Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Aging and Functional Recovery after Stroke
Time:2022-08-01
Number:9290
Author Affiliations

Conditioning Medicine 2022. 5(2):59-69.
Abstract
Stroke incidence rises with aging. Nearly one-third of ischemic stroke events occur in individuals more than 75 years of age, and the elderly tend to suffer higher stroke-related mortality and worse functional outcomes. Chronic low perfusion, blood-brain barrier breakdown, immune alterations, neuronal loss, abnormal demyelination, and impairments in the generation of new neural cells and oligodendrocytes may collectively impede functional recovery after stroke in the aged. In this review, we will summarize functional alterations, including cerebral vascular aging, immune system aging, neurogenesis, and white matter integrity in the aging brain and their impact on brain recovery after cerebral ischemia.
Keywords: Aging, Dementia, Neurodegeneration, Stroke
Abstract
Stroke incidence rises with aging. Nearly one-third of ischemic stroke events occur in individuals more than 75 years of age, and the elderly tend to suffer higher stroke-related mortality and worse functional outcomes. Chronic low perfusion, blood-brain barrier breakdown, immune alterations, neuronal loss, abnormal demyelination, and impairments in the generation of new neural cells and oligodendrocytes may collectively impede functional recovery after stroke in the aged. In this review, we will summarize functional alterations, including cerebral vascular aging, immune system aging, neurogenesis, and white matter integrity in the aging brain and their impact on brain recovery after cerebral ischemia.
Keywords: Aging, Dementia, Neurodegeneration, Stroke
Introduction
Stroke is the second leading cause of death worldwide, with 50% of survivors suffering from long-term disability (Feigin et al., 2022). Ischemic stroke accounts for 87% of all strokes in the United States (Tsao et al., 2022). The number of stroke incidents is expected to double from 2010 to 2050, with the majority of stroke events occurring among the elderly (> 65 years of age) (Feigin et al., 2022; Tsao et al., 2022). Aging elicits alterations in brain anatomy and physiology and influences the incidence, progression, and treatment of stroke (Cai et al., 2017b; Xu et al., 2019a). Compared to younger patients, elderly stroke victims suffer higher mortality and morbidity and worse neurological outcomes (Feigin et al., 2022; Tsao et al., 2022). The primary interventions against ischemic stroke include antiplatelet and/or statin therapy. According to national guidelines, aspirin is recommended in adults aged 60 to 69 years, but not after 70 years of age (Bibbins-Domingo and Force, 2016). Similarly, the current maximum age for statin therapy is 75 years (Grundy and Stone, 2019), and the first exclusion criteria for recombinant tissue plasminogen activator (rt-PA) administration after stroke onset is an age of 80 years or higher. Positive outcomes of endovascular thrombectomy are dramatically diminished in elderly ischemic stroke patients compared to young patients. An age of higher than 80 years is an independent predictor of postprocedural hemorrhage after endovascular thrombectomy (Alawieh et al., 2019).
Given an increased prevalence of stroke and disability in the elderly, the use of young adult animals in most preclinical studies may have contributed to a failure in the clinical translation of neuroprotectants. Therapies effective in young animals may not exert the same benefits in the aged, as aging results in structural and functional changes that profoundly influence the brain repair and recovery process, the subject of this review.
Cerebral vascular aging and its impact on brain recovery
Arteries deliver oxygen and nutrients to all cells, while waste products are collected into veins and excreted. As the major consumer of bioenergetic supplies, the brain is exquisitely sensitive to the loss of oxygen and nutrients. The occlusion of large arteries feeding the brain results in acute ischemic stroke, whereas loss of function in small arteries and capillaries leads to chronic low perfusion and diffuse small infarcts. Age-related hypertension, hyperlipidemia, and diabetes mellitus contribute to a higher prevalence of cerebrovascular and cardiovascular diseases in the elderly. Vascular aging encompasses all the mechanical and structural changes that occur in the vascular wall with age, including increased collagen content and covalent cross-linking of collagen, over-calcification, and decreased elastin content, and elastin fracture in large arteries, resulting in reduced arterial compliance (Jani and Rajkumar, 2006; Ungvari et al., 2018a). Aging induces an imbalance in the production of endothelin and nitric oxide (NO) in endothelial cells, facilitating vascular smooth muscle growth (Jani and Rajkumar, 2006). In addition to low perfusion due to arterial stiffness, age-related impairments in angiogenesis further deteriorate microcirculation in the aged (Reed and Edelberg, 2004). Impaired angiogenesis may be caused by endothelial dysfunction, reduced NO bioactivity, dysregulated pro-angiogenic and antiangiogenic circulating factors, and weakened intrinsic endothelial angiogenic processes, including endothelial proliferation, adhesion, migration, extracellular matrix turnover, apoptosis, synthesis and release of growth factors and cytokines in endothelial cells, smooth muscle recruitment, and vessel stabilization (Ungvari et al., 2018b).
The production of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and insulin growth factor1 (IGF1) are decreased with aging (Reed and Edelberg, 2004; Ahluwalia et al., 2014). Furthermore, endothelial cells derived from aged individuals display impaired angiogenesis, downregulated VEGF production, and decreased angiogenic responses to VEGF stimulation, which may underlie the limited impact of VEGF treatment on the aged brain after ischemic stroke (Gao et al., 2009; Ahluwalia et al., 2014). VEGF stimulates angiogenesis via signaling pathways that depend upon endothelial nitric oxide synthase (eNOS) (Fukumura et al., 2001). Endothelium-derived NO is not only a critical vasodilator in the regulation of tissue perfusion, but also exerts a pro-angiogenic effect by controlling endothelial cell survival, proliferation, migration, platelet aggregation, adhesion of inflammatory cells to endothelium, and interaction with the extracellular matrix (Ungvari et al., 2018b). However, eNOS uncoupling (monomerization) and increased nitrosylation lead to the reduction of NO levels in aged mice (Yang et al., 2009). A deficiency in eNOS therefore exacerbates brain damage and cognitive dysfunctions after cerebral ischemia (An et al., 2021). Reactive oxygen species (ROS) further inactivate NO and cause oxidative damage to the endothelium.
Pericytes are involved in regulating angiogenesis and preserving the structural and functional integrity of the neurovascular unit (Sweeney et al., 2016). Pericytes are recruited to endothelial cells via the PDGFRβ signaling pathway to regulate endothelial cell proliferation, migration, and stabilization. Disrupted PDGFRβ signaling results in diminished microvascular perfusion and cerebral blood flow responses to brain activation induced by whisker-barrel cortex vibrissal stimulation (Bell et al., 2010; Ribatti et al., 2011). Age-associated vascular damage in pericyte-deficient mice elicits secondary neuronal degeneration and cognitive deficits (Bell et al., 2010). Thus, age-related endothelial dysfunction impairs cerebral angiogenesis and microcirculation remodeling after damage, thereby impeding the oxygen supply and functional recovery following stroke (Black et al., 1989; Ungvari et al., 2018b).
Endothelial cells and affiliated tight junction proteins are major components of the blood-brain barrier (BBB). The BBB regulates the penetration of molecules or cells from the circulation into the central nervous system, while also presenting a physical barrier against direct exposure of the brain to the blood. Our previous studies have shown that BBB breakdown exacerbates long-term neurological deficits induced by cerebral ischemia, whereas preserving the BBB facilitates functional post-stroke recovery in adult rodents (Shi et al., 2017; Jiang et al., 2018; Shi et al., 2020b). In humans, age-dependent BBB breakdown has been observed in the hippocampus, which may contribute to cognitive deficits in the elderly (Montagne et al., 2015). The mechanisms underlying age-related BBB breakdown are poorly understood. The ApoE4 allele accelerates BBB changes with age (Banks et al., 2021) and vascular cell senescence contributes to the age-dependent BBB breakdown (Yamazaki et al., 2016). In addition, pericyte deficits result in hyperpermeability of the BBB and are associated with accumulation of serum proteins and neurotoxic macromolecules within brain parenchyma (Bell et al., 2010). Age-dependent BBB breakdown directly facilitates the infiltration of peripheral immune cells into brain parenchyma. Cerebral endothelial cells also produce pro-thrombotic mediators and cellular adhesion molecules, such as plasminogen activator inhibitor-1, and intercellular adhesion molecule-1, which enhance the penetration of immune cells across the BBB in the aged brain (Finger et al., 2022).
Immune system aging and its function in stroke recovery
Aging is associated with profound changes in immune function (Shaw et al., 2013). The progressive deterioration of immune function with aging increases susceptibility to infections and delays recovery processes after injury. Hematopoietic stem cells (HSCs) are circulating progenitor cells that generate blood cells in mammals. HSCs can differentiate into both myeloid and lymphoid precursors to maintain immune homeostasis throughout the lifespan. HSCs gradually lose their self-renewal and differentiation potentials, which eventually affect immune cell populations with aging (Lee et al., 2019). HSC transplantation from young donors can rejuvenate the immune systems of elderly recipients (Das et al., 2019). Blood rejuvenation with parabiosis has been reported to reverse cognitive decline and synaptic plasticity in old mice (Villeda et al., 2014; Kang et al., 2020). Blood replacement also improves post-stroke outcomes in adult rodents (Ren et al., 2020). Therefore, peripheral blood rejuvenation is a potential therapeutic strategy for ischemic stroke.
Aged HSCs are more prone to differentiate towards the myeloid lineage, thereby reducing the numbers of lymphoid progenitors over the lifespan. For example, B lymphocyte counts fall with aging, and this drop is accompanied by a reduced repertoire of B-cell receptors and lower proliferative capacities (Kogut et al., 2012). A subset of B cells, defined as age-associated B cells (ABCs), have been identified. The number of ABCs is increased in the bone marrow and spleen with aging, and TNF-α released by ABCs contributes to the pro-inflammatory microenvironment and loss of B cells progenitors in the aged bone marrow (Ratliff et al., 2013). The number of circulating B cells is negatively correlated with infarct volume and long-term neurological deficits in stroke patients (Wang et al., 2017). The role of B cells is further confirmed in rodent model of cerebral ischemia. Depletion of B cells results in worse neurological functions and higher mortality, while adoptive transfer of B cells reduces infarct volume (Ren et al., 2011). A recent study further reported that B cells migrate into the ipsilateral and contralateral brain parenchyma to support neuronal viability and dendritic complexity, and to promote neurogenesis in an interleukin (IL)-10-dependent mechanism in young mice (Ortega et al., 2020). However, the impact of B cells on the aged brain after stroke is still unknown.
Aside from the age-related loss of B lymphocyte counts, there is also a decline in the number of functional naïve T lymphocytes, due to cellular senescence of activated cluster of differentiation (CD)8+ cells and an increase in memory and effector T cells with aging. This change in T cell subpopulations restricts their ability to respond to new infections in the elderly (Goronzy et al., 2015). Our previous studies demonstrated that transplantation of regulatory T cells (Tregs) markedly stimulates behavioral recovery and brain repair after ischemic stroke in young mice (Zhang et al., 2018; Shi et al., 2021). However, Treg changes during aging remain controversial. Garg et al. (2014) reported that the number and immunosuppression of Tregs are higher in aged mice, while another study revealed that Tregs are more likely to become senescent compared to other T cells, thereby losing their immunosuppressive efficacies with aging (Guo et al., 2020). These discrepancies deserve further investigation in aged animals, given the therapeutic promise of Tregs.
Natural killer (NK) cells are also a critical lymphoid population. NK cells are classified according to expression levels of the adhesion protein CD56, into CD56bright cells and CD56dim proficient cytotoxic cells (Gounder et al., 2018). In addition to cytokine production, NK cells are responsible for the clearance of senescent cells. During aging, the relative proportion of CD56bright cells is decreased compared to CD56dim cells, leading to the abnormal accumulation of senescent cells in the organism (Almeida-Oliveira et al., 2011). Inhibition of cytotoxic activation of NK cells may restore NK cellular function to block the abnormal accumulation of senescent cells to reverse or slow aging (Solana et al., 2014; Andre et al., 2018).
In contrast to shrinkage of the lymphoid progenitor pool, the total number of myeloid cells is relatively stable during aging. However, functional deficits are widely detected in myeloid cells with aging, including loss of cytokine production, reduced phagocytosis, and lower metabolism (De Maeyer and Chambers, 2021). Aged neutrophils exhibit impaired migration and lower pro-inflammatory activities in vitro, and an overly active subset of neutrophils express enhanced αΜβ2 integrin activation and neutrophil extracellular trap formation with aging in vivo (Fulop et al., 2004; Zhang et al., 2015a). Mechanistically, neutrophil aging is driven by the microbiota via Toll-like receptors (TLRs) and the myeloid differentiation factor 88-mediated signaling pathway (Zhang et al., 2015a). After stroke onset, neutrophils are rapidly recruited to the infarct region, and recent evidence has implied that aging may alter neutrophil functions after stroke. Compared to young mice, aged mice have higher mortality and morbidity, increased neutrophil-activating cytokines levels, and elevated generation of ROS in neutrophils. Depletion of neutrophils leads to long-term benefits in functional outcomes in aged animals of both sexes, supporting the potential benefit of neutrophil-target therapies in the elderly after ischemic stroke (Roy-O'Reilly et al., 2020).
As the major antigen-presenting cells (APCs), macrophages play critical roles in clearing infectious agents and cleaning up debris in tissue. Aging leads to a plethora of phenotypic, metabolic, and functional changes in macrophages. Macrophages express a lower level of TLRs and major histocompatibility complex (MHC) II in aging (Renshaw et al., 2002). Consistent with the inflammatory state in the elderly, aged macrophages display pro-inflammatory features of the M1-phenotype. However, the release of anti-inflammatory cytokines, such as IL-10 is also increased to prevent excessive tissue injury (Salminen, 2021). Nicotinamide adenine dinucleotide+ synthesis in macrophages is lowered with age, which may affect their responses to inflammatory stimuli. Wound healing is also delayed in the elderly due to the decreased phagocytic activity of aged macrophages (De Maeyer and Chambers, 2021). Accumulation of macrophages in the ischemic hemisphere is associated with injury development in acute stroke. Classically activated macrophages express CD16 and CD86 at high levels and release pro-inflammatory cytokines, while alternatively activated macrophages release anti-inflammatory factors such as IL-10 to facilitate the clearance of cellular debris and tissue repair (Hu et al., 2012; Kim and Cho, 2016).
Microglia originate from embryonic myeloid progenitors in the yolk sac and are specialized resident immune cells of the central nervous system. Microglia maintain homeostasis and immune responses by interacting with neurons and other glial cells in the brain. As organ-specific macrophages, microglia possess similar functional activities as macrophages, such as release of cytokines and chemokines and clearance of cellular debris via phagocytosis. However, unlike macrophages, microglia display increased proinflammatory properties during aging, with increased production of proinflammatory cytokines in response to stimuli, but diminished phagocytosis and chemotactic activities. Morphologically, microglia display enlarged processes, cytoplasmic hypertrophy, and a less ramified appearance in the aged brain (Conde and Streit, 2006). The dysfunction of aged microglia has been associated with age-related neurodegenerative diseases, such as Alzheimer’s disease (AD) (Leng and Edison, 2021) and Parkinson’s disease (Bartels et al., 2020). Aged microglia also play a critical role in acute brain injury and modulate the recovery process after stroke (Li et al., 2020; Marino Lee et al., 2021). Chronic depletion of microglia initiated at the early-stage of AD has been shown to improve cognitive functions in 5XFAD mouse models (Sosna et al., 2018); however, pre-injury microglia depletion exacerbates stroke outcomes within 72 hours after ischemic insult in 19-month-old mice, supporting the importance of microglial function in the aging-related neurological diseases (Marino Lee et al., 2021). However, the effect of microglia/macrophage depletion was only examined in the acute injury phase. In the latter report, neurological functions were not reported (Marino Lee et al., 2021). Thus, further studies are warranted to investigate the role of microglia in brain damage and repair process in the aging brain.
Neurogenesis in the aging brain
Neurons are terminally differentiated and postmitotic cells, and maintenance of their integrity with age is critical for retaining neurological functions. The number of neurons decreases gradually with age (Morrison and Hof, 1997). According to the work of Pakkenberg and Gunderson (1997), approximately 10% of all neocortical neurons are lost over the lifespan of 20-90 years in both sexes and the degree of neuronal loss varies across different brain regions. Compared to the aging hippocampus, the frontal and temporal cortices shrink more, with subsequent expansion of the ventricular system (Fjell and Walhovd, 2010). Neuronal loss in the hippocampus may be partially compensated by neurogenesis in the subgranular zone (SGZ). In the adult mammalian brain, neural stem cells (NSCs) contribute to neurogenesis in two areas, the subventricular zone (SVZ) adjacent to the lateral ventricles and the SGZ within the dentate gyrus of the hippocampus (Ming and Song, 2011). Neural progenitor cells (NPCs) in the SVZ travel along the rostral migratory stream towards the olfactory bulb to mature into interneurons, while SGZ-derived progenitor cells differentiate and integrate into preexisting hippocampal circuits to assist hippocampus-dependent cognitive functions (Drapeau and Nora Abrous, 2008; Kozareva et al., 2019). As in rodents, neurogenesis appears to be sustained throughout life in the human hippocampus but is impaired in AD patients (Boldrini et al., 2018; Moreno-Jimenez et al., 2019). On the other hand, there are conflicting reports that neurogenesis drops to negligible levels during adulthood (Sorrells et al., 2018). These discrepancies warrant further studies.
In the SVZ of the adult brain, NSCs are composed of quiescent and activated populations (Yabut and Pleasure, 2014). Activated NSCs of the adult brain are further classified into three types (early, mid, and late activation status), based on single-cell transcriptomic analyses (Dulken et al., 2017). Quiescent NSCs (glial fibrillary acidic protein [GFAP]+ and prominin/CD133+) give rise to activated NSCs (epidermal growth factor receptor [EGFR]+) and, in turn, differentiate into neuroblasts to generate new neurons, oligodendrocytes, and astrocytes (Lim and Alvarez-Buylla, 2016). Adult quiescent NSCs are tightly regulated. The inhibitor of DNA binding protein Id4 is enriched in quiescent NSCs to maintain their dormancy, whereas loss of Id4 results in the accumulation of Asc1 protein and NSC activation (Blomfield et al., 2019). On the other hand, LRIG1 primes quiescent NSCs to enter the cell cycle and increases EGFR protein expression, leading to activation and differentiation of NSCs (Marques-Torrejon et al., 2021).
The rate of neurogenesis declines with age, potentially contributing to olfactory and cognitive disorders in rodents and neurodegenerative diseases in humans (Bizon et al., 2004; Enwere et al., 2004; Tobin et al., 2019). The age-related decline in neurogenesis includes but is not limited to loss of NSCs, decreased NSC self-renewal, increased NSC dormancy, and loss of commitment to the neuronal lineage (Hattiangady and Shetty, 2008; Leeman et al., 2018). The NSC transcriptome in the SVZ of young versus aged mice has been evaluated by RNA-sequencing techniques. Few transcriptomic differences were detected in activated NSCs from old versus young mice. However, quiescent NSCs underwent more transcriptional changes during aging. Leeman et al. (2018) reported an abnormality of lysosomes in quiescent NSCs. An accumulation of protein aggregates blocked NSC activation, whereas enhancement of lysosome function was able to rejuvenate NSCs in the aging brain. Inflammation of the SVZ through exposure to cytokines such as interferon alpha and interferon gamma may also force NSCs into quiescence. Peripheral inflammatory factors and immune cells contribute to regulation of hippocampal neurogenesis, but it is not known if peripheral factors regulate neurogenesis via inhibition of quiescence.
Recently, single cell RNAseq technologies were applied to individual cells in the SVZ niche. The numbers of quiescent and activated NSCs are both dramatically decreased in the SVZ of the aging brain (Kalamakis et al., 2019). Old NSCs become resistant to regeneration upon brain injury, perhaps contributing to poor outcomes after ischemic stroke in the aging brain. Surprisingly, once activated, young and old NSCs display similar proliferation and differentiation activities (Kalamakis et al., 2019). Newly generated neurons in the aging brain display normal migration and survival, but neuronal maturation and dendritic growth are diminished (Rao et al., 2005). NSC activation forms part of a critical defense system against neurodegeneration during aging and contributes to functional recovery after stroke. Aside from age-related death of neurons, atrophic shrinkage of neurons and reduction in synaptic spines may account for reductions in gray matter (van der Zee, 2015). We have limited knowledge of the molecular regulation of neuronal shrinkage and loss of synaptic spines during aging. Using single-cell whole-genome sequencing, Lodato et al. (2018) discovered an accumulation of somatic mutations with age in postmitotic human neurons of the prefrontal cortex and hippocampus and reported age-related molecular signatures in brain disorders. Further investigations are needed to identify intrinsic and extrinsic triggers of NSC self-renewal and differentiation during aging.
Neurogenesis in the post-stroke aging brain
Stroke stimulates the proliferation and differentiation of NSCs in the SVZ. Neurogenesis induced by stroke is a critical step for brain recovery after an ischemic insult (Lindvall and Kokaia, 2015; Ceanga et al., 2021; Rahman et al., 2021). Neuroblasts generated from the neurogenic niche migrate along blood vessels towards the infarct and peri-infarct parenchyma and differentiate into functional neurons to replace lost cells, thereby facilitating sensorimotor recovery (Jin et al., 2001; Zhang et al., 2001; Zhang et al., 2015b). As discussed above, neurogenic activity is significantly decreased in the aging brain, which may delay stroke recovery and worsen outcomes in the elderly. Although increased doublecortin+ neuroblasts have been observed in the contralateral hemisphere of aged stroke mice, whether these neuroblasts can be differentiated into mature neurons remains controversial (Arvidsson et al., 2002; Darsalia et al., 2005; Adamczak et al., 2017). In our studies, newly generated neurons were rarely detected in 19-month-old aged mice up to 2 months after permanent ischemic stroke, whereas doublecortin+ neuroblasts were still present. Thus, despite shrinkage of the neurogenic niche with aging, we believe that NSCs still can generate neuroblasts after stroke injury in the aged brain. However, these neuroblasts may not differentiate into mature neurons.
Omega-3 polyunsaturated fatty acids are essential for brain health during developmental and adult stages and may stabilize quiescent NSCs, as NSC dormancy is coupled to high glycolytic and lipid metabolism (Stoll et al., 2015; Lo Van et al., 2019). We found that omega-3 polyunsaturated fatty acids robustly stimulate neurogenesis in aged mice after ischemic stroke (Cai et al., 2017a; Jiang et al., 2019). Our studies support neurogenesis as a promising therapeutic target for brain repair and recovery after stroke. Aside from identifying the regulatory factors in aging-related neurogenesis, the protection of remaining mature neurons and existing synapses, stimulation of new synapses, and modulation of inflammation also deserve further study.
White matter integrity in the aging brain
White matter (WM) paves the routes for neuronal communication and signal integration between different brain regions and across hemispheres. WM constitutes only about 14% of total brain volume in rodents, but reaches almost 50% of total brain volume in humans (Zhang and Sejnowski, 2000). WM is composed of neuronal axons wrapped by mature oligodendrocytes. Based on the presence or absence of the myelin sheath, axons are divided into myelinated and unmyelinated types, respectively. Oligodendrocytes provide metabolic support to enwrapped axons, and the intact myelin sheath facilitates fast transmission of electrical impulses along axons (Funfschilling et al., 2012; Lee et al., 2012). Abnormalities of WM are observed in many neurological diseases, such as multiple sclerosis, acute disseminated encephalomyelitis, stroke, and AD. In contrast to mild and regionally heterogeneous neuronal loss in aging brains, the length of myelinated axons is dramatically decreased with aging, up to nearly 50% (Terao et al., 1994; Tang et al., 1997). With aging, ranging from 30 to 90 years old, there is up to 26% loss of WM in the cerebral hemispheres of humans (Jernigan et al., 2001). Mechanically, the chronic decline of white matter blood perfusion results in the disruption of myelin integrity in aging brain (Bouhrara et al., 2020b). Furthermore, a reduction in white matter volume leads to loss of connectivity between brain regions of individuals greater than 75 years old (Vernooij et al., 2008).
Most of our knowledge on WM structural and functional connectivity is derived from diffusion tensor imaging (DTI). DTI is widely used to study cortical dysconnectivity in aging and neurological diseases (Bennett and Madden, 2014). DTI measures diffusion or movement of water molecules in various brain tissues as an estimation of structural integrity. Within fluid-filled spaces of the brain, such as blood vessels and ventricles, water diffusion is almost unbounded and thus non-directional. Diffusion of water molecules within gray matter is also relatively non-directional, whereas the parallel organization of axons in the WM and myelin sheaths restricts the directionality of water movement (Stahon et al., 2016). Diffusion of water molecules is faster in the direction parallel to the WM fibers than in the perpendicular direction. Hence, WM can be evaluated by several parameters, including fractional anisotropy (FA), mean diffusivity (MD), axial diffusivity, and radial diffusivity (RD). FA reflects the restricted proportion of total diffusion; thus, higher FA indicates higher WM integrity. MD is the average rate of non-directional diffusion; thus, higher MD values reflect lower WM integrity (Bennett and Madden, 2014). Axial diffusivity elevation is sensitive to axonal disruptions, while RD is sensitive to myelin breakdown (Bennett and Madden, 2014). A large body of work has characterized an age-related decline of WM volume and integrity, as evidenced by decreased FA and increased MD values (Lebel et al., 2012; Bennett and Madden, 2014). An increase in RD, especially in the genus of the corpus callosum, appears to be a reliable index of impaired WM during aging (Burgmans et al., 2011). It is also important to note that WM connectivity in old brains is positively correlated with cognitive functions (Bennett and Madden, 2014).
Aside from DTI, electron microscopy is commonly used to examine the ultrastructure of WM. Age-related alterations of myelinated nerve fibers, such as loss of myelinated nerve fibers and abnormal morphology and composition of myelin sheaths, have been detected in the primate and human brain (Meier-Ruge et al., 1992; Sandell and Peters, 2003). A progressive degeneration of myelinated nerve fibers mainly accounts for shrinkage of WM during aging. There are two types of myelin-degeneration conditions in the aging brain; some myelin sheaths degenerate because of degeneration of the enwrapped axons, and some myelin sheaths degenerate while their axon is still intact. The major age-related degeneration alteration is an accumulation of dark cytoplasm between the lamellae (Peters, 2009). Myelin balloons also occur with age-related structural alterations in the aging cortex. The balloon-like fluid-filled cavities are formed by splits in the intraperiod line of the affected sheaths, whose outer faces forming plasma membranes come into apposition. However, the thickness of the sheaths is unaffected and no abnormality is observed in the periodicity of the myelin lamellae (Peters, 2009). Notably, the presence of dense cytoplasm or myelin balloons in aging is correlated with cognitive decline (Peters, 2009). In addition to myelin degeneration, there is continued formation of myelin with aging and abnormally thicker sheaths, because of increased lamellae and formation of redundant sheaths. For example, small axons with thicker myelin sheaths are observed in the aged mouse optic nerve and old monkeys (Peters et al., 2001; Stahon et al., 2016).
Paranodes lie at the junction between the Node of Ranvier and compact myelin and assist in the rapid transmission of electrical impulses. Thus, preserving a stable paranodal length is critical for axonal function. In the aged brain, the efficacy of paranodal reformation is severely decreased, resulting in loss of junctional components (Shepherd et al., 2012). On the other hand, due to thicker myelin sheaths with aging, paranodes may lose contact with the axolemma, thereby hampering conduction velocity, which may contribute to cognitive deficits in the aged brain (Hinman et al., 2006). In addition to changes in conduction velocity, an increase in the frequency of paranode profiles has been reported in the aged monkey brain, due to a lengthening of the paranodes and shortening of internodal lengths, which may also slow impulse conduction rates (Peters and Sethares, 2003; Peters, 2009). Consistent with the age-related increase in remyelination, the overall number of mature oligodendrocytes is elevated in aging rodents and primates. Newly generated oligodendrocytes are mainly derived from precursor cells that are widely distributed across the central nervous system (Rivers et al., 2008; Hill et al., 2018). Thus, myelin formation occurs throughout life, and the increase in oligodendrocytes with aging may be a protective response to WM degeneration. Aging oligodendrocytes still possess the capability to develop new myelin sheaths, but this endogenous response is not sufficient to prevent age-related WM loss.
White matter injury and repair in the post-stroke brain
WM injury is the major cause of neurological disability in cerebrovascular disease (Marin and Carmichael, 2019). Like the cells within gray matter, WM is also highly vulnerable to ischemic insult (Pantoni et al., 1996). Myelination is initiated in humans at 30 weeks after gestation and achieves a stable level at 5 years of age (Yeung et al., 2014). Neonatal oligodendrocytes are extremely vulnerable, and perinatal hypoxia-ischemia reduces the survival and maturation of immature oligodendrocytes and delays axonal myelination, potentially leading to permanent WM injury and disabilities (Xu et al., 2019b). In the adult brain, occlusion of large intracranial arteries interrupts the blood supply to both gray matter and white matter, resulting in neuronal injury, synapse loss, and axonal dysfunction (Marin and Carmichael, 2019). Perhaps for these reasons, neuron-targeted therapeutic strategies that ignore WM injury have largely failed in clinical trials.
Cerebral ischemia induces severe WM injury through (but not limited to) energy deprivation, oxidative stress, and proinflammatory cytokines, which are all further increased in aged individuals (Xu et al., 2019a). WM recovery becomes more challenging when the injury is compounded by age-related WM loss. WM recovery consists of axon sprouting and remyelination of demyelinated and/or newly generated axons. In postmortem samples of WM from human stroke victims, axons within the peri-infarct region appear to be relatively intact (Hinman et al., 2015). The sprouting of spared axons is thought to contribute to the repair of neuronal connections and functional recovery (Xu et al., 2019a). A variety of molecules, such as growth factors, cell adhesion factors, axonal guidance cues, and cytoskeleton-modifying factors may be involved in the neuronal regrowth program after stroke. Of note, there are dramatic differences between young and aged rodents in cytokines/chemokines, growth factors, axonal guidance cues, bone morphogenic proteins, and cell adhesion molecules (Li et al., 2010). Among these factors, ephrin type-A receptor 4 and Lingo1 are two receptors for axonal growth-inhibitory proteins and are upregulated with aging in sprouting neurons and may retard recovery (Li et al., 2010).
Aside from the age-related loss of axonal sprouting, loss of paranode reformation, shrinkage of WM, and impairment of remyelination may also hinder WM recovery after stroke. Oligodendrocyte precursor cells (OPCs) are resident progenitor cells that differentiate into mature myelinating oligodendrocytes in the adult brain. In a model of demyelination, an age-related decline in remyelination efficiency is associated with impaired recruitment of OPCs and insufficient differentiation into myelinating oligodendrocytes (Sim et al., 2002). Upon demyelination, factors that restrict oligodendrocyte differentiation are downregulated before the synthesis of new myelin in the young adult brain, while accumulation of transcriptional inhibitors prevents expression of myelin formation-related genes (Shen et al., 2008). Although OPCs are defined as precursors to oligodendrocytes, they can also differentiate into neurons and astrocytes (Kondo and Raff, 2000). In ischemic stroke models, OPCs accumulate in peri-infarct regions, where axonal sprouting also occurs. However, OPCs can fail to mature into myelinating oligodendrocytes that wrap intact axons and may differentiate into astrocytes in young and aged mice.
NgR1, a glycosylphosphatidylinositol (GPI)-anchored protein, forms a receptor complex with p75 and LINGO-1. The latter complex binds to myelin-associated growth inhibitory molecules, such as NogoA, myelin-associated protein (MAG), and oligodendrocyte myelin glycoprotein (OMgp) to restrict axon growth (Mi et al., 2004). In oligodendrocytes and OPCs, the Nogo receptor1 (NgR1) helps to regulate OPC maturation and myelin formation after ischemic WM injury (Chong et al., 2012). Stroke induces NgR1 ligands and downregulates NgR1 inhibitors (such as Lgi1, Lotus, Adam22) to block OPC maturation towards the oligodendrocyte lineage, whereas an NgR1 antagonist switches OPC-to-astrocytic fate toward the generation of more oligodendrocytes. It is also important to note that NgR1 antagonism dramatically stimulates WM repair and encourages motor recovery in aged mice (Chong et al., 2012; Sozmen et al., 2016).
In addition to downregulation of oligodendrogenesis with aging, growing evidence suggests that failure to clear myelin debris also limits WM repair in the aging brain after stroke (Rosenzweig and Carmichael, 2013). Microglia/macrophages are professional phagocytes and play a major role in clearing myelin debris in the aging brain and after stroke. However, age-related alterations in microglia morphology and functions are found in the aging brain of rodents and humans (Safaiyan et al., 2016; Koellhoffer et al., 2017; Spittau, 2017). Microglial expression of MHCII (VanGuilder et al., 2011), CD68 (Griffin et al., 2006), and TLRs (Letiembre et al., 2007) is upregulated with age. Age-dependent microglial activation is characterized by accumulation of lipofuscin inclusions, reduced complexity of microglial processes, increased expression of proinflammatory (IL-1β, tumor necrosis factor α) and anti-inflammatory (IL-10, transforming growth factor β) cytokines, and diminished production of neuroprotective factors (Safaiyan et al., 2016; Spittau, 2017; Shi et al., 2020a). The efficiency of myelin debris clearance decreases with age, thereby hampering OPC differentiation and remyelination (Ruckh et al., 2012; Safaiyan et al., 2016). TREM2 is a key regulator of age-related microglial phagocytic functions (Kim et al., 2017; Schoch et al., 2021). In the aging brain, stroke adds to the burden of myelin debris and increases the engulfment load of microglia/macrophages. Aged microglia are also prone to proinflammatory activation upon stimulation (Niraula et al., 2017). In turn, proinflammatory cytokines induced by ischemic stroke exacerbate secondary damage to OPCs, oligodendrocytes, as well as neurons and NSCs. A chronic inflammatory microenvironment in aging slows functional recovery after an ischemic insult. Microglia/macrophage-targeted therapies may therefore show promise against age-related neurogenerative diseases and acute brain injuries.
A limitation of our discussion in this review is the binary treatment of the young versus the aged, based largely on the lower cost of researching only two groups in the laboratory. In real life, aging lies along a multidimensional continuum, and the biological responses to aging are often best fitted by parabolas or skewed J-shaped or heavy-tailed (Pareto-like) curves, rather than simple linear regressions. For example, myelin water fraction, a measure of myelin content, is not linear but U-shaped over the human lifespan (Bouhrara et al., 2020a) and the observed curve can be fitted with a quadratic equation (Arshad et al., 2016). WM estimations over the lifespan may also contribute to nonlinear changes in cognitive/executive function with age (Filippi et al., 2020; Ferguson et al., 2021). Thus, cognitive decline in humans may only be evident toward the end of life, typically after exiting the workforce (Fisher et al., 2017).
A second limitation of our discussion is that the evidence has not been stratified according to biological sex. Sex is a determinant of lifespan and influences aging-associated post-stroke recovery (Rexrode et al., 2022). For example, sex differences in the responses to injury are apparent even in neonatal ischemia, suggesting sex-chromosomal differences can be manifested early in life (Charriaut-Marlangue et al., 2017). Adult female rodents also display smaller infarcts compared with age-matched males. However, the female-skewed protection against injury is reversed after menopause in human beings (Gasbarrino et al., 2022; Rexrode et al., 2022). Female sex hormones such as estrogen are well-established cytoprotective molecules in ischemic stroke (Maioli et al., 2021). Although biological sex may not predict mortality after adjustment by age and pre-stroke functions, sex biases in functional recovery have nonetheless been reported in clinical and preclinical studies (Bushnell et al., 2014; Feigin et al., 2022). It is therefore necessary to include aged animals of both sexes in preclinical studies to investigate the pathophysiology of ischemic stroke and to test therapeutics.
In summary, aging is an independent and critical risk factor for ischemic stroke. Elderly stroke victims suffer severe outcomes because of retarded recovery functions, including age-dependent endothelial dysfunction, BBB breakdown, neuronal loss, and alterations in the functions of progenitor and immune cells (Figure 1). Therapeutic strategies that target these age-related processes may alleviate long-term neurological deficits after stroke.
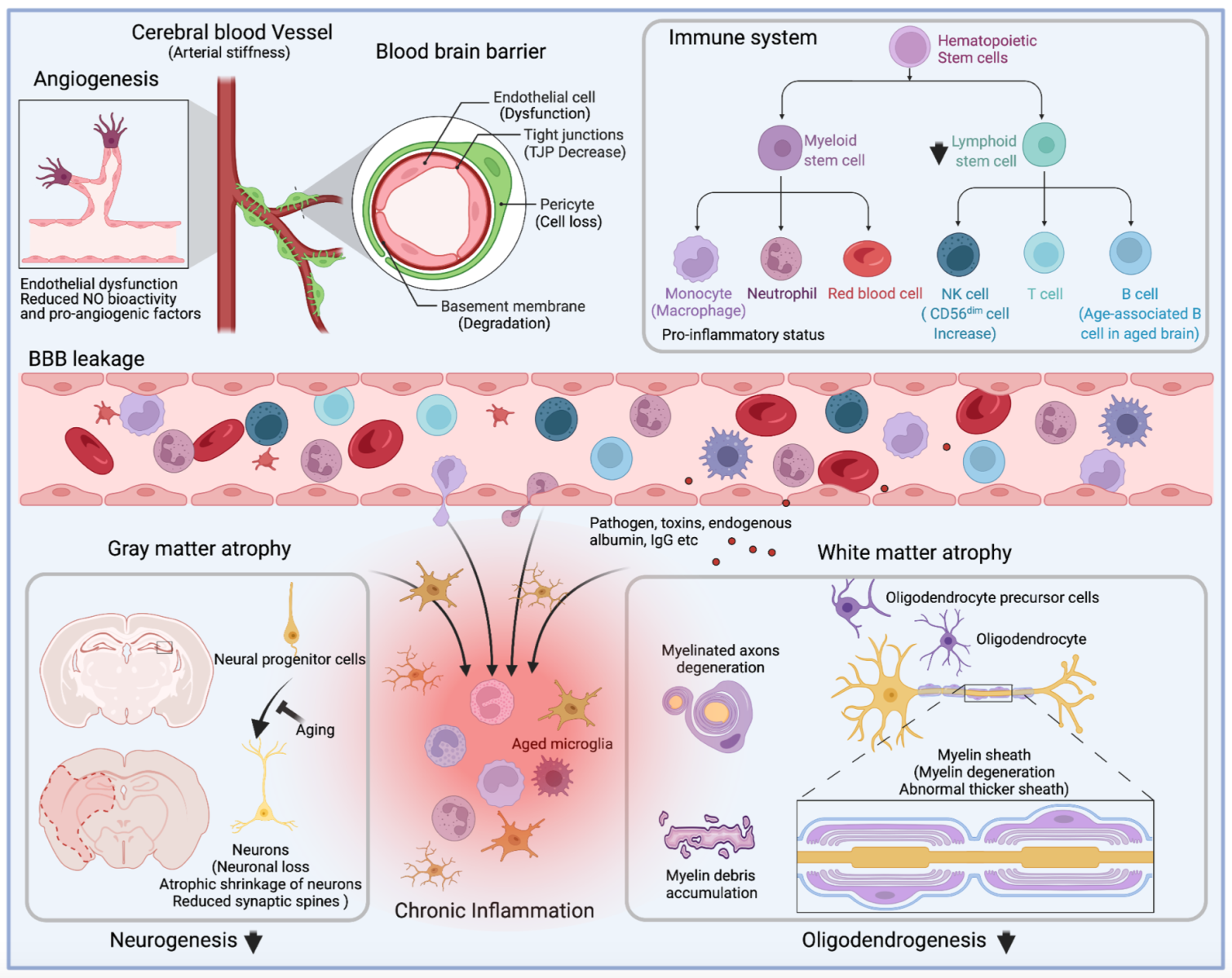
In a new window | Download PPT
Figure 1: Alterations of cerebral vessels, immune system, and brain during aging. Depiction of the main alterations that affect angiogenesis, immune cell population and function, blood-brain barrier permeability, and gray matter and white matter integrity during aging. Briefly, aging ultimately results in the senescence of mature and progenitor cells in the central nervous and the circulation systems. In circulation, age-related arterial stiffness leads to low perfusion of cerebral blood flow, and angiogenesis impairment further exacerbates the brain's supply of nutrients and oxygen. Besides weakened angiogenesis, senescent endothelial cells contribute to the blood-brain barrier's breakdown, accompanied by decreased expression of tight junction proteins, degeneration of basement membrane, and loss of pericytes. The pro-inflammatory immune system, characterized by excessive production of inflammatory cytokines and loss of lymphocytes, enhances BBB damage during aging. In the brain, aging-associated gray matter and white matter atrophy is attributed to senescent neural progenitor cells, loss of mature neurons, and degeneration of myelin sheath, resulting in functional deficits in the elderly. Meanwhile, the leakage of circulation proteins (endogenous albumin and IgG), toxins, pathogens, and peripheral immune cells disrupt the stability of the aging brain. Brain atrophy is aggravated by the pro-inflammatory factors produced by aged microglia and the infiltrated immune cells.
The conflicts of interest
None
Acknowledgments
We thank Patricia Strickler for administrative support. This project was supported by the University of Pittsburgh School of Medicine. J.C. is the Richard King Mellon Professor of Neurology and a recipient of a VA Senior Research Career Scientist Award (821-RC-NB-30556). J.C. is also supported by VA Merit Review Grants I01BX005290 and I01BX003377 and NIH grant NS0105430.
Dr. Jun Chen, who serves on the Publication Committee for Conditioning Medicine, did not participate at any level in the editorial review of this manuscript.
References
Wenting Zhang1,2
1Geriatric Research, Education and Clinical Center, Veterans Affairs Pittsburgh Health Care System, Pittsburgh, PA 15261, USA 2Pittsburgh Institute of Brain Disorders & Recovery and Department of Neurology, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Rehana K. Leak3
3Graduate School of Pharmaceutical Sciences, School of Pharmacy, Duquesne University, Pittsburgh, PA 15282, USA.
Jun Chen1,2*
1Geriatric Research, Education and Clinical Center, Veterans Affairs Pittsburgh Health Care System, Pittsburgh, PA 15261, USA 2Pittsburgh Institute of Brain Disorders & Recovery and Department of Neurology, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Corresponding author:
Dr. Jun Chen
Email: chenj2@upmc.edu
Handling editor: Derek J. Hausenloy, MD, PhD
In a new window | Download PPT
Figure 1: Alterations of cerebral vessels, immune system, and brain during aging. Depiction of the main alterations that affect angiogenesis, immune cell population and function, blood-brain barrier permeability, and gray matter and white matter integrity during aging. Briefly, aging ultimately results in the senescence of mature and progenitor cells in the central nervous and the circulation systems. In circulation, age-related arterial stiffness leads to low perfusion of cerebral blood flow, and angiogenesis impairment further exacerbates the brain's supply of nutrients and oxygen. Besides weakened angiogenesis, senescent endothelial cells contribute to the blood-brain barrier's breakdown, accompanied by decreased expression of tight junction proteins, degeneration of basement membrane, and loss of pericytes. The pro-inflammatory immune system, characterized by excessive production of inflammatory cytokines and loss of lymphocytes, enhances BBB damage during aging. In the brain, aging-associated gray matter and white matter atrophy is attributed to senescent neural progenitor cells, loss of mature neurons, and degeneration of myelin sheath, resulting in functional deficits in the elderly. Meanwhile, the leakage of circulation proteins (endogenous albumin and IgG), toxins, pathogens, and peripheral immune cells disrupt the stability of the aging brain. Brain atrophy is aggravated by the pro-inflammatory factors produced by aged microglia and the infiltrated immune cells.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 9290 | 16 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA