroscience 166:1110-1118.
Pan-Montojo F, Funk RH (2012) Implications of Parkinson's disease pathophysiology for the development of cell replacement strategies and drug discovery in neurodegenerative diseases. CNS Neurol Disord Drug Targets 11:907-920.
Pearce RK, Hawkes CH, Daniel SE (1995) The anterior olfactory nucleus in Parkinson's disease. Movement disorders : official journal of the Movement Disorder Society 10:283-287.
Pellicano C, Benincasa D, Pisani V, Buttarelli FR, Giovannelli M, Pontieri FE (2007) Prodromal non-motor symptoms of Parkinson's disease. Neuropsychiatr Dis Treat 3:145-152.
Perez-Pinzon MA, Lutz PL, Sick TJ, Rosenthal M (1993) Adenosine, a "retaliatory" metabolite, promotes anoxia tolerance in turtle brain. J Cereb Blood Flow Metab 13:728-732.
Perry TL, Godin DV, Hansen S (1982) Parkinson's disease: a disorder due to nigral glutathione deficiency?
Neurosci Lett 33:305-310.
Pincus JH, Barry K (1987a) Influence of dietary protein on motor fluctuations in Parkinson's disease. Arch Neurol 44:270-272.
Pincus JH, Barry KM (1987b) Dietary method for reducing fluctuations in Parkinson's disease. Yale J Biol Med 60:133-137.
Quigney DJ, Gorman AM, Samali A (2003) Heat shock protects PC12 cells against MPP+ toxicity. Brain Res 993:133-139.
Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, Joosten LAB, Xavier RJ, van der Meer JWM, Stunnenberg HG, Netea MG (2012) Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12:223-232.
Rakshi JS, Uema T, Ito K, Bailey DL, Morrish PK, Ashburner J, Dagher A, Jenkins IH, Friston KJ, Brooks DJ (1999) Frontal, midbrain and striatal dopaminergic function in early and advanced Parkinson's disease A 3D [(18)F]dopa-PET study. Brain 122 ( Pt 9):1637-1650.
Reimer KA, Murry CE, Yamasawa I, Hill ML, Jennings RB (1986) Four brief periods of myocardial ischemia cause no cumulative ATP loss or necrosis. Am J Physiol 251:H1306-1315.
Reinoso G, Allen JC, Jr., Au WL, Seah SH, Tay KY, Tan LC (2015) Clinical evolution of Parkinson's disease and prognostic factors affecting motor progression: 9-year follow-up study. Eur J Neurol 22:457- 463.
Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson's disease. J Exp Med.
Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, Lee VM, Brundin P (2017) Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol.
Rinne JO (1993) Nigral degeneration in Parkinson's disease. Mov Disord 8 Suppl 1:S31-35.
Ristow M, Schmeisser K (2014) Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 12:288-341.
Roemmich RT, Nocera JR, Stegemoller EL, Hassan A, Okun MS, Hass CJ (2014) Locomotor adaptation and locomotor adaptive learning in Parkinson's disease and normal aging. Clin Neurophysiol 125:313- 319.
Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A, Robledo G, Hmadcha A, Delgado M, Dobson CM, Pozo D (2013) Preconditioning of microglia by alpha-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One 8:e79160.
Roth S, Li B, Rosenbaum PS, Gupta H, Goldstein IM, Maxwell KM, Gidday JM (1998) Preconditioning provides complete protection against retinal ischemic injury in rats. Invest Ophthalmol Vis Sci 39:777-785.
Savica R, Rocca WA, Ahlskog JE (2010) When does Parkinson disease start? Arch Neurol 67:798-801.
Schallert T, Kozlowski DA, Humm JL, Cocke RR (1997) Use-dependent structural events in recovery of function. Adv Neurol 73:229-238.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD (1989) Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1:1269.
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54:823-827.
Schell H, Boden C, Chagas AM, Kahle PJ (2012) Impaired c-Fos and polo-like kinase 2 induction in the limbic system of fear-conditioned alpha-synuclein transgenic mice. PLoS One 7:e50245.
Schenkman M, Moore CG, Kohrt WM, Hall DA, Delitto A, Comella CL, Josbeno DA, Christiansen CL, Berman BD, Kluger BM, Melanson EL, Jain S, Robichaud JA, Poon C, Corcos DM (2017) Effect of High- Intensity Treadmill Exercise on Motor Symptoms in Patients With De Novo Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol.
Schmidt C, Schneble N, Wetzker R (2014) The fifth dimension of innate immunity. J Cell Commun Signal 8:363-367.
Schneider DS (2011) Tracing personalized health curves during infections. PLoS Biol 9:e1001158.
Schumacher L, Urbach A, Lutzenburg M, Bidmon HJ, Witte OW (2014) Bihemispheric ischemic tolerance induced by a unilateral focal cortical lesion. Brain Res 1570:54-60.
Schwarting RK, Huston JP (1996) The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol 50:275-331.
Segura-Aguilar J, Paris I, Munoz P, Ferrari E, Zecca L, Zucca FA (2014) Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem 129:898-915.
Selye H (1950) Stress and the general adaptation syndrome. Br Med J 1:1383-1392. Selye H (1975) Stress without distress. Philadelphia: Signet.
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD (1994) Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol 36:348-355.
Simon RP (2013) Tolerance, Historical Overview. New York: Springer.
Simon RP, Niiro M, Gwinn R (1993) Prior ischemic stress protects against experimental stroke. Neurosci Lett 163:135-137.
Simuni T, Sethi K (2008) Nonmotor manifestations of Parkinson's disease. Ann Neurol 64 Suppl 2:S65-80.
Sofic E, Lange KW, Jellinger K, Riederer P (1992) Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson's disease. Neurosci Lett 142:128-130.
Sokolova E, Reiser G (2008) Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: localization, expression and participation in neurodegenerative diseases. Thromb Haemost 100:576-581.
Song DD, Haber SN (2000) Striatal responses to partial dopaminergic lesion: evidence for compensatory sprouting. J Neurosci 20:5102-5114.
Southam CM, Ehrlich J (1943) Effects of extracts of western red-cedar heartwood on certain wood- decaying fungi in culture. Phytopathology 33:517–524.
Stetler RA, Leak RK, Gan Y, Li P, Zhang F, Hu X, Jing Z, Chen J, Zigmond MJ, Gao Y (2014) Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog Neurobiol 114:58-83.
Storzer L, Butz M, Hirschmann J, Abbasi O, Gratkowski M, Saupe D, Vesper J, Dalal SS, Schnitzler A (2017) Bicycling suppresses abnormal beta synchrony in the Parkinsonian basal ganglia. Ann Neurol 82:592-601.
Sulzer D (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends in neurosciences 30:244-250.
Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18:101-113.
Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT (2011) The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in Parkinson's disease. Neuroscience 198:221-231.
Tai KK, Truong DD (2002) Activation of adenosine triphosphate-sensitive potassium channels confers protection against rotenone-induced cell death: therapeutic implications for Parkinson's disease. In: J Neurosci Res, pp 559-566.
Tai KK, Truong DD (2010) (-)-Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, reduces dichlorodiphenyl-trichloroethane (DDT)-induced cell death in dopaminergic SHSY-5Y cells. Neuroscience letters 482:183-187.
Takahashi J (2017) Strategies for bringing stem cell-derived dopamine neurons to the clinic: The Kyoto trial. Prog Brain Res 230:213-226.
Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM (2004) Aggresomes formed by alpha- synuclein and synphilin-1 are cytoprotective. J Biol Chem 279:4625-4631.
Tapia PC (2006) Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: "Mitohormesis" for health and vitality. Med Hypotheses 66:832-843.
Thannickal TC, Lai YY, Siegel JM (2007) Hypocretin (orexin) cell loss in Parkinson's disease. Brain 130:1586- 1595.
Titler AM, Posimo JM, Leak RK (2013) Astrocyte plasticity revealed by adaptations to severe proteotoxic stress. Cell and tissue research 352:427-443.
Ungerstedt U (1968) 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol 5:107-110.
Ungerstedt U, Arbuthnott GW (1970) Quantitative recording of rotational behavior in rats after 6-hydroxy- dopamine lesions of the nigrostriatal dopamine system. Brain Res 24:485-493.
Unnithan AS, Choi HJ, Titler AM, Posimo JM, Leak RK (2012) Rescue from a two hit, high-throughput model of neurodegeneration with N-acetyl cysteine. Neurochemistry international 61:356-368.
Unnithan AS, Jiang Y, Rumble JL, Pulugulla SH, Posimo JM, Gleixner AM, Leak RK (2014) N-acetyl cysteine prevents synergistic, severe toxicity from two hits of oxidative stress. Neurosci Lett 560:71-76.
Valdes P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C (2014) Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A 111:6804-6809.
van der Mark M, Nijssen PC, Vlaanderen J, Huss A, Mulleners WM, Sas AM, van Laar T, Kromhout H, Vermeulen R (2014) A case-control study of the protective effect of alcohol, coffee, and cigarette consumption on Parkinson disease risk: time-since-cessation modifies the effect of tobacco smoking. PLoS One 9:e95297.
Van Laar VS, Berman SB (2013) The interplay of neuronal mitochondrial dynamics and bioenergetics: implications for Parkinson's disease. Neurobiol Dis 51:43-55.
Venuto CS, Potter NB, Dorsey ER, Kieburtz K (2016) A review of disease progression models of Parkinson's disease and applications in clinical trials. Mov Disord 31:947-956.
Verbaan D, Marinus J, Visser M, van Rooden SM, Stiggelbout AM, van Hilten JJ (2007) Patient-reported autonomic symptoms in Parkinson disease. Neurology 69:333-341.
von Papen M, Fisse M, Sarfeld AS, Fink GR, Nowak DA (2014) The effects of 1 Hz rTMS preconditioned by tDCS on gait kinematics in Parkinson's disease. J Neural Transm (Vienna) 121:743-754.
Vu TC, Nutt JG, Holford NH (2012) Progression of motor and nonmotor features of Parkinson's disease and their response to treatment. Br J Clin Pharmacol 74:267-283.
Wang J, Yang QX, Sun X, Vesek J, Mosher Z, Vasavada M, Chu J, Kanekar S, Shivkumar V, Venkiteswaran K, Subramanian T (2015) MRI evaluation of asymmetry of nigrostriatal damage in the early stage of early-onset Parkinson's disease. Parkinsonism Relat Disord 21:590-596.
Weilnau JN, Carcella MA, Miner KM, Bhatia TN, Hutchison DF, Pant DB, Nouraei N, Leak RK (2017) Evidence for cross-hemispheric preconditioning in experimental Parkinson's disease. Brain Struct Funct.
Whone AL, Moore RY, Piccini PP, Brooks DJ (2003) Plasticity of the nigropallidal pathway in Parkinson's disease. Ann Neurol 53:206-213.
Woodlee MT, Kane JR, Chang J, Cormack LK, Schallert T (2008) Enhanced function in the good forelimb of hemi-parkinson rats: compensatory adaptation for contralateral postural instability? Experimental neurology 211:511-517.
Wu PF, Xie N, Zhang JJ, Guan XL, Zhou J, Long LH, Li YL, Xiong QJ, Zeng JH, Wang F, Chen JG (2012) Resveratrol preconditioning increases methionine sulfoxide reductases A expression and enhances resistance of human neuroblastoma cells to neurotoxins. The Journal of nutritional biochemistry.
Wu T, Hallett M (2013) The cerebellum in Parkinson's disease. Brain 136:696-709.
Xiao-Qing T, Jun-Li Z, Yu C, Jian-Qiang F, Pei-Xi C (2005) Hydrogen peroxide preconditioning protects PC12 cells against apoptosis induced by dopamine. Life Sci 78:61-66.
Yuan H, Sarre S, Ebinger G, Michotte Y (2005) Histological, behavioural and neurochemical evaluation of medial forebrain bundle and striatal 6-OHDA lesions as rat models of Parkinson's disease. J Neurosci Methods 144:35-45.
Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60:337- 341.
Zeevalk GD, Razmpour R, Bernard LP (2008) Glutathione and Parkinson's disease: is this the elephant in the room? Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 62:236-249.
Zhang C, Li C, Chen S, Li Z, Ma L, Jia X, Wang K, Bao J, Liang Y, Chen M, Li P, Su H, Lee SM, Liu K, Wan JB, He C (2017) Hormetic effect of panaxatriol saponins confers neuroprotection in PC12 cells and zebrafish through PI3K/AKT/mTOR and AMPK/SIRT1/FOXO3 pathways. Sci Rep 7:41082.
Zhang J, Yang ZJ, Klaus JA, Koehler RC, Huang J (2008) Delayed tolerance with repetitive transient focal ischemic preconditioning in the mouse. Stroke 39:967-974.
Zhou C, Huang Y, Przedborski S (2008) Oxidative stress in Parkinson's disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci 1147:93-104.
Zhou W, Barkow JC, Freed CR (2017) Running wheel exercise reduces alpha-synuclein aggregation and improves motor and cognitive function in a transgenic mouse model of Parkinson's disease. PLoS One 12:e0190160.
Zhu X, Raina AK, Perry G, Smith MA (2004) Alzheimer's disease: the two-hit hypothesis. Lancet neurology 3:219-226.
Zhu X, Lee HG, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: an update. Biochimica et biophysica acta 1772:494-502.
Zigmond MJ (1997) Do compensatory processes underlie the preclinical phase of neurodegenerative disease? Insights from an animal model of parkinsonism. Neurobiol Dis 4:247-253.
Zigmond MJ, Acheson AL, Stachowiak MK, Stricker EM (1984) Neurochemical compensation after nigrostriatal bundle injury in an animal model of preclinical parkinsonism. Arch Neurol 41:856- 861.
Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM (1990) Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci 13:290-296. Zigmond MJ, Cameron JL, Leak RK, Mirnics K, Russell VA, Smeyne RJ, Smith AD (2009) Triggering endogenous neuroprotective processes through exercise in models of dopamine deficiency. Parkinsonism & related disorders 15 Suppl 3:S42-45.
References
Rehana K. Leak1
1Graduate School of Pharmaceutical Sciences, Duquesne University, 600 Forbes Ave, Pittsburgh, PA 15282
Corresponding author:
Rehana K. Leak
Email: leakr@duq.edu
In a new window | Download PPT
Figure 1: Nonlinear stress responses as a function of the duration (severity) of Parkinson’s disease. (A) Jankovic noted the existence of inverted U-shaped and negative exponential stress response curves during the progression of Parkinson’s disease. Copied with permission from Jankovic’s editorial (Jankovic, 2005). (B) Wu and Hallet hypothesized a U-shaped compensatory phase in cerebellar function early in the course of Parkinson’s disease. Copied with permission from Wu and Hallet’s review (Wu and Hallett, 2013). (C) Fearnley and Lees reported that overall nigral cell loss assumes a negative exponential profile but that different subregions of the substantia nigra lose cells at varying rates. Phase 1 on the X axis in panel C reflects cases with incidental Lewy body disease (assumed to be prodromal Parkinson’s disease), and the plus signs reflect increasing severity of Parkinson’s disease. Dorsal tier: medial part (DM), lateral part (DL) and pars lateralis (PL). Ventral tier: medial part (VM) and lateral part (Vl-intermediate and VL). Copied legend and graph with permission from Fearnley and Lees’ research report (Fearnley and Lees, 1991). For a graph of the alignment of motor deficits and nigral cell loss as exponential, saturating curves, please also consult Figure 4 in Greffard and colleagues’ work (Greffard et al., 2006).
In a new window | Download PPT
Figure 2: Cross-hemispheric preconditioning mitigates motor deficits and nigrostriatal somata and terminal loss in an animal model of Parkinson’s disease. (A) Schematized illustration of the experimental design. Mice were infused in the right hemisphere with 4 μg 6-hydroxydopamine (6-OHDA) or an equal volume of saline with 0.02% ascorbic acid (preconditioning 1st hit). Three days later, animals were infused in the left hemisphere with 4 μg 6-OHDA or an equal volume of saline with 0.02% ascorbic acid (challenging 2nd hit). One and two weeks later, animals were placed in a transparent cylindrical enclosure and spontaneous behavior was videotaped. (B) A blinded observer measured the number of spontaneous turns to the left or right side one week after the second hit. (C-F) Two weeks following the second hit, coronal brain sections through the forebrain were immunostained for two markers of the dopaminergic phenotype—the rate-limiting enzyme for dopamine biosynthesis, tyrosine hydroxylase (TH) and the dopamine transporter (DAT). A blinded observer traced the anatomical boundaries of the dorsal striatum and measured the immunofluorescent signal within the region of interest using an ultrasensitive, low- resolution imager. (G-H) Two weeks following the second hit, coronal brain sections through the midbrain were immunostained for the dopaminergic marker TH and viewed by higher-resolution fluorescence microscopy. The numbers of TH+ cells were counted in the substantia nigra by a blinded observer. Panel H contains representative stitched photomontages of the ventral midbrain in all four groups. A second blinded observer confirmed key behavioral and histological data (not shown). Tissue from all groups was stained simultaneously with the same stock solutions and photographed in parallel at equivalent camera settings. Data are presented as the mean and SEM from 5-6 mice per group. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 left versus right hemisphere; a p ≤ 0.05, aa p ≤ 0.01, aaa p ≤ 0.001 versus group a; b p ≤ 0.05, bb p ≤ 0.01, bbb p ≤ 0.001 versus group b; two-way ANOVA followed by Bonferroni post hoc correction. Legend and figure copied with permission from main text or supplemental files of our recent report (Weilnau et al., 2017).
In a new window | Download PPT
Figure 3: Linear correlations between structural and functional outcomes of cross-hemispheric preconditioning in experimental Parkinson’s disease. Mice were preconditioned with 6-OHDA, as described in the legend of Figure 2, and behavioral and histological outcomes were measured, as described in our previous report (Weilnau et al., 2017). Raw striatal TH or DAT measurements (arbitrary fluorescence units) were employed as indicators of the structural integrity of nigrostriatal axon terminals and plotted as a function of the percentage of left versus right forelimb contacts with a vertical wall (A-D), left versus right forelimb landings on the floor (E-H), and left versus right spontaneous turns (I-L), as measured 7 or 14 days after the second hit (data from Weilnau et al., 2017). Two-tailed Pearson correlation analyses were applied to all the data. The Pearson correlation coefficient r and the corresponding p value are shown. Statistically significant results are presented in red font. Each animal is represented as one dot and each group is represented by a unique color and symbol. Note that some of the values are so similar that the dots overlap. n = 5-6 mice per group.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13254 | 53 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA