Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Stem cell and extracellular vesicle therapy in Huntington’s disease
Time:2024-09-17
Number:10232
Author Affiliations

Conditioning Medicine 2023. 6(6): 177-196.
Abstract
Huntington’s disease (HD) manifests as a debilitating neurodegenerative disorder characterized by a genetic mutation in the huntingtin (HTT) gene, leading to motor deficits, cognitive impairments, and psychiatric symptoms. HD's major influence on patients' daily living warrants the development of new, safe, and effective treatment strategies beyond symptomatic management and disease modification. We systematically explore the preclinical studies and clinical trials focusing on the application of cell-based therapy and extracellular vesicle therapy in HD. The review aims to map the current landscape of cell and extracellular vesicles (EVs) therapy research, pinpointing the successes in ameliorating disease phenotypes and mechanisms, assessing safety and efficacy, and identifying the challenges and limitations encountered. Moreover, we highlight significant gaps in knowledge and propose areas for future research, emphasizing the need for more targeted studies to fully understand the mechanisms of action in the hope of more effective treatments for HD.
Keywords: Huntington’s disease, cell therapy, stem cell therapy, regenerative medicine, extracellular vesicles, exosome
Abstract
Huntington’s disease (HD) manifests as a debilitating neurodegenerative disorder characterized by a genetic mutation in the huntingtin (HTT) gene, leading to motor deficits, cognitive impairments, and psychiatric symptoms. HD's major influence on patients' daily living warrants the development of new, safe, and effective treatment strategies beyond symptomatic management and disease modification. We systematically explore the preclinical studies and clinical trials focusing on the application of cell-based therapy and extracellular vesicle therapy in HD. The review aims to map the current landscape of cell and extracellular vesicles (EVs) therapy research, pinpointing the successes in ameliorating disease phenotypes and mechanisms, assessing safety and efficacy, and identifying the challenges and limitations encountered. Moreover, we highlight significant gaps in knowledge and propose areas for future research, emphasizing the need for more targeted studies to fully understand the mechanisms of action in the hope of more effective treatments for HD.
Keywords: Huntington’s disease, cell therapy, stem cell therapy, regenerative medicine, extracellular vesicles, exosome
Highlights
This paper presents recent advances in cell- and cell-free regenerative medicine approaches for Huntington’s disease (HD), which is a debilitating neurodegenerative disorder with life-threatening motor, cognitive, and psychiatric symptoms. Here, we discuss the therapeutic potential of stem cells and their secreted extracellular vesicles. We review the scientific evidence that both stem cells and extracellular vesicles capture a novel approach relevant to conditioning medicine, in that their treatment intervention in HD may not only retard disease progression but also modify the disease pathology by specifically combating the genetic mutation in the huntingtin gene. Hence, we advance the concept that stem cell and extracellular vesicle therapy is a new, safe, and effective conditioning medicine strategy for symptomatic management and disease-modification of HD.
Introduction
Huntington’s disease (HD) is a progressive neurodegenerative disorder manifesting as a triad of motor, cognitive, and psychiatric symptoms. It impacts populations worldwide, with a prevalence rate of 4 cases per 100,000 individuals. Typically, HD emerges in adulthood, with most diagnoses occurring between the ages of 35 and 44 (Medina et al., 2022). This condition follows an autosomal dominant inheritance pattern stemming from a mutation in exon 1 of the huntingtin (HTT) gene (Bates et al., 2015). The mutation is characterized by an expanded CAG trinucleotide repeat in the HTT gene on chromosome 4, leading to the accumulation of an abnormal form of the huntingtin protein, known as mutant huntingtin (mHTT). The number of CAG repeats is inversely proportional to the age at onset and directly correlates with the disease's severity (Walker, 2007). Accumulation of mHTT in neurons precipitates cellular dysfunction and apoptosis, predominantly affecting the striatum and cortex. The disease's pathogenesis is marked by disruptions in protein folding and degradation, mitochondrial dysfunction, excitotoxicity, and altered gene expression (Li and Li, 2004). The symptomatic spectrum of HD includes involuntary “chorea” motor actions, learning and memory impairments, and psychiatric alterations (Ross and Tabrizi, 2011). Despite extensive research, treatments remain symptomatic, with no current therapy able to alter the disease's progression, highlighting the need for novel therapeutic strategies (Frank, 2014).
This review comprehensively assesses the efficacy and safety of cell therapy and extracellular vesicle (EV) therapy in HD across preclinical studies and clinical trials (Figure 1). We seek to pinpoint research gaps that warrant further investigation, guiding future scientific research in this field.
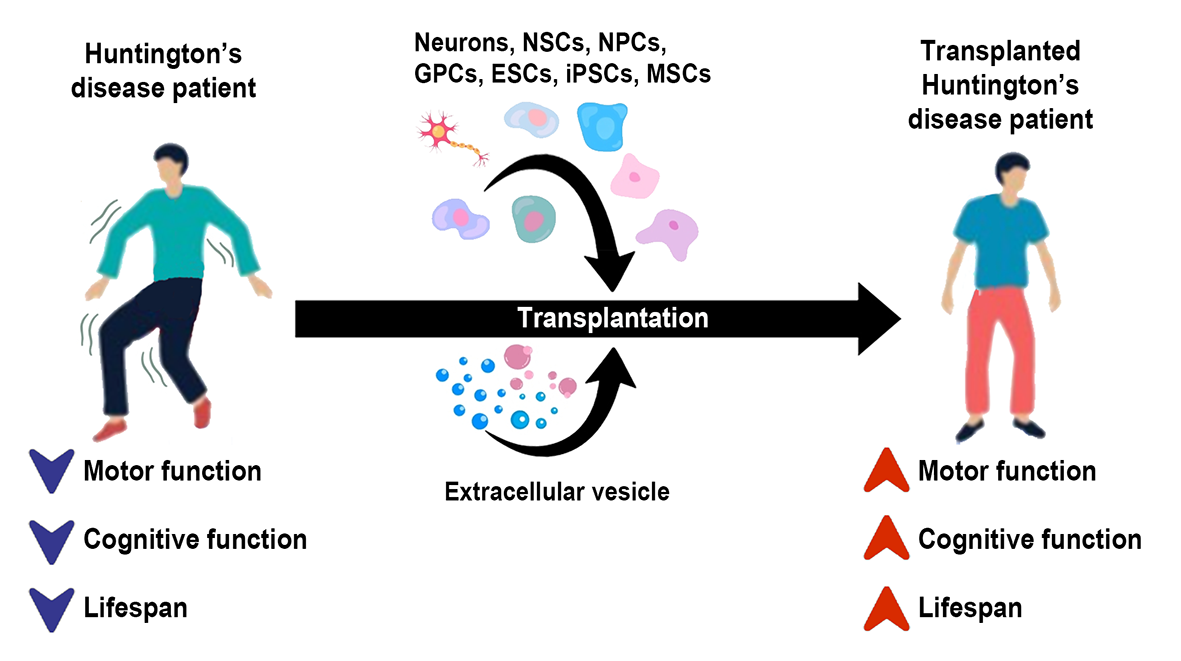
In a new window | Download PPT
Figure 1. HD is a neurodegenerative disorder characterized by motor and cognitive dysfunctions and a shortened lifespan. Novel therapies, including cell and extracellular vesicle therapy, can improve HD phenotypes and clinical symptoms, opening a new conditioning medicine opportunity for HD treatment.
Pathogenesis of HD
Mutant Huntingtin Protein
The pathogenesis of HD is characterized by protein misfolding due to a polyglutamine expansion, leading to oligomer formation (DiFiglia et al., 1997; Cooper et al., 1998; Hoffner et al., 2005; Tabrizi et al., 2020). These oligomers serve as precursors for protofibrils and intracellular inclusions. Contrary to previous assumptions that mHTT inclusions were the main contributors to pathology, recent studies suggest that these inclusions may not be directly responsible for cell death (Ross, 1997; Saudou et al., 1998; Arrasate et al., 2004; Hoffner et al., 2005; Slow et al., 2005), and might even be protective (Arrasate et al., 2004; Nucifora et al., 2012). The current hypothesis is that mHTT toxicity could be largely due to N-terminal fragments containing the toxic exon 1 of the HTT gene produced by proteolytic cleavage of mHTT or CAG length-dependent aberrant splicing, with the toxicity of oligomers potentially reduced by their assembly into larger inclusions (Nagai et al., 2007; Takahashi et al., 2008; Lajoie and Snapp, 2010; Miller et al., 2011; Nucifora et al., 2012; Pieri et al., 2012; Sahl et al., 2012; Leitman et al., 2013). In animal models of HD, polyglutamine-containing N-terminal fragments of mHTT accumulate in the brain more rapidly than the full-length mHTT (Wang et al., 2008; Castiglioni et al., 2012; The Hd iPsc Consortium, 2012).
Furthermore, evidence suggests that mHTT can be transferred between cells through tunneling nanotubes and extracellular vesicles, indicating a potential mechanism for its propagation within the brain. In vitro models of HD have demonstrated that cells can absorb polyglutamine peptides from both the culture media and co-cultured cells (Yang et al., 2002; Herrera et al., 2011; Costanzo et al., 2013; Monsellier et al., 2016). A study in Drosophila showed that mHTT is released from synaptic terminals and subsequently endocytosed by adjacent neurons (Babcock and Ganetzky, 2015). However, evidence of intercellular spreading in humans is currently limited to post-mortem analyses, with inclusion bodies found in the extracellular matrix of striatal transplanted grafts, suggesting the release of mHTT by neurons (Cicchetti et al., 2014).
Ubiquitin-Proteasome System
Perturbation of the ubiquitin-proteasome system, which affects cellular protein degradation (Lin et al., 2011; Cortes and La Spada, 2014), is also found in HD. Evidence showed that mHTT interferes with this system by depleting important proteins such as vasolin-containing protein (also known as p97), ubiquitin fusion degradation protein, nuclear protein localization protein, ubiquitin-specific protease 14, and activating transcription 5, leading to failure in the endoplasmic reticulum stress response (D'Egidio et al., 2023). Moreover, the accumulation of toxic proteins due to the altered ubiquitin-proteasome system strengthens the toxicity inside affected cells, eventually stressing organelles such as mitochondria, thereby elevating oxidative stress. In this view, the induction of autophagy, a process facilitating the clearance of damaged or unnecessary cellular components, has demonstrated promise in reducing HD phenotypes and enhancing the clearance of mHTT in animal models (Ravikumar et al., 2004).
Mitochondria Function
Mitochondrial function is compromised in HD. Analysis of post-mortem brain specimens reveals a reduction in ATP production in HD human (Browne and Beal, 2004) and mouse model brains (Mochel et al., 2012) compared to normal brains. Alterations in mitochondrial structure, quantity, and enzymatic activity have been documented (Goebel et al., 1978; Gu et al., 1996; Browne et al., 1997; Kim et al., 2010; Johri et al., 2013). Brain imaging studies frequently demonstrate downregulated glucose metabolism and upregulated lactate concentration in HD patients, suggesting diminished mitochondrial metabolic function (Jenkins et al., 1993; Antonini et al., 1996; Feigin et al., 2001; Reynolds et al., 2005). Research in HD animal models has identified disruptions in mitochondrial mobility, both anterograde and retrograde, which blocks mitochondrial distribution (Trushina et al., 2004; Orr et al., 2008; Shirendeb et al., 2011; Shirendeb et al., 2012). Moreover, the expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a key regulator of mitochondrial biogenesis, is significantly reduced in HD models (Cui et al., 2006; Johri et al., 2013). However, impairments of mitochondrial fission and fusion have also been observed (Jurcau and Jurcau, 2023). Moreover, evidence suggests that mHTT disrupts the mitochondrial outer membrane, inducing calcium release that leads to cell death, and compromises the inner membrane, obstructing protein transport (Panov et al., 2002; Choo et al., 2004; Yano et al., 2014; Yablonska et al., 2019).
Somatic Instability
In addition to the toxicity of the mHTT protein, the RNA associated with HD is also implicated in cellular toxicity. Studies in animal models of HD demonstrate neurodegeneration even in the absence of CAG repeat translation (Martí, 2016). Various animal models featuring knock-in CAG repeats have highlighted the toxicity of RNA foci (Li et al., 2008; Hsu et al., 2011; Wang et al., 2011). Research involving individuals with HD has shown a correlation between CAG repeats and disease onset and severity, supporting the idea that CAG repeat instability contributes to disease pathogenesis (Swami et al., 2009; Lee et al., 2005). A predictive model indicates that motor symptoms manifest when the CAG repeat count surpasses 115 units and a significant number of cells become vulnerable (Squitieri et al., 2006; Kaplan et al., 2007). The extent of somatic instability varies across tissues, with the pattern of tissue sensitivity aligning with HD neuropathology (Telenius et al., 1993; Aronin et al., 1995; La Spada, 1997; Shelbourne et al., 2007). Repeat-associated non-ATG (RAN) translation has been observed in the brains of HD patients in a CAG repeat-dependent fashion (Bañez-Coronel et al., 2015; Gao et al., 2017). However, the impact of monopeptide aggregates resulting from this unconventional translation process remains to be fully elucidated.
Stem Therapy in HD
Neurons and Other Non-Stem Cells
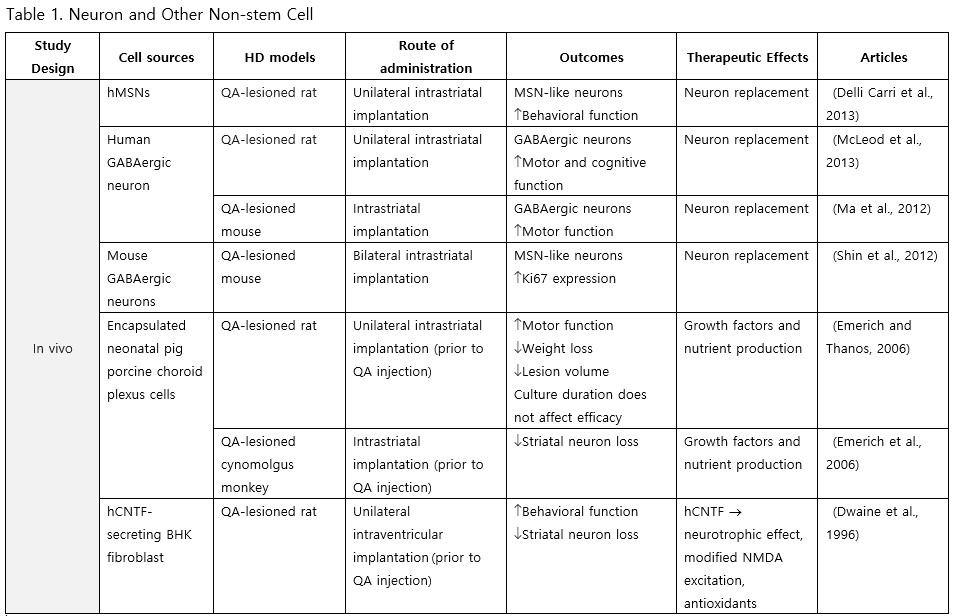
Neurons primarily harvested from embryonic stem cells (ESCs) and neural precursor cells (NPCs) are anticipated to replace degenerated striatal neurons in HD transplantation. Delli Carri et al. (2013) successfully induced differentiation of human ESCs into medium spiny neurons (MSNs), known to be the most susceptible type of neurons in HD, and upon transplantation into the striatum of quinolinic acid (QA)-lesioned rats, the grafted neurons persisted and committed along the DARPP-32 positive neuronal lineage, integrating with the host brain, altogether dampening the apomorphine-mediated rotational behavior. Furthermore, McLeod et al. (2013) demonstrated that γ-aminobutyric acid (GABA)-ergic cells differentiated from human NPCs (hNPCs) could significantly improve motor and memory deficits following transplantation. Additionally, the transplantation of the choroid plexus has shown to confer benefits: pig porcine choroid plexus encapsulated in alginate microcapsules and grafted into the striatum of QA-lesioned rats, reduced weight loss and motor impairment, as well as neural loss and striatal atrophy when transplanted prior to QA injection (Emerich and Thanos, 2006; Emerich et al., 1996).
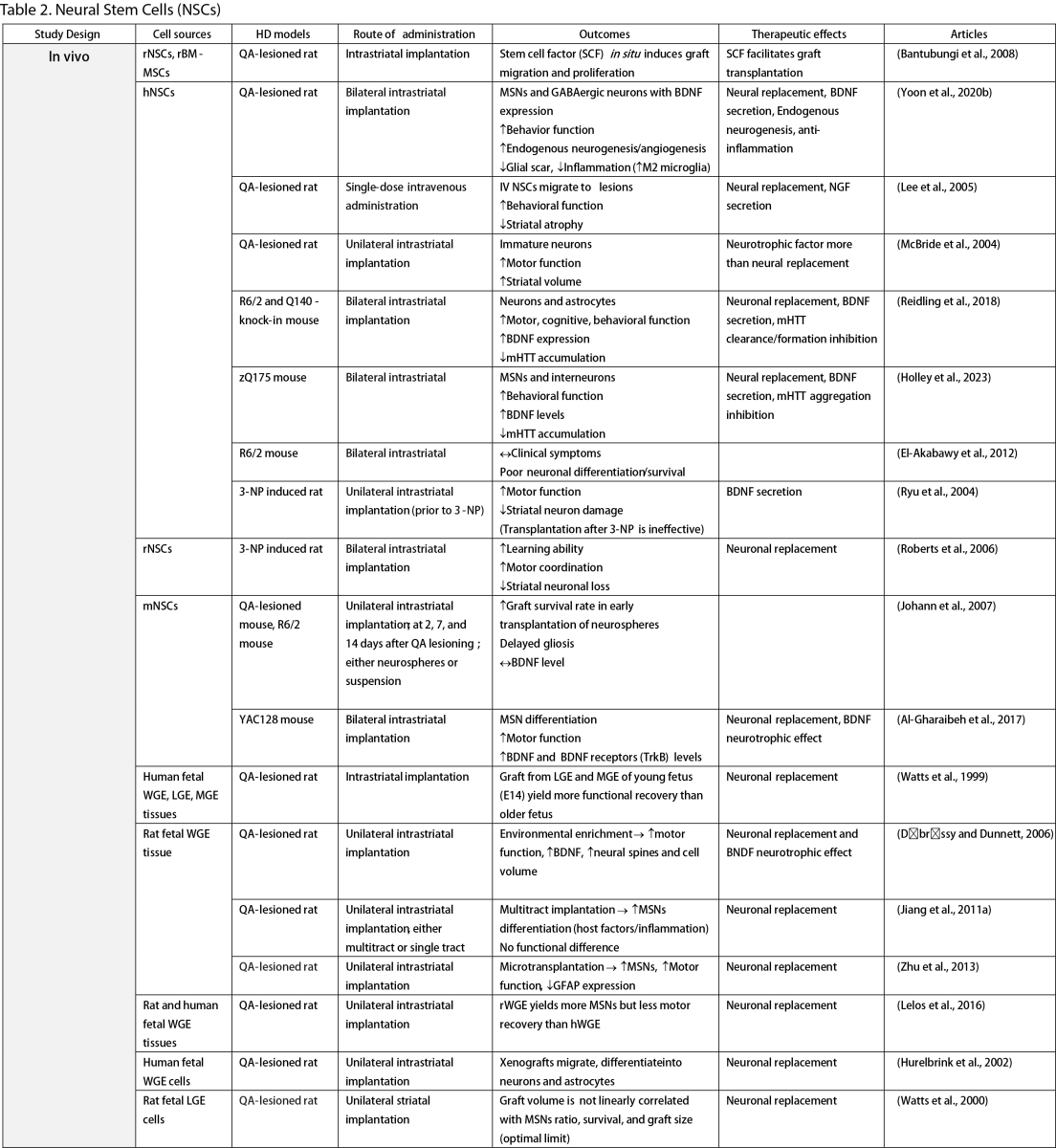
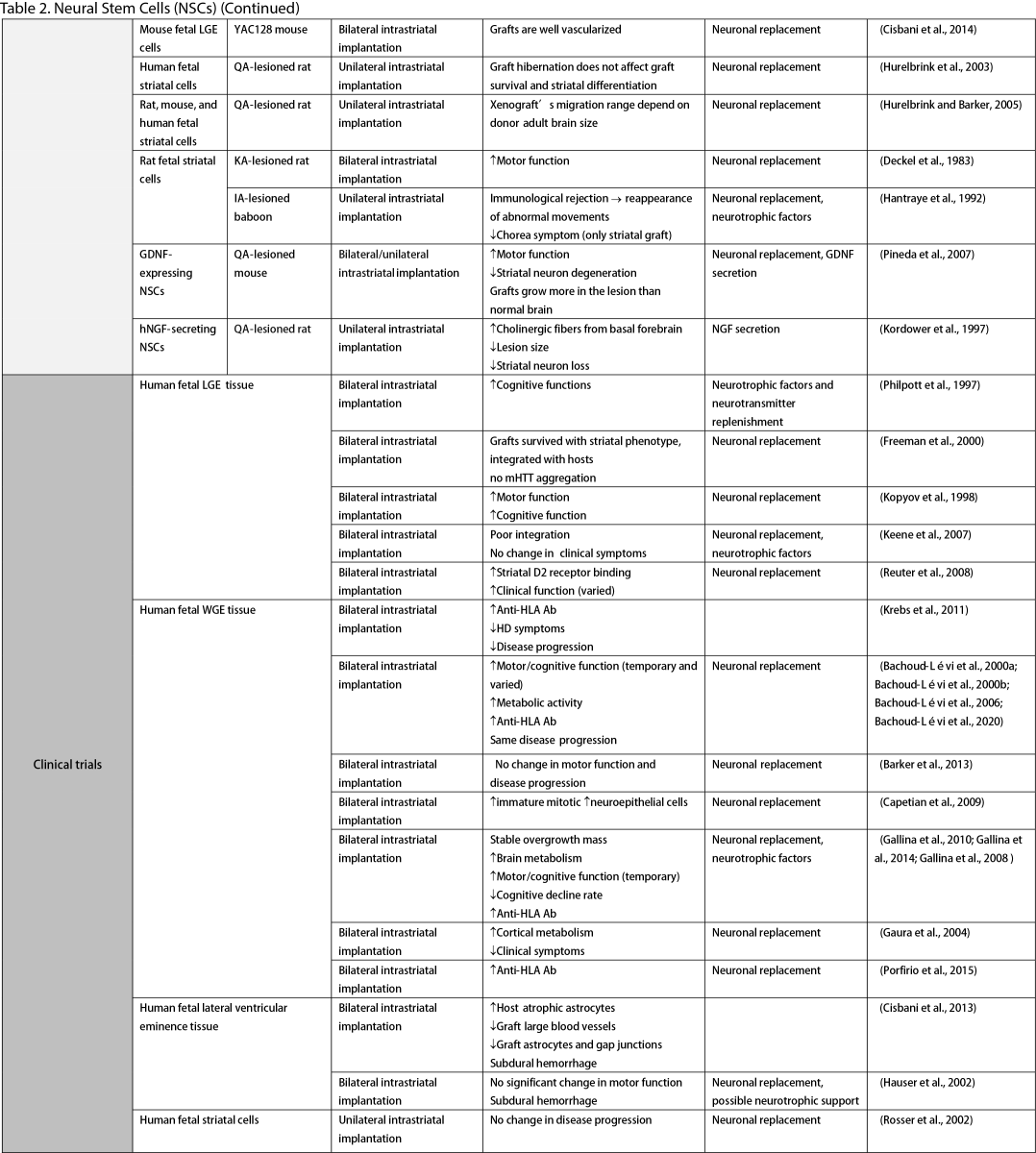
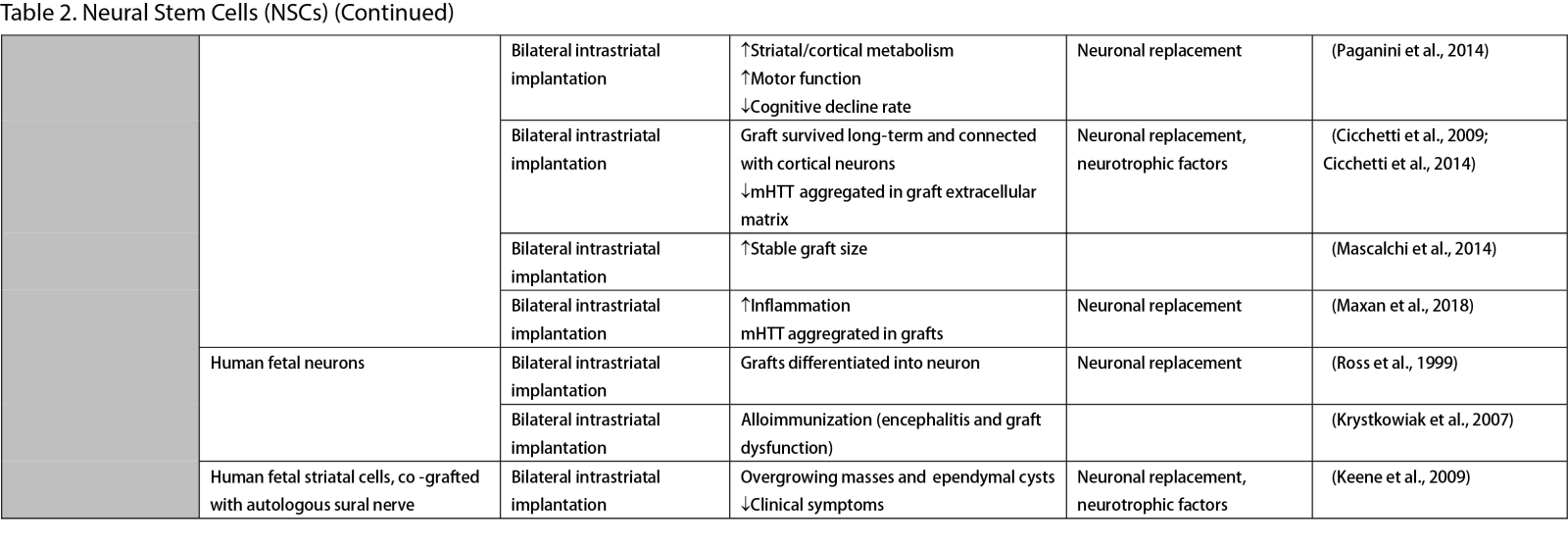
Neural Stem Cells (NSCs)
NSCs have garnered significant interest for transplantation due to their dual role in neuron replacement and neurotrophic factor secretion (Tuazon et al., 2019). The pioneering study by Deckel et al. (1983) demonstrated the potential of this approach. Indeed, rat fetal striatal tissues transplanted into the bilateral striatum of kainic acid (KA1)-injected rats showed notably fewer behavioral abnormalities and well-differentiated grafts with reduced striatal atrophy. Subsequent research predominantly focused on fetal striatal tissue, especially the subventricular zone (SVZ), whole ganglionic eminence (WGE), medial ganglionic eminence (MGE), and lateral ganglionic eminence (LGE), consistently demonstrating the amelioration of HD symptoms and robust neural differentiation. NSCs derived from ESCs and induced pluripotent stem cells (iPSCs) have shown a similar impact across various mouse models of HD (Al-Gharaibeh et al., 2017; Holley et al., 2023). However, some studies have reported no significant effects from NSC transplantation, highlighting the need for further investigation into optimal regimens (Hurelbrink et al., 2003; Jiang et al., 2011a; El-Akabawy et al., 2012). Various experiments have aimed to enhance the therapeutic effects of NSCs, including multitract implantation, optimization of transplantation timing, exploration of NSC sources, and graft storage impact (Watts et al., 1999; Hurelbrink et al., 2003; Hurelbrink and Barker, 2005; Johann et al., 2007; Kelly et al., 2007; Lelos et al., 2016). Pineda et al. (2007) and Kordower et al. (1997) engineered NSCs to overexpress glial cell line-derived neurotrophic factor (GDNF) and human nerve growth factor (NGF), achieving rescue of striatal degeneration and improvement in motor functions (Kordower et al., 1997; Pineda et al., 2007).
Clinical trials of stem cell therapy in HD have primarily involved fetal striatal tissue transplantation, with neurons derived from the WGE and SVZ harvested from elective abortions. The first pilot study of cellular transplantation in HD patients occurred in 1995 (Madrazo et al., 1995), with subsequent trials conducted in locations including Cuba, Czechoslovakia, the United Kingdom, Florida, California, and France. These trials generally reported improved cognitive and motor functions, brain metabolic activity, and disease progression rates. Post-mortem analysis also indicated robust graft survival, striatal neuron differentiation, and host-brain integration (Freeman et al., 2000). Nevertheless, some studies have shown that the benefits of neural stem cell therapy can be temporary (Bachoud-Lévi et al., 2006; Gallina et al., 2014) or even yield no significant improvement (Hauser et al., 2002; Keene et al., 2007; Barker et al., 2013; Bachoud-Lévi et al., 2020), underscoring the importance of long-term follow-up and alternative regimens that allow for continuous treatment. Despite the therapeutic effects of stem cell transplantation, several studies have reported complications, including the development of anti-human leukocyte antibodies antibodies leading to encephalitis and graft dysfunction, as well as concerns about the tumorigenesis potential of stem cells, with some patients developing overgrowing masses causing clinical deterioration (Krystkowiak et al., 2007; Gallina et al., 2008; Keene et al., 2009; Gallina et al., 2010; Krebs et al., 2011; Gallina et al., 2014; Porfirio et al., 2015; Bachoud-Lévi et al., 2020). Cisbani et al. (2013) reported that fetal striatal tissue transplantation decreased blood vessels, astrocytes, and gap junctions in grafts, raising concerns about impaired blood-brain barrier integrity resulting from stem cell therapy. Despite a well-established protocol for intrastriatal implantation, some patients have experienced procedural complications, such as subdural hemorrhage and infection, highlighting the need for careful consideration of these risks (Bachoud-Lévi, 2017; Cisbani et al., 2013).
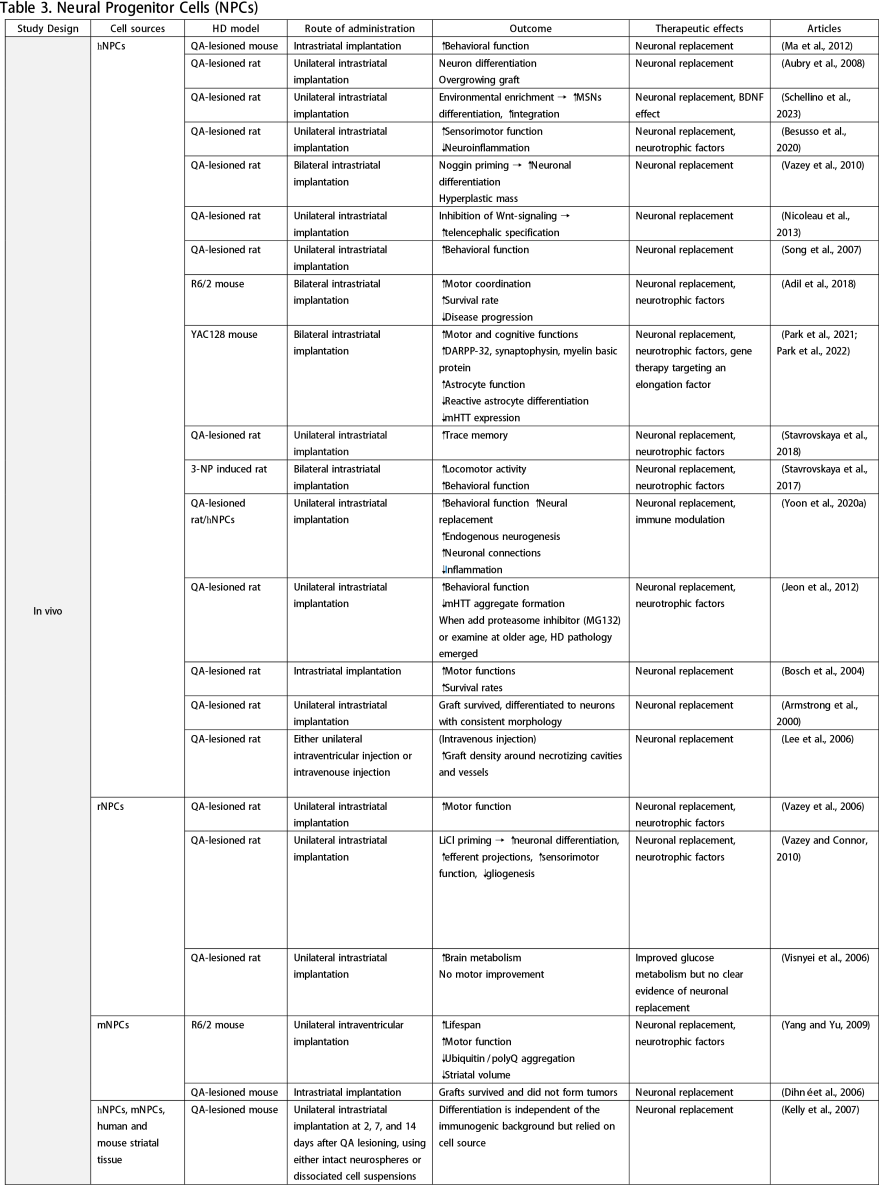
Neural Progenitor Cells (NPCs)
NPCs correspond to brain progenitor cells responsible for generating glial and neuronal cells. Unlike NSCs, NPCs do not give rise to non-neural cells. Numerous in vivo studies have utilized NPCs derived from ESCs, iPSCs, or fetal brain
tissue, demonstrating that NSC transplantation can improve clinical manifestations of HD, reduce neuroinflammation and mHTT accumulation, and enhance MSN differentiation (Aubry et al., 2008; Vazey et al., 2010; Nicoleau et al., 2013; Park et al., 2021; Park et al., 2022; Schellino et al., 2023). Various protocols have been employed to augment the therapeutic effects of NPCs, including priming with lithium chloride (LiCl) (Vazey and Connor, 2010), noggin priming (Vazey et al., 2010), combination therapy (Lee et al., 2006), graft forms (Johann et al., 2007; Kelly et al., 2007), and routes of administration (Lee et al., 2006). A study by Lee et al. (2006) compared intraventricular injection to intravenous administration of human NPCs in a QA-lesion rat model. They found that both methods effectively facilitated graft migration to the lesioned striatum, with the intravenous route resulting in higher graft density. However, concerns about tumorigenesis arose from detecting transplanted cells in other organs following systemic injection, underscoring the need for long-term observation. While some studies have explored NPCs as vehicles for gene therapy (Cho et al., 2019), such applications fall outside the scope of this review.
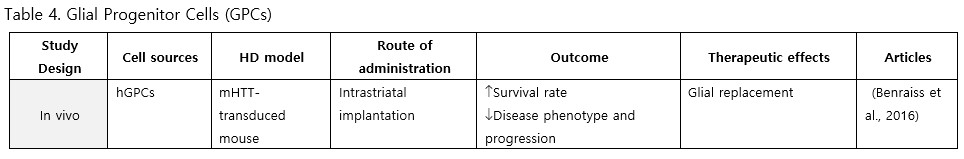
Glial Progenitor Cells (GPCs)
GPCs have received comparatively less attention than neural lineages in HD research. To date, only one study has focused on glial progenitor cells derived from hESCs. Following transplantation into HD chimera mice, the grafts rescued electrophysiological and behavioral phenotypes, maintained potassium homeostasis, decelerated disease progression, and improved survival rates (Benraiss et al., 2016). Since the pathology of HD might involve neuroinflammation, glia and GPCs-based therapy may be worth exploring to gain a complete understanding of HD pathogenesis and treatment.
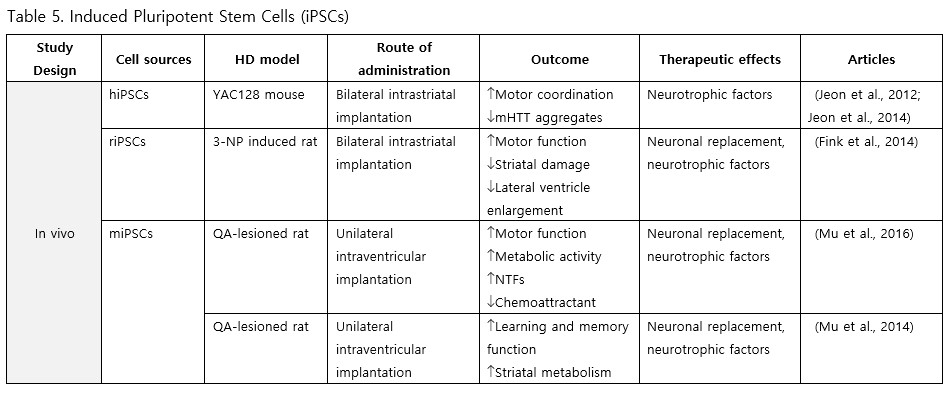
iPSCs
Fibroblast-derived iPSCs represent a viable source for stem cell therapy in HD. Studies involving intrastriatal and intraventricular implantation of these iPSCs across various mouse models have reported enhancements in motor and cognitive functions, metabolic activity, levels of neurotrophic factors, as well as reductions in mHTT aggregation, inflammation, striatal atrophy, and ventriculomegaly (Jeon et al., 2012; Fink et al., 2014; Jeon et al., 2014; Mu et al., 2014; Mu et al., 2016).
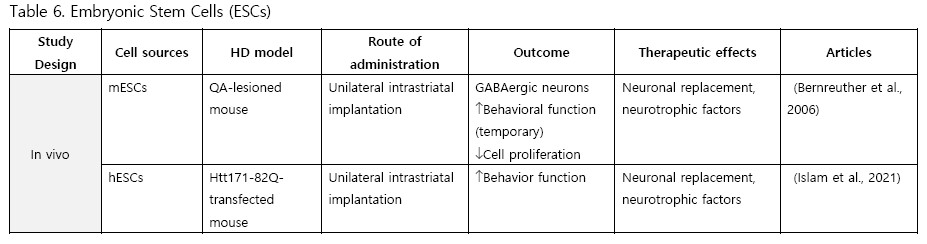
Embryonic Stem Cells (ESCs)
Comparatively few studies have utilized ESCs for transplantation. To date, only two experiments have been conducted with ESCs. Bernreuther et al. (2006) performed the first study in 2006, intrastriatally implanting murine L-1 expressing ESCs into QA-lesioned mice. They found active graft migration, an increase in GABAergic neurons, and temporary behavioral rescue. Islam et al. (2021) conducted another study using human ESCs transplanted into an HTT knock-in mouse model and observing improved behavioral function.
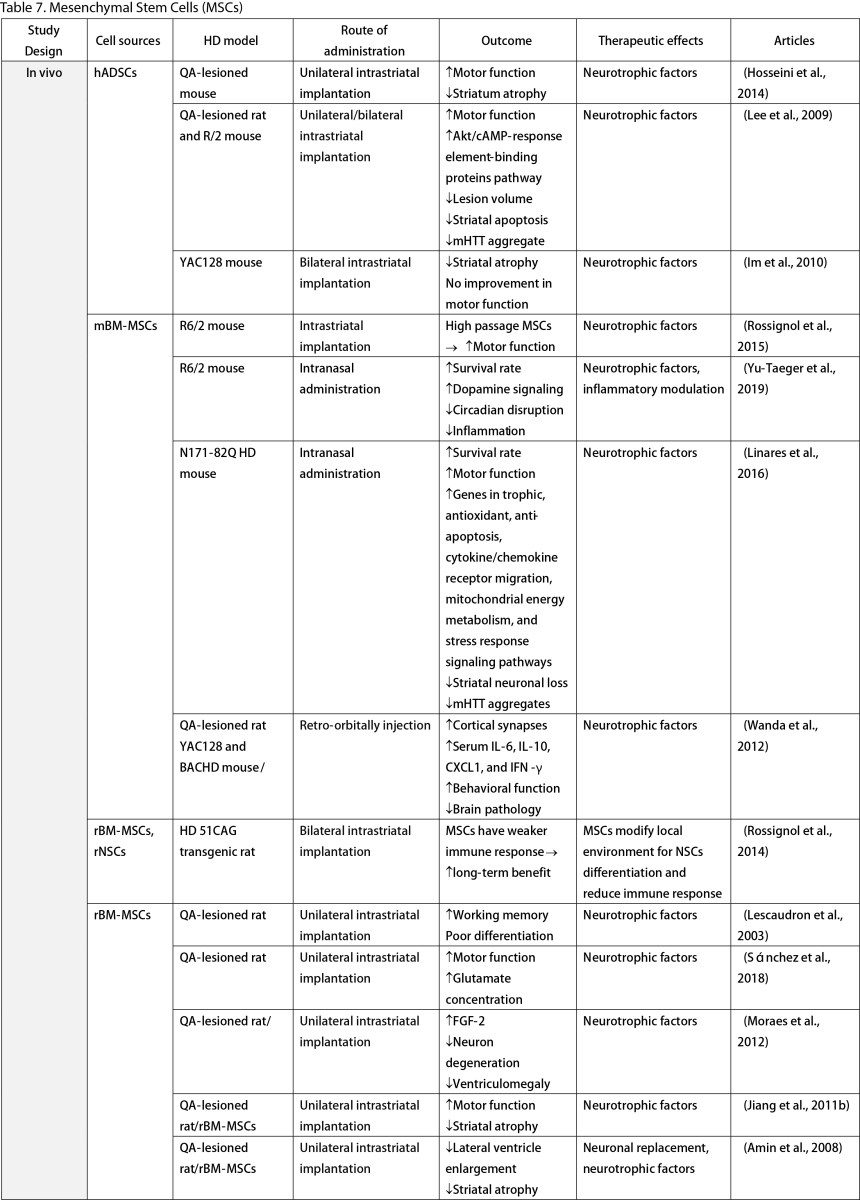
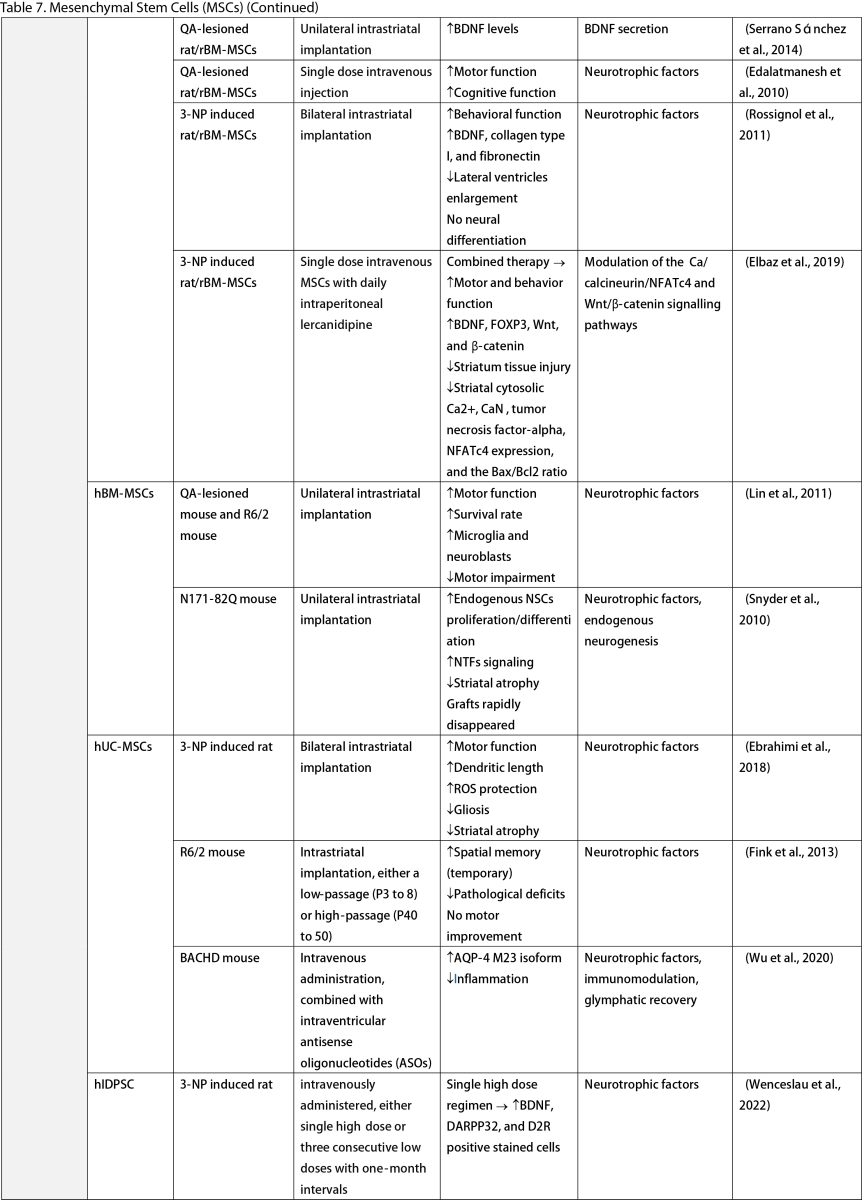
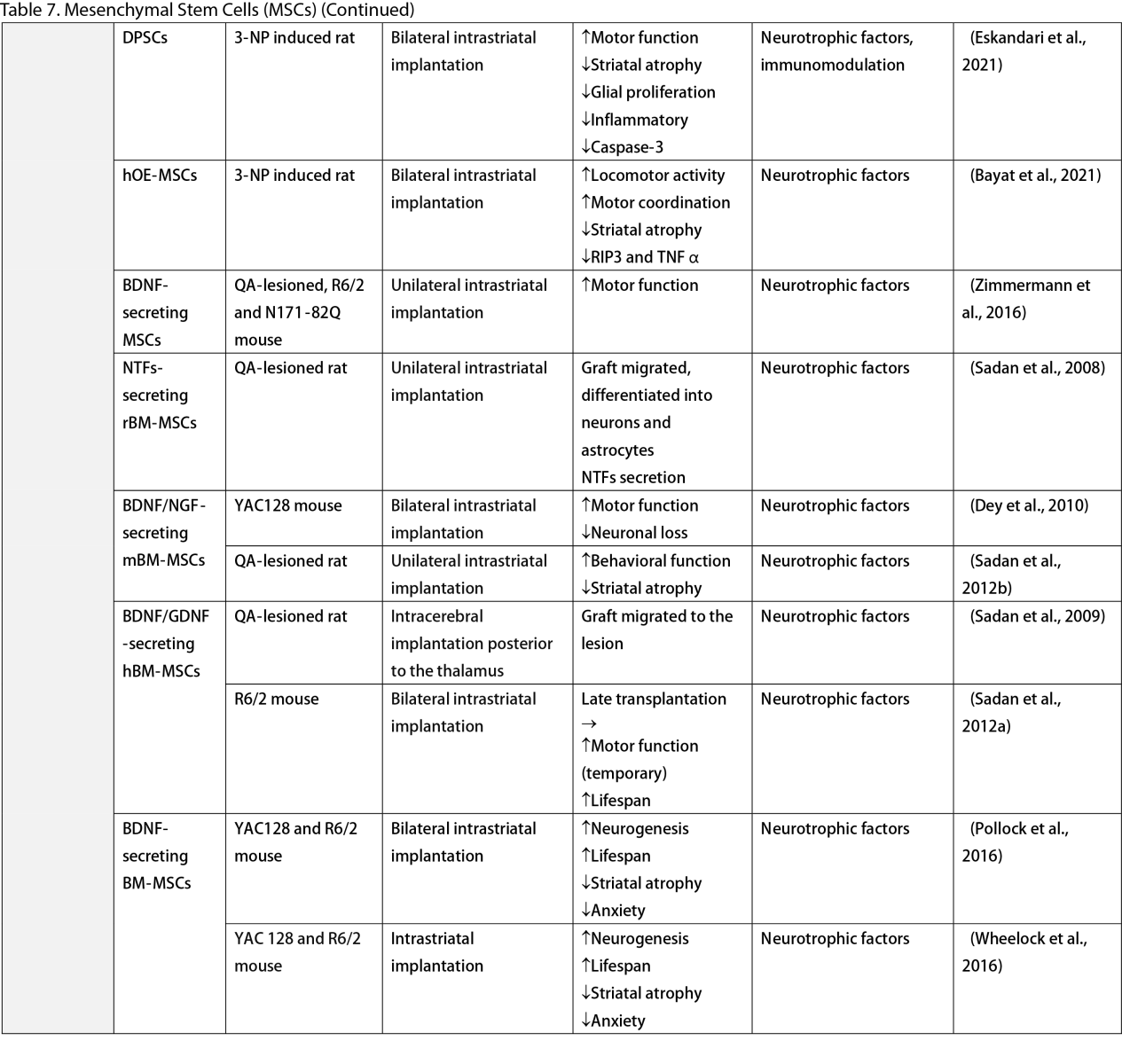
Mesenchymal Stem Cells (MSCs)
MSCs, widely researched for their therapeutic potential, are derived from various tissues such as the adipose, bone marrow, umbilical cord, dental pulp, and olfactory sheath. Research has consistently shown that MSCs, similar to other stem cells, can ameliorate behavioral and memory dysfunctions, mHTT aggregation, striatal atrophy, ventriculomegaly, and enhance neurotrophic factors (Lescaudron et al., 2003; Lee et al., 2009; Edalatmanesh et al., 2012; Moraes et al., 2012; Sánchez et al., 2018; Yu-Taeger et al., 2019). Several injection routes have been explored, including intranasal, intravenous, intrastriatal, and intraventricular. Elbaz et al. (2019) reported positive outcomes from combining intravenous MSCs with intraperitoneal lercanidipine in 3-nitropropionic acid (3-NP) induced rats. The number of cell passages is a critical factor for graft viability, as shown by Fink et al. (2013), where high-passage MSCs reduced pathological deficits and temporarily improved memory function. Wenceslau et al. (2022) found that a single high dose of intravenous human immature dental pulp stem cells significantly increased brain-derived neurotrophic factor (BDNF) levels and DARPP-32 positive neurons compared to a triple low-dose regimen. Lastly, some stem cells are engineered to overexpress neurotrophic factors. Engineering stem cells to overexpress neurotrophic factors like BDNF and GDNF enhanced their therapeutic effects by improving neurogenesis, lifespan, and disease phenotypes (Sadan et al., 2008; Dey et al., 2010; Sadan et al., 2012a; Zimmermann et al., 2016).
Unlike NSCs and NPCs, only a few clinical trials of MSCs in HD patients exist. Human dental pulp stem cells have reached clinical phases I and II, including the SAVE-DH, ADORE-DH, and ADORE-EXT trials (Macedo et al., 2021). These trials indicated that intravenous dental pulp stem cells are well tolerated and lead to significant improvement in motor symptoms in moderate HD patients. The STAR trial, a phase III clinical trial, is currently ongoing. The PRE-CELL trial from the University of California Davis is exploring engineered MSCs to overexpress BDNF in early HD patients (Wheelock et al., 2016) but is still in the participant recruitment stage.
Two primary mechanisms in neural stem cell transplantation for HD amelioration have been identified: the secretion of neurotrophic molecules and neural replacement. Previous studies have found that neural stem cells secrete various neurotrophic factors, such as NGF, BDNF, GDNF, and ciliary neurotrophic factor (CNTF), which enhance endogenous neurogenesis and reduce neuroinflammation, a key pathogenesis of HD (Conforti et al., 2018). The goal of neural replacement is to reconstruct the damaged striatum, focusing on the MSNs of the caudate/putamen, the primary neuronal population degenerating in HD (Ferrante et al., 1985). Research has convincingly shown MSN differentiation and integration into host brain circuits, indicating their regenerative potential. Furthermore, MSCs are free from ethical concerns, unlike ESCs and other fetal tissues (Kim and Park, 2017). However, possible complications highlight the need for further safety profile development.
Extracellular Vesicles (EVs)
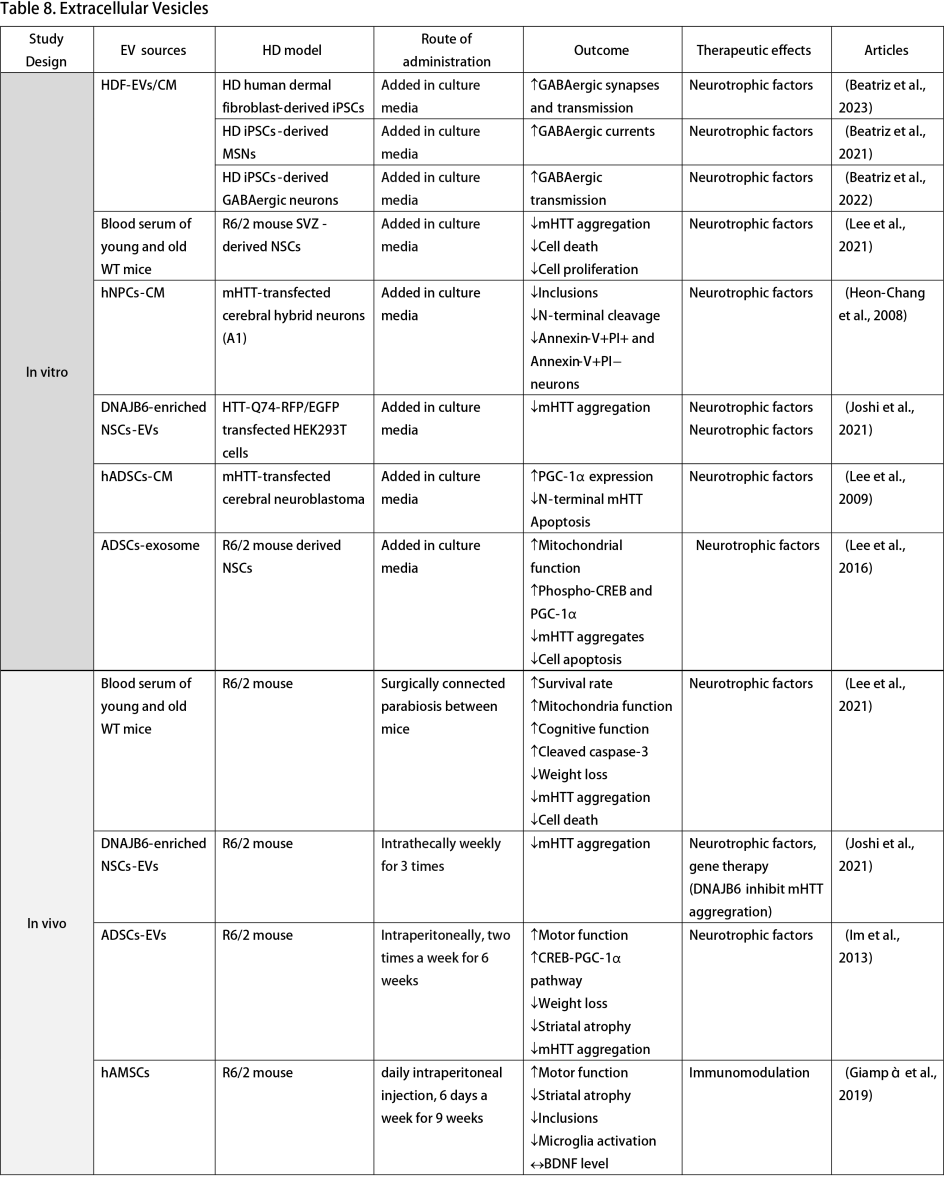
Multiple studies have explored the use of culture media from healthy cells to reverse HD phenotypes in vitro, highlighting the potential of EVs as cell-free alternatives to stem cell therapy. Human fibroblast-derived EVs increase GABAergic synapses and transmission when added to culture media of HD iPSCs and neurons (Beatriz et al., 2021; Beatriz et al., 2023). Culture media from NSCs and NPCs mitigates mHTT aggregation and prevents neuronal apoptosis in HTT knock-in cell models (Heon-Chang et al., 2008; Ma et al., 2012). Furthermore, EVs from adipose tissue-derived stem cells (ADSCs) improve mitochondrial function, phospho-cAMP response element-binding protein, and PGC-1α expression alongside disease phenotypes (Lee et al., 2009; Lee et al., 2016).
In vivo studies further affirm the positive impacts of EVs. Lee et al. (2021) conducted an experiment that established a surgical connection of blood circulation between young wild-type mice, old wild-type mice, and R6/2 HD mouse models. This simulated parasymbiosis demonstrated that blood serum from young healthy mice could enhance survival rates and mitochondrial function while reducing HD symptoms, phenotypes, and cell death (Lee et al., 2021). Extracts from MSCs and ASCs improved disease activity and motor function in the R6/2 mouse model (Im et al., 2013; Giampà et al., 2019). Joshi et al. (2021) advanced this approach by engineering NSCs to overexpress DNAJB6. Post-intrathecal administration of NSCs-derived small EVs in R6/2 mice resulted in decreased mHTT aggregation, a benefit also observed in HTT-Q74 transfected cells (Joshi et al., 2021).
The clinical trial landscape for EV therapy in HD is still in its infancy, with only an observational study by the University of Central Florida investigating the role of EVs as blood-based biomarkers for brain HTT, aiming for application in future HTT-lowering clinical trials (NCT06082713, 2023).
EVs exhibit several properties that make them particularly suitable for treating neurodegenerative diseases (D'Egidio et al., 2024). Firstly, EVs can naturally traverse the blood-brain barrier thanks to their phospholipid composition (Alvarez-Erviti et al., 2011). EVs can also protect their cargo from enzymatic degradation, ensuring that therapeutic molecules remain biologically active upon reaching their target cells. Secondly, EVs demonstrate low immunogenicity and toxicity. They can be administrated intravenously, significantly reducing the risk of procedural complications. Additionally, the potential for tumor growth is minimized because EVs primarily facilitate the delivery of neurotrophic factors instead of actual stem cells. Lastly, EVs can be specifically engineered to target distinct cells or tissues, thereby increasing the specificity and effectiveness of the treatment. These benefits set EV therapy apart from direct stem cell transplantation, presenting a cell-free option that reduces risks inherent in cell-based therapies while leveraging the advantageous effects of stem cell secretomes.
Conclusion
HD represents a profound neurological challenge, currently without effective treatment options. Cell and EV therapies have emerged as promising avenues as treatment for neurodegenerative diseases such as Alzheimer’s disease (Duan et al., 2023; Garcia-Contreras et al., 2023), Parkinson’s disease (Upadhya et al., 2021; Shastry et al., 2023), multiple sclerosis (Islam et al., 2023; Barabadi et al., 2024), and stroke (Park et al., 2020; Zhao et al., 2023). In the HD context, these approaches can potentially shift the focus from mere symptomatic relief to a reversal of HD. The literature of the last decades depicts the effects of cell therapy in HD models well, describing the complex interconnection between stem cell properties and the cellular and molecular contexts in the recipient. Indeed, the content of the stem cell secretome can positively modulate the diseased environment, causing, for instance, reduced oxidative stress, eventually via mitochondrial transfer, and neuroinflammation via secretion of inflammatory mediators that also potentially impact the surrounding glial cells. Moreover, cell therapy can be exploited to improve neurogenesis, and eventually, the stem cell secretome can improve the functional and electrical integration of neuronal cells. However, cell therapy brings significant risks, including surgical complications, alloimmunization, and the development of overgrowing masses, highlighting the necessity for safer therapeutic alternatives. EV therapy offers a promising cell-free alternative, potentially mitigating the risks associated with cell transplantation while leveraging the benefits of cellular communication for therapeutic purposes. In fact, EVs represent the principal actors mainly responsible for the assessed therapeutic effects within all the components of the stem cell secretome. Moreover, the possibility of optimizing engineered EVs as carriers of therapeutic molecules underscores their potential as therapeutic instruments in HD and beyond. Although there are many stem cell transplantation studies, clinical trials for EVs have not yet been performed. More studies of EVs' safety profile and efficacy in HD are needed before moving to the next stage.
The journey towards effective treatments for HD is complex and requires further extensive research to address these gaps. As research continues, there is hope for developing therapies that can manage or even cure HD, offering new possibilities for those affected by this debilitating condition.
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
Napasiri Putthanbut1,2
1Center of Aging and Brain Repair, Department of Neurosurgery, University of South Florida. 2Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University.
Francesco D'Egidio3
3Department of Life, Health and Environmental Sciences, University of L'Aquila.
Jea-Young Lee1
1Center of Aging and Brain Repair, Department of Neurosurgery, University of South Florida.
Napasiri Putthanbut and Francesco D'Egidio contributed equally to this article.
Corresponding author:
Jea-Young Lee
Email: jeayoung@usf.edu
In a new window | Download PPT
Figure 1. HD is a neurodegenerative disorder characterized by motor and cognitive dysfunctions and a shortened lifespan. Novel therapies, including cell and extracellular vesicle therapy, can improve HD phenotypes and clinical symptoms, opening a new conditioning medicine opportunity for HD treatment.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 10232 | 24 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA