Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The interferon response as a common final pathway for many preconditioning stimuli: unexpected cross-talk between hypoxic adaptation and antiviral defense
Time:2018-06-30
Number:16197
Author Affiliations

Conditioning Medicine, 2018. 1(4):163-170.
Abstract
Despite major advances in understanding how the brain goes awry in disease, identification of therapeutics for neuroprotection in stroke remains an unsolved challenge. A promising strategy to delineate endogenous mechanisms of neuroprotection is to understand adaptive homeostatic transcription induced by sublethal ischemia. Homeostatic adaptation is defined as the body’s restorative responses to stress. Activating adaptive homeostatic pathways can lead to transcription of a panoply of genes involved in cell survival and repair, can suppress pro-death signaling, and can stimulate metabolic changes congruent with survival. All of these mechanisms have been shown to be operative in protection induced by sublethal stress. In this context, central mediators of cellular adaptation to hypoxic and viral stress have been implicated in preconditioning. Here we present data that suggest an unexpected convergence in the pathways triggering adaptation to hypoxia and viral infection leading to preconditioning neuroprotection in the CNS.
Abstract
Despite major advances in understanding how the brain goes awry in disease, identification of therapeutics for neuroprotection in stroke remains an unsolved challenge. A promising strategy to delineate endogenous mechanisms of neuroprotection is to understand adaptive homeostatic transcription induced by sublethal ischemia. Homeostatic adaptation is defined as the body’s restorative responses to stress. Activating adaptive homeostatic pathways can lead to transcription of a panoply of genes involved in cell survival and repair, can suppress pro-death signaling, and can stimulate metabolic changes congruent with survival. All of these mechanisms have been shown to be operative in protection induced by sublethal stress. In this context, central mediators of cellular adaptation to hypoxic and viral stress have been implicated in preconditioning. Here we present data that suggest an unexpected convergence in the pathways triggering adaptation to hypoxia and viral infection leading to preconditioning neuroprotection in the CNS.
Introduction
Stroke remains a clinical problem with few neuroprotective treatments. Stroke-related ischemia that occurs due to transient or permanent blockade of blood flow results in a decrease in oxygen and glucose supply to cells. Prolonged ischemia results in permanent damage to tissue leading to stroke. While the mortality rate has diminished in recent years, stroke remains the leading cause of disability in the United States. The financial (over 34 billion dollars every year) and human costs are immense (Benjamin et al., 2017). Many people spontaneously recover following a stroke; however, two-thirds of survivors remain with some disability, and despite rehabilitation, they are left with chronic disability. Immense progress has been made in understanding the pathophysiology of stroke; however, few proven alternatives exist for treating this debilitating disorder. One potential therapeutic approach is to augment endogenous adaptive programs in our body to protect and repair the brain following stroke.
Numerous preclinical and clinical data suggest that preconditioning provides durable and robust protection to cells after ischemia (Murry et al., 1986; Schurr et al., 1986; Kitagawa et al., 1991; Kirino et al., 1991; Gidday et al., 1994; Perez-Pinzon et al., 1996, 1997; Chen et al., 1996; Vannucci et al., 1998; Barone et al., 1998; Bernaudin et al., 2002; Dirnagl et al., 2003; Wegener et al., 2004; Sharp et al., 2004; Stenzel-Poore et al., 2003; Dhodda et al., 2004; Nakamura et al., 2006; Koch et al., 2011; Hausenloy et al., 2007; Hess et al., 2015). Preconditioning is a well-defined phenomenon in which a sublethal stimulus confers tolerance to a subsequent more severe insult in the brain (Kitagawa et al., 1991). A preconditioning stimulus acts at many biological levels to engage programs that produce durable (>24 hours) tolerance against ischemic injury. There has been significant interest in understanding the mechanisms by which preconditioning promotes cell survival. In conditions such as ischemia, where the reduction in blood supply results in the decreased supply of oxygen and glucose to the tissues, stress is activated by nutrient deprivation as well as deprivation-induced damage. The reduction in oxygen availability induces hypoxic stress, which then activates homeostatic adaptive mechanisms to assure survival ( Chavez and LaManna, 2002). Homeostatic hypoxic adaptation is mediated, in part, via oxygen sensors, the HIF prolyl hydroxylases (HIF PHDs). HIF PHDs are the enzymatic gatekeepers of the adaptive response to hypoxia and transduce changes in oxygen availability into stabilization of a host of proteins, including hypoxia-inducible factors (HIF). Stabilization of HIF has been believed to play a critical role in this transcriptional response, which is triggered at a cellular, local and systemic level to alleviate the discrepancy in oxygen demand and supply (Ratan et al., 2007; Karuppagounder and Ratan, 2012). Indeed, our initial hypothesis was that HIF PHDs would activate a homeostatic set of genes driven by HIF (e.g., EPO, VEGF, glycolytic enzymes), but recent data suggest that HIF PHD inhibition can precondition the brain and external organs via alternate mechanisms. While we cannot exclude the possibility that HIF PHD inhibition-mediated preconditioning is acting in peripheral cells (immune or vascular) via HIF-dependent gene expression to mediate preconditioning, our current data suggest that preconditioning by HIF PHDs could act by alternative mechanisms, including diversion of metabolic substrates or pro-death gene suppression (Olenchock et al., 2016; Karuppagounder et al., 2016) (Figure 1). What has emerged from these and other studies is that HIF transcription factors are not the primary target for manipulating the hypoxic response to precondition the brain and other organs. Rather, the HIF PHDs, established oxygen sensors, are a validated target as they sense and transduce changes in oxygen into a number of metabolic and transcriptional changes (Figure 1). The ability of oxygen sensors to be tunable modulators of preconditioning leads to a testable, generalizable model whereby a molecular understanding of how stress is converted into adaptive transcriptional responses will foster the understanding of a panoply of new targets (e.g. the sensors themselves) to drive preconditioning. In this review, we discuss this evidence and some novel, if not surprising, evidence that links hypoxia adaptation with antiviral defense.
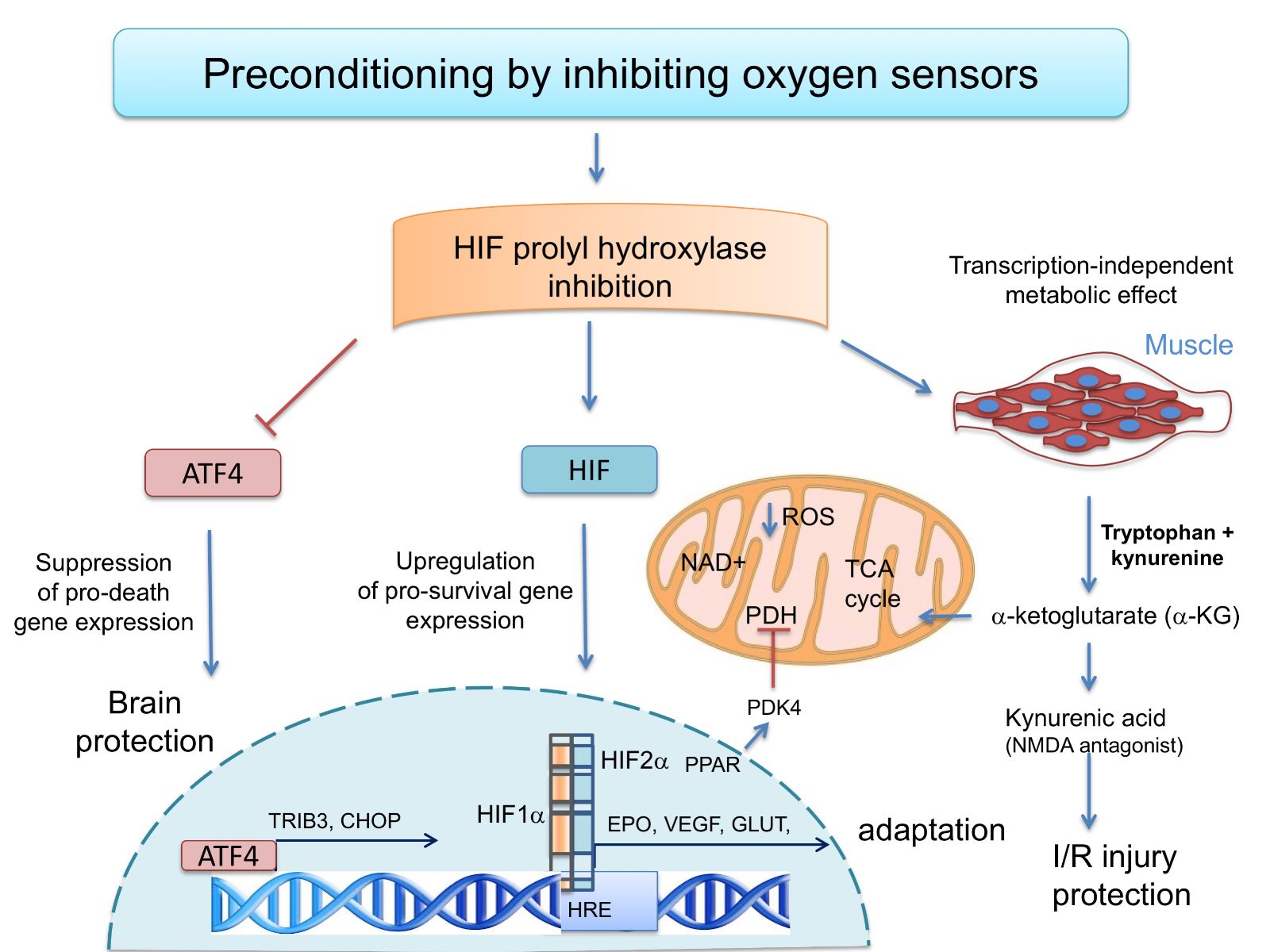
In a new window | Download PPT
Figure 1: The oxygen sensing HIF prolyl hydroxylases modulate transcriptional and metabolic adaptive responses. Preconditioning by HIF PHD inhibition can drive pro-survival HIF-1a-or HIF-2a-dependent gene expression. Alternatively, HIF PHD inhibition can suppress pro-death gene expression and protect the brain via its ability to inhibit ATF4-dependent transcription. Finally, in muscle, HIF PHDs regulate circulatinga-ketoglutarate, and this leads to increases in the hepatic production of kynurenic acid, which is an NMDA antagonist and protects against ischemic injury.
Isoforms of HIF PHD domain enzymes
There are at least three HIF PHD isoforms with diverse expression in various cell types and organs (Schodel et al., 2009). The family originated as a single isoform in flies and worms and diverged to three isoforms: HIF PHD1 (EGLN2), HIF PHD2 (EGLN1) and HIF PHD3 (EGLN3) in humans.HIF PHD1, also known as egg-laying defective nine homolog-2 (EGLN-2), is primarily localized to the nucleus (Metzen et al., 2003). HIF PHD1 contains a nuclear localization signal (NLS), and an actin-regulated importin α/β-dependent extended NLS directs the nuclear import (Steinhoff et al., 2009). Under non-stress conditions, it is highly expressed in testis, moderately in the liver and at low levels in heart, brain, and kidney (Lieb et al., 2002). HIF PHD1 can hydroxylate the N- and C-terminal ODD domain in HIF-1α(Tian et al., 2006). In cultured cortical neurons, HIF PHD1 inhibition but not inhibition of other isoforms regulates normoxic oxidative stress-induced death in an HIF-independent and CREB-independent manner (Siddiq et al., 2009).
HIF PHD2 is also known as EGLN-1 and is primarily localized in the cytoplasm (Metzen et al., 2003)and highly expressed in heart, testis and moderately in brain, kidney, and liver (Lieb et al., 2002). HIF PHD2 is believed to be the primary regulator of the HIF-1atranscription factors (Appelhoff et al., 2004 ; Berra et al., 2003; Epstein et al., 2001; Lieb et al., 2002). Like HIF PHD1, HIF PHD2 hydroxylates both N-and C-terminal ODD in HIF-1α (Appelhoff et al., 2004). HIF PHD2 deletion leads to accumulation of HIF-1α but not HIF-2α in mouse liver and kidney tissue (Takeda et al., 2007).
HIF PHD3, also known as EGLN3, is localized to the cytoplasmic and nuclear compartments (Metzen et al., 2003)and is highly expressed in heart and liver, and moderately in brain and kidney (Lieb et al., 2002). HIF PHD3 expression can be upregulated by hypoxia, hypoxia mimetics, or in sympathetic neurons by growth factor deprivation as a necessary part of the apoptotic program (Lomb et al., 2007; Appelhoff et al., 2004). Further studies suggest that there are other substrates for HIF PHDs including activating transcription factor 4 (ATF4) or NF-kappa B. A study by Koditz et al. reported that ATF4 is stabilized in hypoxia by siRNA-mediated silencing of HIF PHD3, or by mutation of proline residues in the ATF4 protein (Koditz et al., 2007). Remarkably, it is still not known whether one isoform dominates in mediating the preconditioning effects of hypoxia. As there is functional redundancy among isoforms, it may be that all three isoforms must be inhibited to produce an optimal therapeutic effect, although, as discussed below, deletion of single isoforms can mediate biological effects not observed with the other isoforms (Minamishima and Kaelin, 2010; Karuppagounder et al., 2016).
HIF PHD inhibitors as mediators of preconditioning
Subsequent investigations confirmed that the oxygen sensors could be manipulated to mediate preconditioning, and some of these effects required the transcriptional activator, HIF-1a(Baranova et al., 2007). However, evidence that forced expression of HIF-1acould lead to death or survival, and that inhibition of HIF could in some cases be beneficial (Chen et al., 2008) created new questions about the pathway and its role in preconditioning. It was then shown that inhibition of the HIF PHDs, the oxygen sensors,could inhibit the pro-death effects of HIF-1a(( Aminova et al, 2008 )). Another study used a more specific inhibitor of the HIF PHDs developed by Fibrogen to confirm a role for HIF PHD inhibition in preconditioning (Siddiq et al., 2005). More recent studies (Karuppagounder et al., 2016) have raised significant concerns that canonical HIF PHD inhibitors (e.g., desferrioxamine (DFO), 3,4 dihydroxy benzoate) do not penetrate the BBB well, raising the very real possibility that preconditioning mediated by these and other putative HIF PHD inhibitors is the result of effects on a peripheral organ (immune cell) or a peripheral cell type (muscle, endothelial cells) or even on a target independent of the HIF PHDs. Indeed, it is very important to develop molecular evidence supporting the role of HIF PHDs in preconditioning, as many of the established small-molecule inhibitors of this pathway are non-selective and could influence other iron, oxygen and 2-oxoglutarate-dependent dioxygenases including the Jumanji histone demethylases or the ten-eleven translocase (Tet) demethylases (Pogribny et al., 2013; Karuppagounder et al., 2016).
Molecular evidence for HIF PHD inhibition-mediated neuroprotection
Several converging lines of molecular investigation have established HIF PHDs as a bona fidetarget for preconditioning: 1) peptide inhibitors of the HIF PHDs were effective in blocking oxidative death of neurons in vitro(Siddiq et al., 2005); 2) shRNA reduction of HIF PHD1 but not PHD2 or PHD3 leads to neuroprotection from oxidative death in vitro(Siddiq et al., 2009); and 3) molecular reduction of PHD1, PHD2 and PHD3 in astrocytes and neurons in the striatum using Cre recombinase in PHD1/PHD2/PHD3 triple-floxed mice leads to improvements in functional recovery following ICH (Karuppagounder et al., 2016). Molecular reduction of PHD1 in muscle alone preconditions the heart to resist a subsequent myocardial ischemic insult (Olenchock et al., 2016) or preconditions the muscle or liver itself to resist local ischemia (Aragones et al., 2008; Schneider et al., 2010). These peptide and molecular approaches provide confidence that HIF PHDs are a target for protecting the brain and organs other than the brain.
The notion that molecular modulation of HIF PHD activity can trigger adaptation in preconditioning in brain is amplified and supported by subsequent studies by Kunze et al. demonstrating that inactivation of neuronal specific HIF PHD2 reduced infarct size and neuronal apoptosis following ischemic stroke (Kunze et al., 2012). Recently, the same group demonstrated that neuronal-specific deletion of HIF PHD’s increased endogenous adaptive response (Li et al., 2016). As mentioned above, we have recently demonstrated that molecular reduction of all three isoforms of HIF PHD’s selectively in the brain improved functional recovery after hemorrhagic stroke by suppression of ATF4-induced pro-death genes (Karuppagounder et al., 2016). Kaelin's group recently showed that molecular (deletion in muscle) or pharmacological inhibition of EGLN1 could mediate ischemic preconditioning by sparing circulating a-ketoglutarate (a-KG). Uptake of a-KG by the liver leads to increased synthesis and release of kynurenic acid, which can act not only as an NMDA antagonist but also as a precursor for the synthesis of NAD (Olenchock et al., 2016; Lovelace et al., 2016).
Aragones et al., 2008 provided another putative mechanism by which HIF PHD inhibition can lead to preconditioning-induced protection. They demonstrated that muscle devoid of HIF PHD1 was exercise-intolerant with reduced oxygen utilization, but that the muscle was protected from arterial occlusion-induced necrosis (Aragones et al., 2008). In this paradigm, preconditioning was not dependent on HIF PHD2 and HIF-1, but rather via HIF PHD1-mediated stabilization of HIF-2a, leading to induction of PPAR-a, and presumptive induction of pyruvate dehydrogenase kinase 4 (PDK4), an inhibitor of pyruvate dehydrogenase (PDH). PDH inhibition could then lead to resistance to ischemia via its ability to inhibit mitochondrial pyruvate metabolism and mitochondrial ROS production.
Taken together, these studies demonstrate that molecular reduction of HIF PHD’s in a variety of tissues can mediate preconditioning, but that preconditioning may occur via HIF-dependent mechanisms that are transcription-dependent; HIF-independent mechanisms involving transcriptional suppression; and transcription-independent mechanisms involving the diversion of a-KG to kynurenic acid. Collectively, these studies justify a focus on HIF PHD inhibition for ischemic preconditioning in a host of tissues.
Novel and safe pharmacological strategies for preconditioning the brain: Small molecule-driven activation of transcriptional adaptation to hypoxia
Because HIF PHD inhibition is a viable molecular target for preconditioning, we actively embarked upon an FDA-approved screen to identify clinically approved pharmacological agents that could drive the adaptive response to hypoxia. The screen involved looking for clinically approved agents that activate a stably expressed hypoxia response element (the DNA binding domain of HIF-1) luciferase reporter greater to or equal to DFO. DFO is clinically approved, but because it binds free iron, it can diminish metalloenzymes necessary for brain physiology of iron, thus causing toxicity. The screen was intended to identify a small molecule agent that could avoid this potential pitfall and produced a significant number of hits and pharmacophores (Aleyassin et al., 2015). Of note, we found that all anthelminthic benzimidazoles were slightly better than DFO in stabilizing HIF. Mechanistic studies showed that the HIF-stabilizing effects of benzimidazoles were mediated via drug binding to monomeric tubulin, and that those benzimidazoles could be neuroprotective (Aleyasin et al., 2015). Of even greater interest was the small molecule tilorone, which induced the hypoxia response element 3-4-fold greater than other clinically approved activators of the HIF pathway (Ratan et al., 2008; Table 1). Remarkably, tilorone provided 80% reduction in infarct size when delivered 24 hours prior to permanent ischemia in rats, and significant reductions in lesion volume in a rat model of spinal cord injury (Ratan et al., 2008). Tilorone was initially developed as an antiviral immunomodulator, raising the possibility of unexpected cross-talk between hypoxia signaling and antiviral defense. Although tilorone was our best “hit” in driving hypoxia response element-driven luciferase activity, we have not been able to confirm evidence of HIF stabilization or activation of HIF target genes by tilorone (unpublished observations). The results suggest that tilorone likely worked indirectly to drive hypoxia response element-driven transcription.
Table 1: Clinically approved activators of the HIF pathway

Reproduced from Aleyasin et al., 2015, Antioxidants & Redox Signaling.
Tilorone: Unknown target but evidence of cross-talk between hypoxia signaling and Type I antiviral interferon response?
Tilorone was developed as the first orally bioavailable small molecule interferon-inducing agent, and it was marketed as an FDA-approved drug in the mid-1970's (Chandra and Wright, 1977). It was removed from the market because of cell culture evidence that its side chains could induce a form of mucopolysaccharidosis in rodents (Prokopek, 1991). Since its initial discovery, we have learned a great deal about the molecular signaling pathways involved in activating the Type I interferon response, a response involved not only in antiviral defense, but also in tumor survival and preconditioning (Marsh et al., 2009). Indeed, Stenzel-Poore's group has demonstrated that interferon signaling induced by poly dI/dC induces preconditioning via a TLR3-independent pathway that depends on MDAV5, IPS1, and Interferon-a(Gesuete et al., 2016). Tilorone is believed to intercalate into DNA, but its mechanisms for inducing the anti-viral response are unclear (Stringfellow, 1976). We are currently exploring several possibilities. First, it is possible that tilorone stabilizes cytosolic DNA released via nuclear double-strand breaks or via mitochondrial damage (Erdal et al., 2017; Fig. 2). This stabilization could occur via tilorone binding to cytosolic DNA and consequent inhibition of TREX1, a cytosolic DNA endonuclease that can degrade cytoplasmic DNA to reduce cytosolic DNA concentrations (Erdal et al., 2017). Inhibition of TREX activity by tilorone binding to cytoplasmic DNA would lead to activation of the cytosolic DNA sensor, cGMP-cAMP synthase (cGAS synthase). Activation of cGAS leads to accumulation of 2',5' cGAMP, an atypical cyclic dinucleotide second messenger that leads to the activation of the ER scaffold protein, stimulator of interferon genes (STING). Binding of STING by 2',5' cGAMP or other molecules, possibly tilorone, recruits TANK-binding kinase I (TBK1), and phosphorylation and activation of the transcription factor IRF3 (Cavlar et al., 2013; Tanaka and Chen, 2012; Guo et al., 2015). IRF3 activation leads to induction of the innate immune response, including Type I interferon response genes. As tilorone appears to induce an “IFN-related DNA damage resistance signature” (IRDS) (( Weichselbaum et al. 2008 )), a clinical classifier comprised of seven IRDS genes (STAT1, MX1, ISG15, OAS1, IFIT1, IFIT3, and IFI44) that identify patients whose cancers are resistant to chemotherapy and radiotherapy (Erdal et al., 2017; Zhai et al., unpublished), similar genes may be important in preconditioning by tilorone or poly dI/dC. Indeed, future studies will query whether STING, TBK1, and IRF3 are necessary for tilorone's effects and whether these signaling pathways act to induce an IFN-related DNA damage resistance signature (Mathur et al., 2017).

In a new window | Download PPT
Figure 2: Schematic diagram illustrates how tilorone and hypoxia may share common mechanisms in the mediation of the preconditioning response. 1) Tilorone is believed to intercalate into DNA, and its mechanisms for inducing the anti-viral response are unknown. 2) Hypoxia leading to secondary increases in mitochondrial or nuclear DNA damage can result in accumulation of cytosolic DNA. Under normal conditions, TREX1, a cytosolic DNA endonuclease, can degrade accrued cytosolic DNA. However, it is possible that tilorone could stabilize cytosolic DNA by binding to it and thereby suppress TREX1 activity leading to activation of cGAS synthase. Activation of cGAS synthase catalyzes the formation of cGAMP from ATP and GTP. 3) As an alternative to binding to cytosolic DNA, tilorone could be an agonist of the ER adaptor protein STING. STING activation leads to the recruitment of the kinase TBK1 and consequent phosphorylation of IRF3. Phosphorylated IRF3 translocates to nucleus to induce a cassette of interferon genes involved in innate immunity preconditioning. As a DNA binding drug, tilorone could also bind to nuclear DNA to enhance IRF3-mediated transcription downstream of STING.
It is formally possible that a convergence between hypoxia signaling and interferon signaling occurs because hypoxia or hypoxia/ischemia, like a viral infection, can lead to the accumulation of cytosolic nuclear DNA or mitochondrial DNA, thus activating the cytoplasmic DNA sensor (cGAS), thus increasing 2',5' cGAMP. cGAMP is an agonist for activation of the ER resident protein STING, leading to phosphorylation and activation of the transcription factor IRF3. IRF3 phosphorylation then leads to an increase in transcription with an IRSG signature. The model raises important questions about which cell types this pathway must be activated in to optimize the effects of tilorone or other putative small-molecule activators of STING as preconditioning agents. While there is no current evidence that tilorone activates STING directly, we speculate that it might be metabolized in vivo to an activator of STING.
Conclusion
In this review, we highlight the potential value of oxygen sensing as a target for ischemic preconditioning in the brain. We also describe unexpected cross-talk between one novel class of HIF activator (tilorone, an antiviral immunomodulator) and activation of endogenous anti-viral defenses that appear to mediate preconditioning via LPS or poly dI/dC. Understanding whether our bodies engage a host of parallel endogenous adaptive responses to reproduce preconditioning-mediated protection or whether there is a dominant common final pathway is critical to improving outcomes in patients who suffer from debilitating stroke conditions. Our data build on the exciting studies of Hallenbeck and Stenzel-Poore to support the notion that interferon transcription, particularly Interferon-a,may be a common final target of many types of preconditioning stimuli.
Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgements
We acknowledge the support of the Sperling Center for Hemorrhagic Stroke Recovery at Burke Medical Research Institute; the Burke Foundation; Dr. Miriam and Sheldon G Adelson Medical Research Foundation grant to RRR; the National Institutes of Health (Grant P01 NIA AG014930, Project 1 to RRR).
References
Saravanan S. Karuppagounder1,2
1Sperling Center for Hemorrhagic Stroke Recovery, Burke Medical Research Institute;
2Department of Neurology and Neuroscience, Weill Medical College, Cornell University;
Yujia Zhai1,2,3
1Sperling Center for Hemorrhagic Stroke Recovery, Burke Medical Research Institute;
2Department of Neurology and Neuroscience, Weill Medical College, Cornell University;
3Anti-stress and Health Research Center, College of Pharmacy, Jinan University, Guangzhou 510632, China.
Yingxin Chen1,2
1Sperling Center for Hemorrhagic Stroke Recovery, Burke Medical Research Institute;
2Department of Neurology and Neuroscience, Weill Medical College, Cornell University.
RongrongHe3
3Anti-stress and Health Research Center, College of Pharmacy, Jinan University, Guangzhou 510632, China.
Rajiv R. Ratan1,2
1Sperling Center for Hemorrhagic Stroke Recovery, Burke Medical Research Institute;
2Department of Neurology and Neuroscience, Weill Medical College, Cornell University.
Corresponding author:
Dr. Saravanan S. Karuppagounder
Email: ssk2004@med.cornell.edu
and
Dr. Rajiv R. Ratan
Email: rrr2001@med.cornell.edu
In a new window | Download PPT
Figure 1: The oxygen sensing HIF prolyl hydroxylases modulate transcriptional and metabolic adaptive responses. Preconditioning by HIF PHD inhibition can drive pro-survival HIF-1a-or HIF-2a-dependent gene expression. Alternatively, HIF PHD inhibition can suppress pro-death gene expression and protect the brain via its ability to inhibit ATF4-dependent transcription. Finally, in muscle, HIF PHDs regulate circulatinga-ketoglutarate, and this leads to increases in the hepatic production of kynurenic acid, which is an NMDA antagonist and protects against ischemic injury.
In a new window | Download PPT
Figure 2: Schematic diagram illustrates how tilorone and hypoxia may share common mechanisms in the mediation of the preconditioning response. 1) Tilorone is believed to intercalate into DNA, and its mechanisms for inducing the anti-viral response are unknown. 2) Hypoxia leading to secondary increases in mitochondrial or nuclear DNA damage can result in accumulation of cytosolic DNA. Under normal conditions, TREX1, a cytosolic DNA endonuclease, can degrade accrued cytosolic DNA. However, it is possible that tilorone could stabilize cytosolic DNA by binding to it and thereby suppress TREX1 activity leading to activation of cGAS synthase. Activation of cGAS synthase catalyzes the formation of cGAMP from ATP and GTP. 3) As an alternative to binding to cytosolic DNA, tilorone could be an agonist of the ER adaptor protein STING. STING activation leads to the recruitment of the kinase TBK1 and consequent phosphorylation of IRF3. Phosphorylated IRF3 translocates to nucleus to induce a cassette of interferon genes involved in innate immunity preconditioning. As a DNA binding drug, tilorone could also bind to nuclear DNA to enhance IRF3-mediated transcription downstream of STING.
Table 1: Clinically approved activators of the HIF pathway
Reproduced from Aleyasin et al., 2015, Antioxidants & Redox Signaling.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 16197 | 50 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA