Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Inorganic nitrate and nitrite in cardioprotection
Time:2024-03-08
Number:4837
Author Affiliations

Conditioning Medicine 2023. 6(4): 131-137.
Abstract
The advent of percutaneous coronary intervention has significantly reduced the morbidity and mortality from acute myocardial infarction. Paradoxically, reperfusion can cause additional myocardial injury and increase the infarct size beyond that associated with ischemia alone, hence the term ischemia-reperfusion injury (IRI). Endogenous nitric oxide (NO) generation is crucial in cardiovascular homeostasis, and its antioxidant and anti-inflammatory properties appear protective against myocardial IRI. However, in the face of ischemia, NO generation via the endogenous NO synthase pathways is significantly impaired. Thus, substantial interest has grown in interrogating the therapeutic potential of inorganic nitrate and nitrite, which do not have the adverse features seen with their organic counterparts, in delivering this ‘lost’ NO to reduce myocardial IRI. This review will focus on understanding the role of inorganic nitrate and nitrite in myocardial IRI with an overview of the mechanisms in cardioprotection and current pre-clinical and clinical studies.
Keywords: Inorganic nitrate, Inorganic nitrite, Myocardial ischemic reperfusion injury, Cardioprotection, Nitric oxide
Abstract
The advent of percutaneous coronary intervention has significantly reduced the morbidity and mortality from acute myocardial infarction. Paradoxically, reperfusion can cause additional myocardial injury and increase the infarct size beyond that associated with ischemia alone, hence the term ischemia-reperfusion injury (IRI). Endogenous nitric oxide (NO) generation is crucial in cardiovascular homeostasis, and its antioxidant and anti-inflammatory properties appear protective against myocardial IRI. However, in the face of ischemia, NO generation via the endogenous NO synthase pathways is significantly impaired. Thus, substantial interest has grown in interrogating the therapeutic potential of inorganic nitrate and nitrite, which do not have the adverse features seen with their organic counterparts, in delivering this ‘lost’ NO to reduce myocardial IRI. This review will focus on understanding the role of inorganic nitrate and nitrite in myocardial IRI with an overview of the mechanisms in cardioprotection and current pre-clinical and clinical studies.
Keywords: Inorganic nitrate, Inorganic nitrite, Myocardial ischemic reperfusion injury, Cardioprotection, Nitric oxide
Highlights
This review evaluates the potential therapeutic role of inorganic nitrate and nitrite in conditioning medicine, specifically myocardial ischemic-reperfusion injury. Inorganic nitrate, found in high concentrations in green leafy vegetables, is reduced to nitrite and then NO via the enterosalivary circulation. Pre-clinical studies have alluded to the potential mechanisms of cardioprotection afforded by replacing the 'lost' bioavailable NO with inorganic nitrate and nitrite, including anti-platelet effects, anti-inflammatory effects, and improved mitochondrial function and biogenesis. Many animal studies report the positive impact inorganic nitrate and nitrite have on reducing infarct size in myocardial ischemic-reperfusion injury. However, clinical studies assessing inorganic nitrate and nitrite in myocardial ischemic-reperfusion injury are lacking, with two small trials demonstrating no significant benefit in the primary endpoints. Larger randomized controlled clinical trials are still much needed.
Introduction
Ischemic heart disease is the leading cause of death worldwide, accounting for 16% of all deaths globally in 2019 (World Health Organization, 2020). The primary pathological process in ischemic heart disease is atherosclerosis, which culminates in the build-up of plaques in the coronary arteries. These plaques may eventually rupture or erode, causing acute thrombosis and luminal occlusion, resulting in acute myocardial infarction. These changes are accompanied by ST elevation on an electrocardiogram, hence the term ST-elevation myocardial infarction (STEMI). Restoring blood flow by primary percutaneous coronary intervention (PPCI) promptly can limit the size of an infarct. Paradoxically, this reperfusion may result in additional myocardial injury in a phenomenon termed ischemic-reperfusion injury (IRI). The pathophysiology of IRI is characterized by reperfusion-induced arrhythmias, myocardial stunning, microvascular obstruction (MVO), and lethal reperfusion injury (Yellon and Hausenloy, 2007; Hausenloy et al., 2019; Griffiths et al., 2021). A significant body of literature exists that examines potential therapeutic strategies to prevent or reduce IRI by cardioprotection. These range from pharmacological to mechanical options, such as remote ischemic preconditioning, but none have been effectively translated into the clinical arena (Hausenloy et al., 2017). Much interest has been placed on assessing the effects of inorganic nitrate and nitrite and, hence, nitric oxide (NO) to reduce IRI. This review will give an overview of the current evidence for the cardioprotective role of inorganic nitrate and nitrite in myocardial IRI. Readers are directed to Kapil et al. (2020) and Griffiths et al. (2021) for excellent comprehensive reviews.
Nitric oxide in the cardiovascular system
NO plays an important role in the cardiovascular system. Classically, NO derived from the endothelium causes vascular smooth muscle to relax, leading to vasodilatation, a pivotal determinant of blood pressure homeostasis. However, this simple free radical molecule also has diverse effects on biological functions due to its chemical properties, particularly its ability to freely diffuse and its membrane permeability. The pleiotropic effects of NO include platelet inhibition and anti-inflammatory activity (Andreadou et al., 2015). Importantly, NO acts as an antioxidant by reacting with free radical species such as superoxide (O2−·) and nitrogen dioxide. However, NO also reacts with O2−·to form peroxynitrite (ONOO−), a rapidly generating highly reactive oxidant species (Ferdinandy and Schulz, 2003). The interplay between NO, O2−·, and ONOO− is finely balanced in homeostasis, but in pathological conditions such as myocardial IRI, it is tipped towards ONOO−. Moreover, this is exacerbated by co-morbidities, particularly cardiometabolic diseases such as diabetes and hypercholesterolemia, which are chronically NO-deficient states.
Generation of circulatory nitrite and NO
Conventional NO generation occurs through endothelial NO synthase (eNOS) in a healthy endothelium by the oxidation of L-arginine when stimulated by circulating hormones such as insulin and estrogen and from shear stress. NO activates soluble guanyl cyclase, generating the secondary messenger cyclic guanosine monophosphate, subsequently leading to its downstream effect on smooth muscle relaxation and vasodilatation. Through this pathway, basal NO generation from eNOS is critical in maintaining vascular tone (Vallance et al., 1989; Huang et al., 1995). In addition to oxygen, essential co-factors are required, specifically tetrahydrobiopterin and NAPDH. Endothelial dysfunction is common in all circulatory diseases, which is synonymous with the impairment of NO generation by eNOS, partly because of increased ROS in disease states (Ohara et al., 1993; Warnholtz et al., 1999). There is established evidence for the role of NO in IRI, for example, in knockout animal models, a deficiency in NOS can lead to spontaneous myocardial infarction and exacerbation of myocardial IRI (Jones et al., 1999; Nakata et al., 2008). There is also evidence that increased ROS in the presence of cardiometabolic co-morbidities may lead to redox crosstalk, aggravating IRI damage (Andreadou et al., 2021). Replacing this ‘lost’ NO and reducing oxidative and nitrosative stress could theoretically be achieved by exogenous nitrate administration, which is reduced to nitrite and then NO.
Organic synthetic NO donors such as isosorbide mononitrate have the potential to replace “lost” NO; however, repeated and/or long-term administration of these drugs paradoxically leads to endothelial dysfunction manifesting as tolerance and tachyphylaxis (Caramori et al., 1998; Münzel et al., 2014). Conversely, inorganic nitrates from natural sources such as green leafy vegetables do not exhibit this problem, and thus, interest in its therapeutic potential has significantly increased over the last couple of decades.
Inorganic nitrates were previously considered inert, with no biological effects on the cardiovascular system. However, research has now dispelled this dogma (Lundberg and Govoni, 2004) with the identification of the ‘enterosalivary circulation’ as the key non-canonical pathway for nitrate reduction to nitrite and then NO. After ingestion, dietary inorganic nitrate is rapidly absorbed into the circulation from the gastrointestinal tract with almost 100% bioavailability, as first-pass metabolism is bypassed. Approximately 75% of this plasma nitrate is excreted in the urine, with peak excretion occurring around six hours following supplementation (Pannala et al., 2003), with a small proportion excreted through perspiration and in faces. The remaining plasma nitrate (~25%) is secreted into the saliva via the salivary glands, resulting in approximately ten times greater nitrate levels than in plasma (Tannenbaum et al., 1976). Reduction of this nitrate to nitrite occurs by bacteria in the saliva. The nitrite is then swallowed and absorbed into the circulation from the stomach or undergoes acidification to NO (Figure 1). Initial observations (Larsen et al., 2006) showed that dietary nitrate load led to a significant elevation in plasma nitrite levels with a subsequent reduction in blood pressure. Subsequently, the Ahulwalia group demonstrated that delivering inorganic nitrate with beetroot juice reduced blood pressure (BP) in healthy volunteers (Webb et al., 2008) and in patients with hypertension (Kapil et al., 2015).
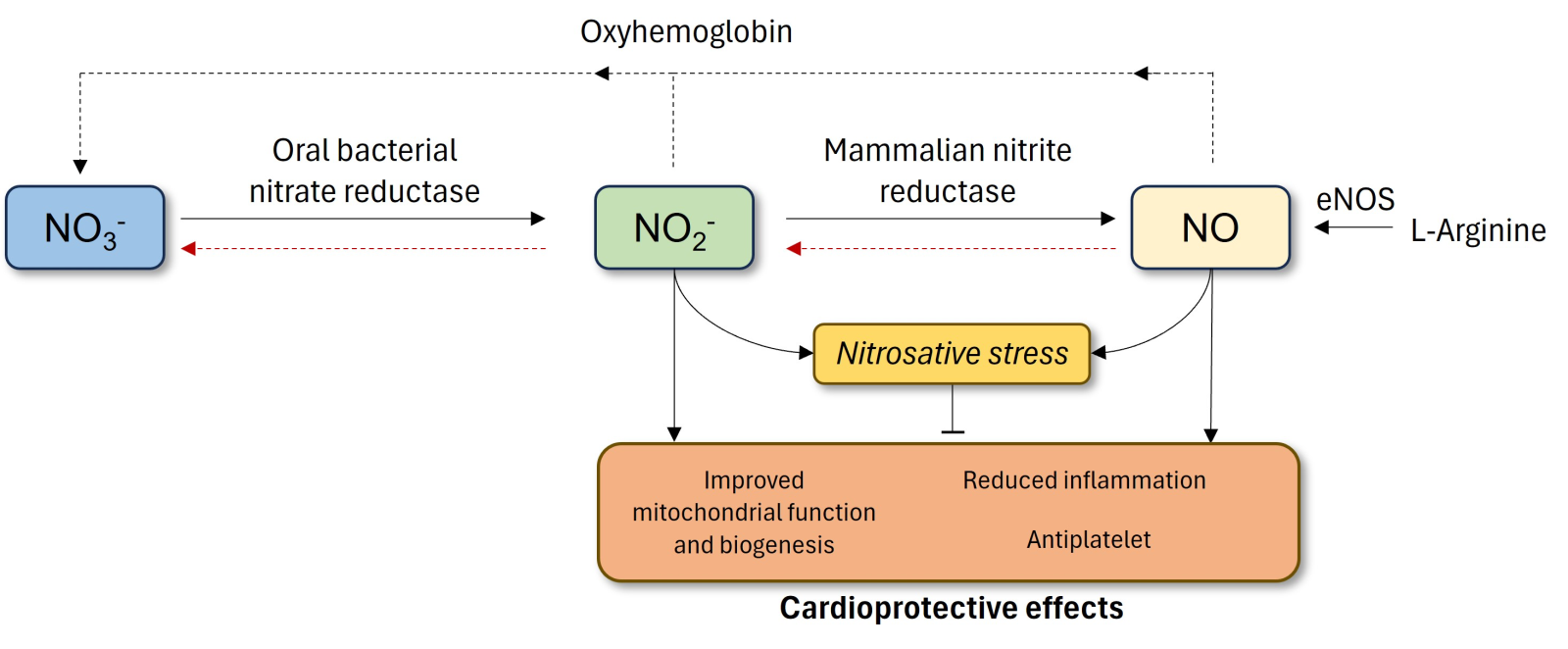
In a new window | Download PPT
Figure 1. Non-canonical and the conventional generation of NO via the NOS pathway. The classical pathway in the endothelium involves conversion of L-arginine to NO by eNOS. The alternative, non-canonical pathway occurs with the ingestion of dietary inorganic nitrate. Absorbed NO3- concentrated in salivary glands is reduced to NO2- by oral bacteria. The NO2- is then swallowed, enters the gastrointestinal tract, where it is reduced to NO in the stomach or absorbed into the circulation. In the latter, mammalian nitrite reductase enzymes reduce NO2- to NO. Both nitrite and NO have potential cardioprotective effects only if not accompanied by increased nitrosative stress. eNOS, endothelial nitric oxide synthase; NO, nitric oxide; NO2-, nitrite; NO3-, nitrate.
Role of nitrite in IRI
There is significant evidence in the literature that the generation of NO from the reduction of nitrite in blood and tissues confers cytoprotective signaling in the cardiovascular system, specifically in myocardial IRI (Andreadou et al., 2015). In the cardiovascular system, ischemia leads to a reduction in pH. Zweier et al. (1995) showed that tissue nitrite released NO in the ischemic Langendorff rat heart despite NOS inhibition. In addition, nitrite reduction occurs via mammalian nitrite reductases, including deoxyhemoglobin, aldehyde dehydrogenase, and xanthine oxidoreductase (XOR) (van Faassen et al., 2009). Specific enzymatic pathways may predominate depending on the tissue environment, e.g., XOR may prevail in hypoxia and ischemic tissue (Millar et al., 1998; Gladwin et al., 2005). Nitrite has been shown to offer cytoprotective effects through NO reduction during ischemia or hypoxemia seen in IRI. These conditions inactivate NO synthase enzymes, with NO generation facilitated by nitrite reductases. The NO scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO) blocks nitrite dependent protection against IRI (Webb et al., 2004; Jung et al., 2006; Tripatara et al., 2007) and this effect does not involve the NOS enzymes (Webb et al., 2004; Lu et al., 2005), suggesting that cytoprotection is not affected by NOS inhibition or eNOS knockout models (Duranski et al., 2005; Milsom et al., 2010).
XOR is a key nitrite reductase in the myocardium of animals and humans. Webb et al. (2004) showed that allopurinol, an XOR inhibitor, significantly reduced NO generation from nitrite in isolated rat hearts and in men using atrial appendages taken from cardiac surgery. In hypoxic conditions, XOR acts as nitrite reductase, increasing NO generation while reducing oxygen tensions (Li et al., 2001) in rats and mouse models of IRI (Tripatara et al., 2007; Milsom et al., 2010) and in pulmonary hypertension (Baliga et al., 2012). XOR requires three different substrates (xanthine, aldehydes, and NADH) to reduce nitrite (Khambata et al., 2015). Indeed, NADH levels increase in hypoxia and IRI, purporting the evidence that NO derived from XOR nitrite reduction predominates in these conditions (Dejam et al., 2007). Similarly, animal studies (Rassaf et al., 2007; Shiva et al., 2007; Hendgen-Cotta et al., 2008) have implicated a role for deoxygenated myoglobin also acting as a nitrite reductase in myocardial IRI.
Potential mechanisms for nitrite-induced cytoprotection in IRI
The mechanism for the positive effect of nitrite in cytoprotection in IRI may be attributable to its improvement in mitochondrial function and anti-platelet and anti-inflammatory effects.
Antiplatelet effects
Platelet activation and aggregation are key events in ischemic events such as acute myocardial infarction. Activated platelets also release mediators, which may contribute to damage seen in IRI, increasing the risk for secondary events (Badimon et al., 2002). In homeostasis, NO suppresses platelet activation and adhesion to intact vascular endothelium (Radomski et al., 1990; Dangel et al., 2010) in addition to platelet aggregation (Radomski et al., 1990). The NITRITE-AMI trial, in which intracoronary nitrite was administered to patients presenting with STEMI at the point of reperfusion after PPCI, ex vivo platelet reactivity (assessed by aggregation and cell activation markers) was reduced acutely and six months after intervention (Jones et al., 2017). The authors suggested that this effect on platelets may contribute to reduced MVO seen in IRI, which can worsen cardiac cell death after reperfusion (Nazir et al., 2016; Niccoli et al., 2019).
Anti-inflammatory effects
Leucocyte recruitment and the proinflammatory effects influence the magnitude of IRI-induced damage (Entman and Smith, 1994). There is evidence that each step of leucocyte recruitment (tethering, rolling, adhesion, and emigration) in inflammation is influenced by NO modulating the interaction of leucocytes with endothelial cells in animal models of IRI (Kubes et al., 1991; Ahluwalia et al., 2004). Nitric oxide exerts anti-inflammatory effects in animal studies of IRI. NO inhibits the expression and activation of essential adhesion molecules, including P-selectin, intercellular adhesion molecule 1, vascular cell adhesion molecule-1, and cluster of differentiation 11b (Gauthier et al., 1994; Lefer and Lefer, 1996; Leite et al., 2009). In the NITRITE-AMI study, neutrophil numbers and activation were reduced following PPCI (Jones et al., 2017). In comparison, an increase in total circulating neutrophil numbers and high-sensitivity C-reactive protein levels were observed after reperfusion, which then decreased over time. Interestingly, in the treatment arm, these changes were reduced for up to six months post PPCI (p < 0.01) with reduced expression of neutrophil cluster of differentiation 11b, plasma CXC motif chemokine ligand 1, CXC motif chemokine ligand 5, and CC motif chemokine ligand 2 levels (p < 0.05) while the number monocytes and lymphocytes and the activation markers expressed by these cells between the treatment groups were similar (Jones et al., 2017; Kapil et al., 2020). These changes were associated with reduced MVO and infarct size (Jones et al., 2017).
Improved mitochondrial function and biogenesis
NO has a crucial role in mitochondrial metabolism and biogenesis. An important pathway is its competitive, reversible binding to cytochrome c oxidase with oxygen in the mitochondrial respiratory chain (Clementi and Nisoli, 2005; Nisoli et al., 2005). This effect, in turn, may preserve high-energy phosphate stores during IRI (Hendgen-Cotta et al., 2008; Murillo et al., 2011). Studies on IRI in the heart and liver have shown that nitrite inhibits complex I by post-translational S-nitrosation, resulting in reduced mitochondrial ROS generation (Gladwin et al., 2005; Shiva et al., 2007). The inhibitory action of nitrite on complex I may also prevent the opening of the mitochondrial permeability transition pore (MPTP) and, therefore, cell death (Fontaine et al., 1998; Nadtochiy et al., 2007), improving mitochondrial tolerance to calcium overload. Cyclophilin D (CypD), a mitochondrial peptidyl-prolyl cis-trans isomerase, accelerates MPTP opening (Nguyen et al., 2011; Amanakis and Murphy, 2020). Nitrite and, hence, NO may have a role in modulating CypD and blocking the opening of MPTP. In a murine model of myocardial IRI, nitrite reduced infarct size; however, this effect was not seen in CypD knockout mice (Rassaf et al., 2014). In mice, low-dose nitroglycerin administered before reperfusion reduced myocardial infarct size in IRI by preserving eNOS function and eNOS-dependent S-nitrosation of CypD (Bibli et al., 2019).
Although NO appears to play a role in modulating mitochondria during IRI (Andreadou et al., 2020), some studies also show that nitrite itself contributes to this effect. Nitrite directly stimulates mitochondrial biogenesis through adenylate kinase activity, resulting in AMP kinase (AMPK) phosphorylation, downstream sirtuin-1 activation, and deacetylation of peroxisome proliferator-activated receptor gamma coactivator 1-α (Mo et al., 2012). In a rat model of carotid injury, two weeks of continuous oral nitrite treatment postinjury prevented smooth muscle cell hyperproliferation. Moreover, there was a nitrite-dependent upregulation of peroxisome proliferator-activated receptor gamma coactivator 1-α and increased mitochondrial number in the injured artery (Mo et al., 2012). Following these findings, the same group showed that pre-conditioning with nitrite in cultured H9c2 cardiomyocytes during normoxia appears to result in cytoprotection with a reduction in cell death in IRI through modulation of the protein kinase A-dynamin-related protein 1-AMPK pathway in mitochondria (Pride et al., 2014).
The potential role of inorganic nitrate and nitrite in cardioprotectio
To understand the potential benefits of dietary nitrate in cytoprotection, it is important to outline the effects of nitrite on IRI (for an additional review see Rassaf et al. (2014)). In a cat model of IRI, intravenous acidified sodium nitrite (12.5 mmol/kg/hr) administered 30 minutes after coronary artery occlusion for 90 minutes reduced infarct size (expressed as a percentage of the area at risk (AAR)) (Johnson et al., 1990).
Webb et al. (2004) administered NaNO2 (10 or 100 µmol/l) in the isolated rat Langendorff heart preparation either prior to or at reperfusion. Sodium nitrite improved lateral ventricle function and coronary perfusion pressure and reduced infarct size after an IRI. These findings were corroborated by other studies. In mice, an intraventricular injection of NaNO2 (2.4-1920 nmol) five minutes before reperfusion and after 30 minutes of occlusion of the left main coronary artery led to dose-dependent decreases in infarct size (Duranski et al., 2005). A similar benefit was seen with dietary nitrite (50 mg/L) administered seven days prior to ischemia when a 48% reduction in infarct size relative to the AAR was observed (Bryan et al., 2007). In rat hearts in vitro and in vivo, intravenous nitrite but not nitrate conferred cardioprotection by reducing the infarct size following IRI (Baker et al., 2007). This was associated with the activation of NADPH oxidase and ATP-dependent potassium channels and increases in the activity of xanthine dehydrogenase and xanthine oxidase during ischemia (Baker et al., 2007). In eNOS knockout mice, NaNO2 supplementation in the drinking water for one week restored NO homeostasis and protected against myocardial IRI (Bryan et al., 2008). In a study of 27 canines, the left anterior descending artery was ligated for 120 minutes with intravenous NaNO2 (0.2 µmol/min/kg for the first 20 minutes and then 0.17 µmol/min/kg for 40 minutes) administered for the last 60 minutes and the last five minutes of ischemia. Nitrite reduced the infarct size to ~23% and ~36%, respectively, compared to 70% in those receiving saline, as assessed by cardiovascular magnetic resonance (Gonzalez et al., 2008). Moreover, there were improvements in microvascular perfusion and contractile function, as well as reduced apoptosis. Although most studies have demonstrated the cardioprotective benefit of nitrite in animal models, the multicenter CAESAR Cardioprotection Consortium of preclinical studies observed a lack of efficacy as nitrite did not attenuate myocardial infarct size per AAR in mice, rabbit, or pig models (Lefer et al., 2014).
Short-term (seven days) supplementation with beetroot juice powder containing nitrate (10 g/L, ~0.7 mM) reduced infarct size by ~30% following occlusion of the left anterior descending artery for 30 minutes and reperfusion for 24 hours in mice (Salloum et al., 2014). In a recent study assessing the long-term effects of dietary nitrate on myocardial IRI, female Wistar rats were randomly divided into untreated and nitrate-treated (100 mg/L sodium nitrate in drinking water for nine months) groups. Rats in the nitrate-treated group had decreased left ventricular end-diastolic pressure (17%, p < 0.001) and infarct size (34%, p < 0.001) compared to untreated rats (Yassaghi et al., 2023).
Clinical models of IR
Endothelial dysfunction
Small clinical studies have demonstrated the benefits of delivering nitrite and nitrate. In healthy volunteers, a dietary nitrate load was able to abrogate the significant (~60%) reduction in brachial artery flow-mediated dilatation (FMD) after IRI (Webb et al., 2008). There is also similar evidence for preconditioning with a low dose of NaNO2 at 1.5 μmol/min for 20 minutes before IRI in the forearm of healthy volunteers (Ingram et al., 2013). In saline-treated participants, FMD decreased by 43% following IRI when subjects received saline, with no significant differences in those treated with nitrite. There was no benefit seen when nitrite was infused during ischemia.
Myocardial ischemia-reperfusion injury
To date, all clinical studies have assessed the benefit of nitrite rather than nitrate in myocardial IRI. In a small study using dobutamine stress echocardiography, NaNO2 infusion (1.5 μmol/min for 20 min) in a double-blind fashion improved functional responses to ischemic myocardium compared to saline placebo (Ingram et al., 2013). Longitudinal myocardial function was quantified by peak systolic velocity (Vs) and strain rate (SR). At peak dobutamine stress, Vs and SR significantly increased in regions exhibiting ischemia (Vs from 9.5 ± 0.5 to 12.4 ± 0.6 cm/s, SR from −2.0 ± 0.2 to −2.8 ± 0.3 s-1) with no change in normally functioning regions (Vs from 12.6 ± 0.4 to 12.6 ± 0.6 cm/s, SR from −2.6 ± 0.3 to −2.3 ± 0.1 s-1). With nitrite treatment, Vs improved only in ischemic segments (+122%, p < 0.001) but not in normal segments.
Phase II clinical studies have assessed the effects of intravenous and intracoronary nitrite in patients presenting with STEMI. In a randomized, double-blind placebo controlled multicenter study, Siddiqi et al. (2014) recruited 229 patients presenting with STEMI randomized to a 70 μmol intravenous dose of NaNO2 (n = 118) or placebo (n= 111) over five minutes immediately prior to PPCI. There was no significant difference in the primary endpoint of infarct size (6–8 days post-PPCI assessed by cardiac magnetic resonance imaging) between the groups, even after adjustment for confounding factors (diabetes mellitus and recruitment center). There were no significant differences in secondary endpoints, including 72-hour plasma troponin I or creatine kinase area under the curves, lateral ventricle volumes or ejection fraction measured at six to eight days, and infarct size at six months follow-up. Overall, this study demonstrated that intravenous nitrite did not derive any benefit in STEMI patients.
In another clinical trial, Jones et al. (2015) recruited 80 patients randomized to receive intracoronary NaNO2 (1.8 μmol in 10 ml) or NaCl (placebo) through an over-the-wire balloon positioned distal to the coronary artery occlusion before balloon inflation. As expected, plasma nitrite significantly increased 30 minutes after NaNO2 administration. There were no significant differences between the groups for the primary outcome of creatine kinase release, troponin T, or infarct size by cardiac magnetic resonance imaging in the patient cohort. Interestingly, there was an improvement in myocardial salvage index (p = 0.05) and a reduction in major adverse cardiac events at 12 months (2.6% vs. 15.8%; p = 0.04) in the nitrite group. Moreover, in a subgroup of 66 patients with thrombolysis in myocardial infarction ≤1 flow, there was a 20% reduction in serum creatine kinase (p = 0.030), 19% reduction in infarct size (p = 0.034), 48% reduction in MVO (p = 0.015), and 30% increase in myocardial salvage index (p = 0.002) in the nitrite group (Jones et al., 2015). As such, in this subgroup of patients, intracoronary nitrite may be beneficial in addition to standard care (Jones et al., 2015). The difference in the two studies suggests that intracoronary administration of nitrite provides high local concentrations (2.5–10 μM) within the myocardium, which may be more beneficial than systemic intravenous administration, whereby very high circulating levels may be required to provide sufficient levels of nitrite for clinical benefit. This is also apparent in a separate study utilizing a rat model of kidney IRI in which local nitrite administration reduced infarct size compared to intravenous administration (Tripatara et al., 2007). Interestingly, a recent study demonstrated that a nitrate-functionalized patch releasing NO locally implanted onto myocardium reduced infarct size and improved lateral ventricle ejection fraction as assessed by cardiac magnetic resonance imaging following IRI in pigs (Zhu et al., 2021).
The cardioprotective effect of inorganic nitrite and nitrate in animal studies has not been observed in clinical trials. The interaction of risk factors, comorbidities, and medications may modulate the response to interventions aimed at reducing myocardial IRI. This phenomenon may underlie the lack of clinical efficacy in clinical trials assessing preconditioning, postconditioning, and remote conditioning (Ferdinandy et al., 2014). More research with larger clinical studies is required to ascertain whether intracoronary nitrite and dietary inorganic nitrate will have therapeutic benefits in IRI and cardioprotection. A recent small randomized controlled clinical trial assessing the effects of oral sodium nitrate prior to coronary artery bypass graft surgery failed to alter troponin T levels 72 hours after surgery compared to saline placebo (Eriksson et al., 2021). Finally, studies targeting the NOS cofactor tetrahydrobiopterin and downstream pathways of guanyl cyclase and cyclic guanosine monophosphate in myocardial IRI have yet to come to fruition clinically (Inserte and Garcia-Dorado, 2015).
Conclusion
Current understanding of the role of inorganic nitrate and nitrite favors a cardioprotective role in the setting of myocardial IRI, particularly in animal models. The increase in bioavailable NO appears to confer anti-inflammatory and anti-platelet effects and stimulates mitochondria metabolism. Moreover, nitrite itself also appears to have direct cardioprotective effects. Unfortunately, clinical studies utilizing nitrite in patients presenting with myocardial infarction have generated mixed results. One important reason for these disparate findings is that increasing bioavailable NO may only be protective if not accompanied by increased nitrosative stress, which might also be influenced by patients’ co-morbidities and medications. Further work in this developing field is needed, particularly in optimizing the route of administration and conducting large phase III randomized clinical trials to determine the overall efficacy of inorganic nitrate and nitrite in cardioprotection.
Conflict of interest
The author has no conflicts of interest to declare.
References
Clement Lau1,2
1William Harvey Research Institute, Queen Mary University of London, London, UK. 2National Heart Research Institute Singapore, National Heart Centre, Singapore, Singapore.
Corresponding author:
Clement Lau
Email: c.lau@qmul.ac.uk
In a new window | Download PPT
Figure 1. Non-canonical and the conventional generation of NO via the NOS pathway. The classical pathway in the endothelium involves conversion of L-arginine to NO by eNOS. The alternative, non-canonical pathway occurs with the ingestion of dietary inorganic nitrate. Absorbed NO3- concentrated in salivary glands is reduced to NO2- by oral bacteria. The NO2- is then swallowed, enters the gastrointestinal tract, where it is reduced to NO in the stomach or absorbed into the circulation. In the latter, mammalian nitrite reductase enzymes reduce NO2- to NO. Both nitrite and NO have potential cardioprotective effects only if not accompanied by increased nitrosative stress. eNOS, endothelial nitric oxide synthase; NO, nitric oxide; NO2-, nitrite; NO3-, nitrate.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 4837 | 7 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA