Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Cardioprotective effects of sglt2 inhibitors through their direct actions on cardiomyocytes
Time:2023-12-01
Number:11920
Author Affiliations

Conditioning Medicine 2023. 6(2): 42-53.
Abstract
Heart failure (HF) is one of the leading causes of death and disability worldwide and it exerts a huge economic burden on the healthcare system. The sodium glucose co-transporter 2 (SGLT2) is one of the major glucose co-transporters involved in glucose reabsorption in the kidney that helps control body glucose levels. Inhibitors of SGLT2 were originally developed to lower blood glucose for the treatment of type-2 diabetes (T2D), but recently they have been associated with positive outcomes in a variety of HF patient cohorts regardless of diabetic status. The salutatory effects of SGLT2 inhibitors on cardiac function have been attributed to a shift in metabolic flow, reduced blood pressure and plasma volume through natriuretic, anti-fibrotic, and anti-inflammatory effects. More recently SGLT2 inhibitors have been linked to direct effects on cardiomyocytes. However, the exact mechanisms through which SGLT2 inhibitors modulate cardiomyocyte function remains elusive due to the lack of consensus on the expression of SGLT2 in the heart, and their complex impact on different organs. In this review, we focus on studies that have evaluated the direct effects of SGLT2 inhibitors on cardiomyocytes and discuss how these actions could achieve cardioprotection. A better understanding of the cardiomyocyte-specific benefits of SGLT2 inhibitors could help to optimize their usage in clinical practice and improve the prognosis of patients with HF.
Keywords: Heart failure, Sodium glucose co-transporter 2 inhibitors, Cardiomyocytes, Sodium-hydrogen exchanger, AMP-activated protein kinase
Abstract
Heart failure (HF) is one of the leading causes of death and disability worldwide and it exerts a huge economic burden on the healthcare system. The sodium glucose co-transporter 2 (SGLT2) is one of the major glucose co-transporters involved in glucose reabsorption in the kidney that helps control body glucose levels. Inhibitors of SGLT2 were originally developed to lower blood glucose for the treatment of type-2 diabetes (T2D), but recently they have been associated with positive outcomes in a variety of HF patient cohorts regardless of diabetic status. The salutatory effects of SGLT2 inhibitors on cardiac function have been attributed to a shift in metabolic flow, reduced blood pressure and plasma volume through natriuretic, anti-fibrotic, and anti-inflammatory effects. More recently SGLT2 inhibitors have been linked to direct effects on cardiomyocytes. However, the exact mechanisms through which SGLT2 inhibitors modulate cardiomyocyte function remains elusive due to the lack of consensus on the expression of SGLT2 in the heart, and their complex impact on different organs. In this review, we focus on studies that have evaluated the direct effects of SGLT2 inhibitors on cardiomyocytes and discuss how these actions could achieve cardioprotection. A better understanding of the cardiomyocyte-specific benefits of SGLT2 inhibitors could help to optimize their usage in clinical practice and improve the prognosis of patients with HF.
Keywords: Heart failure, Sodium glucose co-transporter 2 inhibitors, Cardiomyocytes, Sodium-hydrogen exchanger, AMP-activated protein kinase
Highlights
A key pathophysiological feature of heart failure is aberrant levels of body glucose. Here, the authors implicate the important role of the sodium glucose co-transporter 2 (SGLT2), which stands as a major glucose cotransporter regulating body glucose levels. Although it has been shown that inhibitors of SGLT2 lower blood glucose for the treatment of type 2 diabetes, their use in heart failure has recently gained attention. The authors review the literature in support of SGLT2 inhibitors in modulating cardiomyocytes with emphasis on cardioprotective strategies. In line with Conditioning Medicine’s overarching goal of advancing cardioprotection, this article probes the mechanistic interaction between SGLT2 inhibitors and cardiomyocyte function that could help in the diagnosis and treatment of heart failure.
Introduction
Heart failure (HF) is one of the leading causes of death and disability worldwide affecting 6.2 million individuals in the United States and 37.7 million individuals worldwide according to recent epidemiological reports (Ziaeian and Fonarow 2016; Virani et al., 2020). HF is the most common complication of cardiovascular diseases, and is classified into HF with preserved ejection fraction (HFpEF, when EF > 50%) and HF with reduced EF (HFrEF, when EF < 50%) (Ponokowski et al., 2016; Yancy et al., 2017). Regardless of HF classification, it happens when the heart fails to pump enough blood to meet the requirements of the body, leading to poor quality of life affected negatively by, for example, shortness of breath, exercise intolerance, and swelling in the lower limbs. The healthcare cost for HF is high mainly due to frequent hospitalizations and chronic treatments (Heidenreich et al., 2022), and is estimated to increase from 31 billion USD in 2012 to 70 billion USD by 2030 in America alone (Heidenreich et al., 2013). Thus, there is an unmet need to identify novel treatment approaches to improve clinical outcomes in HF.
The sodium glucose co-transporter 2 (SGLT2) belongs to a sodium/glucose co-transporter gene family (SLC5A), which express a group of sodium-coupled transporters for several different nutrients (Ostrowska et al., 2015; Wright et al., 2017). Among them, four isoforms transport glucose (SGLT1, 2, 4, and 5) (Hediger et al., 1989; Wells et al., 1992). SGLT2 is the predominant SGLT isoform expressed in renal proximal convoluted tubule epithelial cells (RPCTs) and is responsible for the majority of glucose reabsorption in concert with glucose transporter 2 (GLUT2) (Vallon et al., 2011). As improving glycemic control has been the major aim for treating type-2 diabetes (T2D), SGLT2 inhibitors were originally used to lower blood glucose levels for the treatment of T2D (Heise et al., 2013; Seman et al., 2013). Given its cardiovascular benefits reported in clinical trials for T2D (Häring et al., 2013; Kovacs et al., 2014; Chilton et al., 2015), which include reducing arterial stiffness and lowering systemic blood pressure, SGLT2 inhibitors were later proposed as potential therapeutic agents for patients with HF. Among them, empagliflozin has been associated with the most positive outcomes in a number of patient cohorts regardless of diabetic status, including T2D patients with high CV risk (EMPA-REG Outcome) (Zinman et al., 2015), HFrEF (EMPEROR-Reduced) (Packer et al., 2020), and HFpEF (EMPEROR-Preserved) (Anker et al., 2021), by primarily reducing the risk of HF hospitalization. Apart from empagliflozin, dapagliflozin is another SGLT2 inhibitor recently reported to reduce HF hospitalizations and cardiovascular death in patient cohorts with HFrEF (DAPA-HF) Jhund et al., 2021) and HFpEF (DELIVER) (Solomon et al., 2022). Other SGLT2 inhibitors that have shown efficacy in improving cardiovascular function in patients with T2D include canagliflozin, sotagliflozin, and ertugliflozin (Neal et al., 2017; Cannon et al., 2020; Cong et al., 2020; Bhatt et al., 2021). Taken together, there is substantial evidence that SGLT2 inhibition is cardioprotective and these benefits are independent of their glucose-lowering effects.
In this review article, we discuss potential mechanisms through which SGLT2 inhibitors achieve cardioprotective function, with emphasis on their direct benefits on cardiomyocytes. We also discuss the challenges and controversies investigating cardiomyocyte-specific effects of SGLT2 inhibitors. Better understanding of the actions of SGLT2 inhibitors in improving cardiac function could help to optimize their usage in clinical practice, improve the prognosis of patients with HF, and further our understanding of the underlying mechanisms that drive HF.
Mechanisms of Cardioprotection – Do Cardiomyocytes Matter?
Systemic vs. cardiac benefits of SGLT2 inhibitors
It is critical to identify the targets of SGLT inhibitors to understand the mechanisms through which cardiac function is improved. One of the key pathophysiological conditions of HF is sustained activation of compensatory pathways such as the sympathetic neural system (SNS) and the renin-angiotensin-aldosterone (RAAS) system, accompanied by elevated inflammation and oxidative stress, leading to maladaptative remodeling of the myocardium (Eisen 2017; Fathi et al., 2021). Previous HF therapeutics, such as beta blockers (Groenning et al., 2000) or blood pressure (BP) lowering agents (Greenberg et al., 1995; Cicoira et al., 2002), were developed to reverse these compensatory pathways to ameliorate the severity of the disease state. A similar notion was applied to SGLT2 inhibitors as SGLT2 inhibition in the renal system could shift metabolic flow through glucosuria and reduce BP and plasma volume through natriuresis (Heerspink et al., 2016; Cowie and Fisher 2020) that could either improve fuel-usage or reduce preload of the heart to alleviate cardiac stress. Apart from direct inhibition of SGLT2 in the kidney, empagliflozin could also stimulate anti-inflammatory signaling pathways in the immune system that could be beneficial to cardiac function, including augmented M2 marker expression in macrophages, suppressed inducible nitric oxide synthase, interleukin (IL)-6, and IL-10 levels in the blood (Andreadou et al., 2017; Lee et al., 2017; Koyani et al., 2020). However, while the majority of studies focused on indirect cardiac benefits as in other current HF therapeutics, emerging reports also showed SGLT2 inhibitors could act on the heart to improve its function by targeting cardiac fibroblasts, endothelial cells, and cardiomyocytes. As cardiomyocytes are the predominant cell type that governs mechanical and bioenergetic properties of the heart, we will focus on the direct beneficial effects of SGLT2 inhibitors on cardiomyocytes in this review.
Cardiomyocyte-specific effects of empagliflozin
A key area of controversy when investigating direct actions of SGLT2 inhibitors on cardiomyocytes is that there is lack of consensus on whether SGLT2 is expressed in cardiomyocytes. Although the majority of earlier reports have suggested that SGLT2 is absent in the heart (Chen et al., 2010; Vrhovac et al., 2015), emerging evidence has shown SGLT-2 expression in cardiomyocytes, particularly in aging or diseased conditions. In 2017, Hammoudi el al. (2017) reported increased SGLT2 expression in the heart tissue of a genetic model of T2D mice (ob/ob), which trended downwards with empagliflozin treatment. This result was supported by a later study showing that high glucose (HG)-treated human induced pluripotent stem cells (hiPSCs) induced a dramatic increase in SGLT2 expression, which was reversed by empagliflozin (Ng et al., 2018). Later, Olgar el al. (2020) demonstrated the appearance of SGLT2 expression at the protein level in cardiomyocytes isolated from 24-month-old Wistar male rats. Similar results have been found in the heart tissue of murine models of myocardial infarction (Lee et al., 2021; Li et al., 2021). Nonetheless, a recent study reported that the protective effect of empagliflozin on myocardial infarction remained intact in whole-body SGLT2 KO mouse, suggesting a mechanism independent of SGLT2 inhibition (Chen et al., 2023). As SGLT2 does not appear to be the predominant SGLT isoform nor the major sodium/glucose transporters in cardiomyocytes, pre-clinical studies have focused on off-target effects of empagliflozin. These identified cardiomyocyte-specific effects of SGLT2 inhibitors include inhibition of sodium proton exchanger 1 (NHE1) (Baartscheer et al., 2017), activation of AMP-activated protein kinase (AMPK) (Lu et al., 2020; Kondo et al., 2021), inhibition of the late Na+ current (Philippaert et al., 2021), and inhibition of Ca2+/calmodulin-dependent kinase II (CaMKII) (Mustroph et al., 2018). As improved cardiomyocyte function in an in vivo animal model could result from systemic benefits instead of cardiomyocyte-specific actions, we will focus on in vitro effects of SGLT2 inhibitors on cardiomyocytes. We will mainly discuss results from isolated adult or neonatal cardiomyocytes from rats or mice, and cardiomyocytes differentiated from hiPSCs. We will also include results using H9c2 or HL-1 cells, as a significant portion of the studies in this field are still at the stage of using immortalized cell lines for cardiac phenotyping. Information provided from this review will help to clarify the exact actions of SGLT2 inhibitors at the cardiomyocyte level and how these could be linked to cardioprotection.
NHE1 Activities and Sodium Homeostasis
NHE1 is the predominant isoform of NHE and major regulator of Na+ influx in cardiomyocytes, and its activity has been shown to increase in HF (Karmazyn 1988; Yokoyama et al., 2000; Engelhardt et al., 2002). Elevated NHE1 activity increases cytosolic [Na+] and [Ca2+], leading to cardiomyocyte injury and cardiac hypertrophy (Nakamura et al., 2008; Despa and Bers 2013). Clinical trials using specific NHE1 inhibitors to treat acute myocardial infarction, however, have mostly been unsuccessful because of serious side effects such as increased incidence of cerebrovascular events leading to strokes, despite their cardioprotective effects (Mentzer Jr et al., 2008; Karmazyn 2013). The role of NHE1 as a potential mediator of the effects of SGLT2 inhibitors in cardiomyocytes was first described by Baartscheer et al. (2017) who demonstrated that empagliflozin directly inhibited NHE1 in rat and rabbit cardiomyocytes. The same group reported that while empagliflozin treatment reduced cytosolic [Na+], and both diastolic and systolic [Ca2+], mitochondria [Ca2+] was increased (Baartscheer et al., 2017). This is likely due to the secondary effect of reduced cytosolic [Na+], leading to Na+ efflux from mitochondria through the mitochondrial sodium calcium exchanger (NCLX) in exchange for Ca2+ influx (Pogwizd et al., 2003; Liu and O'Rourke 2008). In terms of the inhibitory mechanism on NHE1, the same group showcased that empagliflozin inhibited NHE1 likely through binding to its Na+ binding site (Uthman et al., 2018). This finding from Baartscheer et al (2017) was later supported by a follow-up study from another group (Trum et al., 2020). Since then, several studies have shown empagliflozin attenuated cytosolic Na+ overload, improved mitochondrial function, and reduced reactive oxygen species (ROS) production in adult mouse cardiomyocytes from different disease models (Lee et al., 2019; Uthman et al., 2019; Peng et al., 2023). In a recent study, Jiang et al. (2022) showed that empagliflozin treatment could reduce cytosolic [Na+] and [Ca2+] and protected neonatal mouse cardiomyocytes and H9c2 cells from glucose deprivation-induced autosis. These effects could be mimicked by NHE1 knockout and nullified by NHE1 overexpression (Jiang et al., 2022). In immortalized cell lines, NHE1 expression was restored when dapagliflozin or empagliflozin protected H9c2 cells from high glucose-triggered apoptosis or angiotensin II-induced cellular hypertrophy, respectively. (Shih et al., 2021; Abdulrahman et al., 2022).
Despite plenty of promising evidence that SGLT2 inhibitors are cardioprotective through NHE1 inhibition, this mechanism has been questioned by some publications (Chung et al., 2021; Li et al., 2021; Baker et al., 2022). Also, the underlying mechanism by which NHE1 inhibition with SGLT2 inhibitors provides cardioprotective effects is still not well understood. The major hypothesis as to how NHE1 inhibition is beneficial to HF is through reduction in cytosolic [Na+], which has the secondary consequence of lowering cytosolic [Ca2+], resulting in multiple favorable effects for HF, as previously reviewed in detail elsewhere (Chen et al., 2022). One possible outcome of these effects is increased mitochondrial [Ca2+]. Although mitochondrial Ca2+ overload is known to be detrimental, basal mitochondrial Ca2+ is required for normal function of the tricarboxylic acid cycle and antioxidant network (Cortassa et al., 2003; Liu and O'Rourke 2008; Kohlhaas et al., 2010; Liu et al., 2014; Kohlhaas et al., 2017). Apart from NHE1 inhibition, several recent studies have demonstrated that the late sodium current (INa,late), which is the sustained component of Na+ current during the plateau of the action potential that controls Na+ entry homeostasis of the cardiomyocytes, was suppressed by empagliflozin treatment (Philippaert et al., 2021; Hegyi et al., 2022; Mustroph et al., 2022). Since INa,late is known to be elevated in different types of HF (Horvath and Bers 2014; Makielski 2016) as seen in NHE1 activity, SGLT2 inhibitors likely play a broader regulatory role apart from NHE1 inhibition for the rebalancing of cytosolic Na+ in response to HF. Interestingly, a recent study demonstrated that in vitro treatment with empagliflozin could directly increase ATP levels in mouse cardiomyocytes partly through NHE1 inhibition and INa,late inhibition (Choi et al., 2023), suggesting a direct link between sodium homoeostasis and mitochondrial function. Furthermore, the role of SGLT2 inhibitors in down-regulating INa,late likely explain why they were also recently demonstrated to have beneficial clinical outcomes in patients with arrhythmias (Li et al., 2021).
CaMKII Activities and Calcium Handling
Calcium/calmodulin-dependent kinase II (CaMKII) is a serine/threonine-based phosphokinase that is found in almost every organ (Braun and Schulman 1995), with its δ form being the predominant cardiac isoform (Schworer et al., 1993; Mayer et al., 1995). CaMKII is activated when there is increased intracellular [Ca2+], and when activated in cardiomyocytes, it phosphorylates key regulators of Ca2+ homeostasis and excitation-contraction coupling (ECC) including ryanodine receptor (RyR), phospholamban, and L-type Ca2+ channel (Yuan and Bers 1994; Lokuta et al., 1995; Karczewski et al., 1997; Dzhura et al., 2000). In the cardiomyocytes of the failing hearts, there is up-regulation of CaMKII activity, leading to defective ECC, enhanced diastolic sarcoplasmic reticulum (SR) Ca2+ leak, and reduced SR Ca2+ sequestration, resulting in impaired contraction and relaxation (Hoch et al., 1999; Kirchhefer et al., 1999). With regard to the interaction with SGTL2 inhibitors, Mustroph et al. (2018) demonstrated that 24 hour exposure to empagliflozin (1 µM) reduced CaMKII activity in adult cardiomyocytes isolated from HF patients and mice subjected to transverse aortic constriction. Consistent with CaMKII activity, they also found decreased CaMKII-dependent RyR phosphorylation and reduced spontaneous diastolic Ca2+ leaks from the SR, and most importantly, increased SR Ca2+ transient in these cardiomyocytes, suggesting that increased contractile function at the level of the cardiomyocytes might account for the beneficial effects of empagliflozin in HF. The ability of SGLT2 inhibitors to regulate Ca2+ homeostasis was also observed when empagliflozin was shown to reduce elevated Ca2+ transient and improve [Ca2+] re-uptake in hiPSCs derived cardiomyocytes with HG administration (Ng et al., 2018). Later, Olgar et al. (2020) showed that treatment with dapagliflozin abated elevated intracellular [Ca2+] and Ca2+ leak from SR in cardiomyocytes from aging mice (Olgar et al., 2020). These results, however, were challenged by a report that observed no changes in either CaMKII activity or and Ca2+ transient after hiPSC cardiomyocytes were subjected to long-term (2 and 8 weeks) administration of empagliflozin (0.5 µM) (Pabel et al., 2020). One possible explanation is that healthy cardiomyocytes do not have impaired Ca2+ handling or CaMKII, so the effect of SGLT2 inhibitors was minimized. Interestingly, in a recent report, Mustroph et al. (2022) demonstrated that reduced INa,late observed in adult mouse cardiomyocytes treated with empagliflozin was dependent on CaMKII inhibition. Thus, there is the likelihood of crosstalk between Ca2+ and Na+ homeostasis in response to SGLT2 inhibitors at the level of cardiomyocytes, and their potential mechanisms still need to be determined. Current knowledge regarding the role of SGLT2 inhibitors to control Ca2+ and Na+ homeostasis is summarized in Table 1.

AMPK – Nutritional Homeostasis and Autophagy
Another well-reported target of SGLT2 inhibitors at the cardiomyocyte level is AMPK and its associated autophagy/mitophagy pathways. AMPK is a master regulator of energy homeostasis, which maintains proper cellular function in response to energetic stress (Heidrich et al., 2010; Steinberg and Carling 2019). One of the key allosteric activators of AMPK is elevated the ADP/ATP ratio during energy deprivation, leading to the shift from anaerobic to catabolic pathways in favor of energy production (Davies et al., 1995; Hardie et al., 1999), thus AMPK acts as both a nutrient sensor and responder. In HF, there is a surplus of nutrients such as glucose or long chain fatty acids in the cytosol owing to impaired mitochondrial function that reduces oxidative metabolism (Sharma et al., 2004), leading to the suppression of AMPK (Wang et al., 2018).
Restoring the activity of AMPK and the energy production machinery has been one of the major benefits of SGLT2 inhibitors in earlier publications, as deduced mostly from in vivo studies (Inoue et al., 2019; Sayour et al., 2019; Lu et al., 2020). However, these studies could not rule out the likelihood that AMPK is activated through systemic mechanisms such as reduced blood glucose levels, which is a well-known consequence of SGLT inhibitor administration. Interestingly, short-term treatment of empagliflozin did not appear to have effects on AMPK signaling of isolated diabetic mouse hearts (Zhang et al., 2020). It was not until 2020 that a series of publications demonstrated that treatment with SGLT2 inhibitors in vitro directly activated AMPK and its mitoprotective actions for energy production in adult mouse cardiomyocytes (Lu et al., 2020; Sun et al., 2020) and HL-1 cells (Koyani et al., 2020) in response to ischemia-reperfusion injury, LPS-induced inflammation, or in vivo high fat diet treatment. The activation of AMPK pathway with SGTL2 inhibitor also relieved the adverse effects of oxidative stress at the level of cardiomyocytes by restoring PGC-1α (Tsai et al., 2021) or down-regulating NADPH oxidase through Rac1 signaling (Kondo et al., 2021). The study by Kondo el al. (2021) also demonstrated that canagliflozin blunted glucose uptake by inhibiting SGLT1, leading to elevated AMP/ATP ratio, which activated AMPK. This proposed mechanism for how canagliflozin (IC50 = 694-910nM to SGLT1) activates AMPK, however, does not really fit other SGLT2 inhibitors such as empagliflozin as the later has a much lower affinity to SGLT1 (IC50 = 8.3µM) and its inhibitory effect on SGLT1 is negligible (Cong et al., 2022). With regard to regulation of nutrition uptake, GLUT1 expression in adult mouse cardiomyocyte was shown to be increased with empagliflozin treatment, resulting in increased glucose influx (Mustroph et al., 2019), but the finding has not been confirmed by others. In the latest studies, it was found that the activation of AMPK and its anti-oxidative action also alleviated ferroptosis, an iron-dependent programmed cell death resulting from accumulation of lipid peroxides (Min et al., 2023; Zhang et al., 2023).
Apart from the regulatory role in nutritional homeostasis, AMPK also mediates downstream signaling pathways that are involved in the cellular housekeeping process of autophagy, such as activating sirtuin (SIRT) 1, SIRT3, or mammalian target of rapamycin (mTOR) suppression to ensure the quality of the cellular machinery when stress appears (Packer 2023), including mitophagy and mitobiogenesis (Wu and Zou 2020). In neonatal mouse cardiomyocytes, it was found that empagliflozin relieved doxorubicin-perturbed autophagic flux through a Beclin1-Toll-like receptor 9-SIRT3 pathway, which was nullified with SIRT3 siRNA (Wang et al., 2020). Also, ischemia/reperfusion injury-triggered NLRP3 inflammasome activation was limited by dapagliflozin through autophagy activation (Yu et al., 2022). In immortalized cell lines, treatment with SGLT2 inhibitors activated AMPK phosphorylation and blocked the mTOR/hypoxia inducible factor-1 pathway to activate autophagy in H9c2 and HL-1 cells treated with sunitinib or palmitic acid (PA), respectively (Ren et al., 2021; Sun et al., 2021). Apart from SIRT3, SIRT1 was also shown to play regulatory roles in the beneficial effects of SGLT2 inhibitors on cells treated with hypertrophic stimulation or insult from ethanol (Tian et al., 2021; Ren et al., 2022). Interestingly, in a recent study investigating the effects of mitophagy on the efficiency of empagliflozin on mice subjected to myocardial infarction, Parkin was required for empagliflozin-induced mitochondria-related energetics, but not for empagliflozin’s protective effects against adverse cardiac remodeling, suggesting the existence of additional mechanisms (Song et al., 2021). Current studies reporting the function of SGLT2 inhibitors in mediating energy homeostasis and autophagic flux in cardiomyocytes are summarized in Table 2.

Antioxidative/Mitoprotective Effects
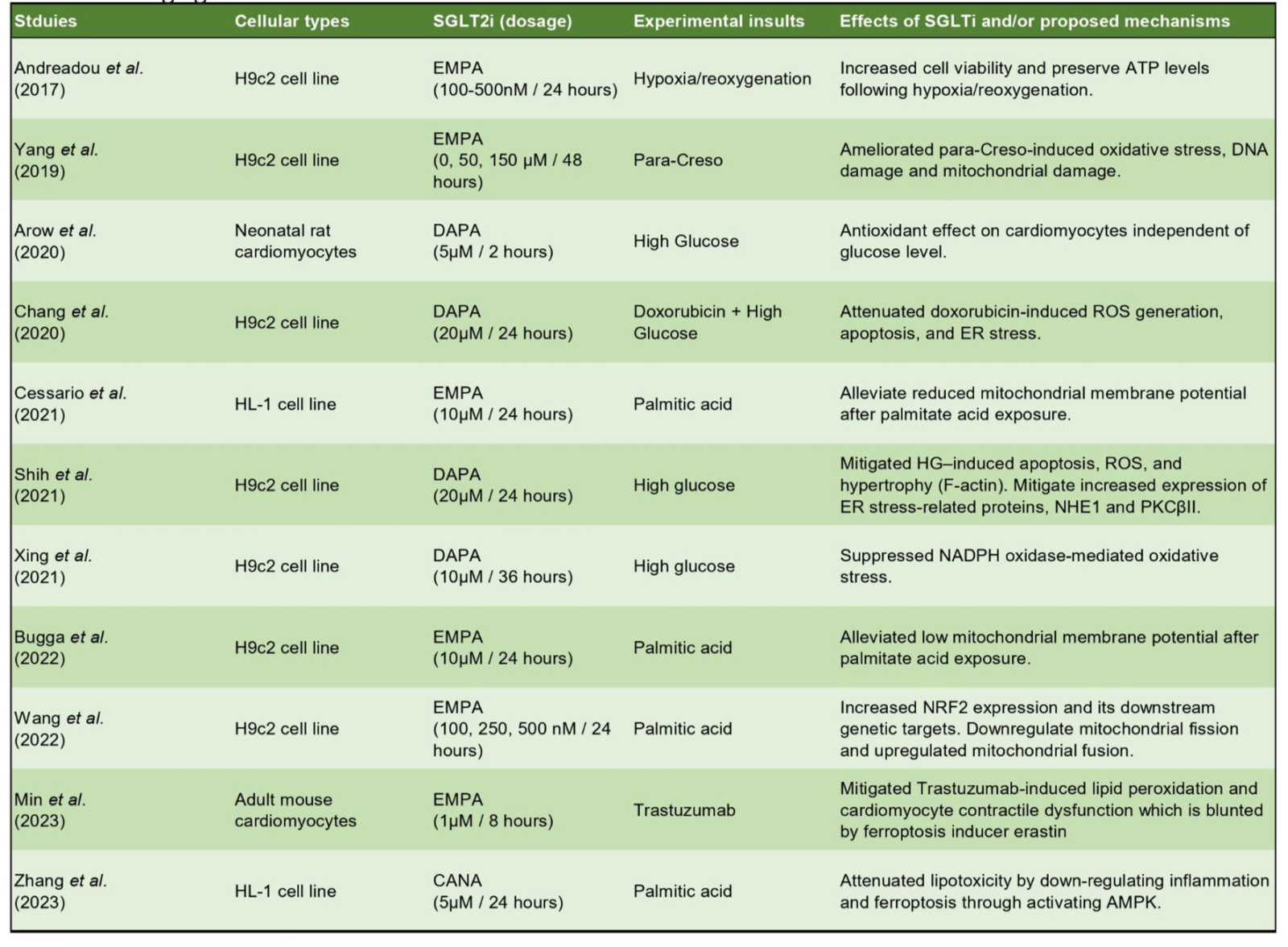
Another driving factor of the pathophysiology of cardiac remodeling and HF is elevated oxidative stress, which happens when there is excess production of ROS compared with the antioxidant capability of the cells. The major intracellular sources of ROS in the cells include mitochondria, NADPH oxidase, xanthine oxidase, and uncoupled nitric oxide synthase (Tsutsui et al., 2011). Thus, the maintenance of mitochondrial function and its crosstalk with other compartments play a pivotal role in preventing a chronic increase of ROS, which is detrimental to the organelles of the cells, including mitochondria, SR, and the contractile machinery of cardiomyocytes. As SGLT2 inhibitors were shown to have mitoprotective effects potentially attributed to their role in promoting mitobiogenesis and mitophagy through AMPK pathways, it was not surprising that they could serve as antioxidative agents. In neonatal rat cardiomyocytes, Arow et al. (2020) demonstrated that elevated intracellular ROS induced by HG treatment + angiotensin II could be alleviated with dapagliflozin treatment. ROS and improved mitochondria function were also seen in immortalized HL-1 and H9c2 cells treated with either HG or PA (Andreadou et al., 2017; Chang et al., 2021; Liu et al., 2021; Shih et al., 2021; Xing et al., 2021; Bugga et al., 2022). Current studies that have evaluated the antioxidative and mitoprotective effects of SGLT2 inhibitors are summarized in Table 3.
Post-Translational Modifications on Myofilaments
Myofilaments are the basic contractile unit that occupy the largest portion of cardiomyocytes (Gerdes 2012). Contraction and relaxation of myofilaments are mediated by alternating sliding actions between the thin and thick filaments toward or away from the M-band (Huxley 1957), while the structural support of myofilament is mainly provided by titin. The stiffness of titin is regulated by its isoform composition (Neagoe et al., 2002; Wu et al., 2002) or post-translational modifications (Anderson and Granzier 2013; Loescher et al., 2022). In a rat model of HFpEF where the myocardium had relaxation impairment, protein kinase G (PKG) was shown to be phosphorylated by titin at the N2Bus element, resulting in reduced stiffness of the skinned cardiomyocytes (Hamdani et al., 2013; Kovacs et al., 2016), which could be beneficial to myocardial relaxation. In recent years, it was reported that acute treatment of empagliflozin alleviated titin stiffening by reducing oxidative stress, leading to increased bioavailability of nitric oxide, which activated the soluble guanylyl cyclase/cyclic guanosiine monophosphate/PKG pathway in skinned human and rat HFpEF myocardium (Pabel et al., 2018; Kolijn et al., 2021), suggesting SGLT2 inhibitors target diseased myofilaments and improve their compliance. However, whether nitric oxide is increased via the direct effects of empagliflozin on cardiomyocytes is unclear. Furthermore, as energy deprivation could impair both contractile and relaxation mechanics of isolated single myofibrils (Tesi et al., 1999; Tesi et al., 2002), it is possible that the energy restoring effects of SGLT2 inhibitors on diseased cardiomyocytes facilitated contraction and relaxation of the myofilament.
Conclusions and Future Perspectives
The emergence of SGLT2 inhibitors, empagliflozin in particular, sheds new light on therapeutics in the treatment of HF for several reasons: (1) SGLT2 inhibitors appear to have beneficial effects in a broad range of cardiovascular diseases supported by plenty of clinical trials, including HF (regardless of ejection fraction) (Packer et al., 2020; Anker et al., 2021), acute decompensation of chronic HF (Kambara et al., 2019; Bhatt et al., 2021), HF after acute myocardial infarction (Tripolt et al., 2020), hypertension (Kario et al., 2018; Papadopoulou et al., 2021), and arrhythmias (Li et al., 2021). (2) SGLT2 inhibitors (empagliflozin and dapagliflozin) were the first therapeutics to demonstrate beneficial effects across a broad range of patients suffering from HFpEF, which is poised to be the predominant cause of HF in the near future. (Oktay et al., 2013; Ambrosy et al., 2014) Therapeutic approaches for HF were limited prior to the advent of SGLT2 inhibitors due to its complex pathophysiology and diverse clinical phenotypes (Shah et al., 2016; Lam et al., 2018; Yap et al., 2022). As HFpEF is a multi-systemic disorder triggered by a wide spectrum of clinical risk factors and comorbidities (Lam et al., 2018; Mishra and Kass 2021; Withaar et al., 2021; Yap et al., 2022), the multi-systemic regulatory roles in their cardioprotective effects are likely the reason why SGLT2 inhibitors have shown promise for the treatment of HFpEF. Interestingly, despite all the positive outcomes, the exact reasons why SGLT2 inhibitors are superior to other HF drugs, especially at the level of cardiomyocytes, are still elusive and require further investigation.
SGLT2 inhibitors have been reported in in vitro studies to have direct effects on cardiomyocytes but whether this is achieved via SGLT2 or off-targets effects remains unclear. There is the likelihood that these different effects interact with each other. As summarized in Fig. 1, NHE1 inhibition leads to reduced intracellular [Na+], which leads to (1) reduced intracellular [Ca2+] that might contribute to blunted CaMKII activities with SGLT2 inhibitor treatment (Mustroph et al., 2018) and (2) restored mitochondrial [Ca2+] required for normal function of the tricarboxylic acid cycle and antioxidant network to improve mitochondrial function. As mitochondria are the major antioxidant system in the cells, NHE1 inhibition could reduce oxidative stress, further improving mitochondria function. (3) Reduced oxidative stress could enhance nitric oxide bioavailability of the cell and PKG, which leads to titin phosphorylation at its N2Bus element, and reduced titin stiffness and improved myocardial relaxation. This NHE-1-mitoprotection-antioxidation linkage has not been addressed in the literature. Despite extensive reports demonstrating cardioprotection with SGLT2, to date, the underlying pathways through which SGLT2 inhibitors mediate their beneficial effects in HF have not yet been addressed, apart from targeting SNS and RAAS to indirectly ameliorate heart function (Brann et al., 2019; Pellicori et al., 2020). One important point to note regarding the cardiomyocyte-specific effects of SGLT2 inhibitors is that a variety of dosages have been used in these studies (as seen in Table 1-3), and it is important to use clinically relevant dosages when undertaking in vitro studies (e.g. 1 µM or lower (Brand et al., 2012; Laffel et al., 2018) to reduce the risk of off-target effects. Furthermore, despite an abundance of studies working on the potential mechanisms through which SGLT2 inhibitors directly activate AMPK and enhance autophagic flux, a number of these in vitro studies were performed in immortalized cell lines, and therefore need to be verified using isolated cardiomyocyte or cardiomyocyte derived from hiPSCs. Knowledge obtained from these studies could help in the identification of more specific targets to optimize therapeutic strategies to efficiently treat HF using SGLT2 inhibitors.

In a new window | Download PPT
Figure 1. Summary of potential mechanistic actions of SGLT2 inhibitors at the level of cardiomyocytes. Major targets of SGLT2 inhibitors at the level of cardiomyocyte to commit cardioprotective effects are labelled with solid black blocks: Inhibitions of NHE1 and late sodium current, inhibition of CaMKII, activation of AMPK for energy production pathways and reduction of ROS. Potential outcomes for cardioprotection are labelled with solid white blocks: Improved mitochondria function, increased energy production and improved contractility and relaxation of the myofilaments. Red arrows with questions mark stand for potential cross-talks among previously reported effects of SGLT2 inhibitors which has not been investigated in depth. NO: nitric oxide; LTCC: L-type calcium channel. [Na+]i: cytosolic sodium concentration; [Ca2+] m: mitochondrial calcium concentration.
Acknowledgements
Ying-Hsi Lin is supported by Singapore Ministry of Health’s National Medical Research Council under its Open Fund Young Individual Research Grant (MOH-OFYIRG22jul-0006). Chrishan Ramachandra is supported by the Goh Cardiovascular Research Award (Duke-NUS-GCR/2022/0027). Derek Hausenloy is supported by the Duke-NUS Signature Research Programme funded by the Ministry of Health, Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research Investigator Award (MOH-STaR21jun-0003), Centre Grant scheme (NMRC CG21APR1006), and Collaborative Centre Grant scheme (NMRC/CG21APRC006). This article is based upon work supported by the COST Actions EU-CARDIOPROTECTION IG16225 and EU-METAHEART CA22169 supported by COST (European Cooperation in Science and Technology).
Conflicts of interest
The authors declare that they have no conflicts of interest. Derek J. Hausenloy and Chrishan J. Ramachandra, who serve as EIC and as an Editorial Board member for Conditioning Medicine, respectively, have not participated at any level in the editorial review of this manuscript.
References
Ying-Hsi Lin1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Shuo Cong1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Yitong Zeng2
2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Chrishan J. Ramachandra1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Derek J. Hausenloy1-4
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore. 3Yong Loo Lin School of Medicine, National University Singapore, Singapore. 4The Hatter Cardiovascular Institute, University College London, London, UK.
Corresponding author:
Ying-Hsi Lin
Email: ying-hsi.lin@duke-nus.edu.sg
In a new window | Download PPT
Figure 1. Summary of potential mechanistic actions of SGLT2 inhibitors at the level of cardiomyocytes. Major targets of SGLT2 inhibitors at the level of cardiomyocyte to commit cardioprotective effects are labelled with solid black blocks: Inhibitions of NHE1 and late sodium current, inhibition of CaMKII, activation of AMPK for energy production pathways and reduction of ROS. Potential outcomes for cardioprotection are labelled with solid white blocks: Improved mitochondria function, increased energy production and improved contractility and relaxation of the myofilaments. Red arrows with questions mark stand for potential cross-talks among previously reported effects of SGLT2 inhibitors which has not been investigated in depth. NO: nitric oxide; LTCC: L-type calcium channel. [Na+]i: cytosolic sodium concentration; [Ca2+] m: mitochondrial calcium concentration.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11920 | 48 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA