Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Insights into the selective tolerance of oculomotor units to degeneration in ALS
Time:2024-11-20
Number:6107
Author Affiliations

Conditioning Medicine 2024. 7(1): 22-25.
Abstract
Neurodegenerative diseases are characterized by selective loss of specific neuronal populations, with the lack of the corresponding function defining the patient’s clinical outcome. Among these, motor neuron diseases (MNDs) are progressive and multifarious pathologies caused by the loss and death of upper motor neurons (MNs), lower motor neurons, or both. Therefore, patients display progressive and prominent muscle weakness, wasting, fasciculation, loss of skeletal muscle movements, cramping, and spastic paralysis (Maranzano et al., 2023). MNDs provide an example of selective vulnerability at multiple levels. Indeed, even if a single neuronal class is mainly affected, a widely varying degree of degeneration can be noticed within different subpopulations of the same functional class. Amyotrophic lateral sclerosis (ALS) represents a typical MND characterized by such multi-level differential vulnerability. In ALS, both upper and lower motor neurons die, but individual motor pools – motor neurons innervating extraocular, pelvic sphincter, and slow limb muscles – are relatively resistant to degeneration (Kaplan et al., 2014). In particular, ocular motor neurons and corresponding extraocular muscle sparing is a common feature of ALS and other MNDs, such as spinal muscular atrophy, spinobulbar muscular atrophy, and many forms of muscular dystrophy (Hedlund et al., 2010; Kallestad et al., 2011). In light of these premises, identifying intrinsic characteristics that confer motor units’ tolerance to degeneration could provide insight into the pathogenetic mechanisms underlying these pathologies and an important cue to prospect novel therapeutic intervention strategies.
Keywords: Neurodegeneration, ALS, Ocular motor neurons, Extraocular muscles, Tolerance
Abstract
Neurodegenerative diseases are characterized by selective loss of specific neuronal populations, with the lack of the corresponding function defining the patient’s clinical outcome. Among these, motor neuron diseases (MNDs) are progressive and multifarious pathologies caused by the loss and death of upper motor neurons (MNs), lower motor neurons, or both. Therefore, patients display progressive and prominent muscle weakness, wasting, fasciculation, loss of skeletal muscle movements, cramping, and spastic paralysis (Maranzano et al., 2023). MNDs provide an example of selective vulnerability at multiple levels. Indeed, even if a single neuronal class is mainly affected, a widely varying degree of degeneration can be noticed within different subpopulations of the same functional class. Amyotrophic lateral sclerosis (ALS) represents a typical MND characterized by such multi-level differential vulnerability. In ALS, both upper and lower motor neurons die, but individual motor pools – motor neurons innervating extraocular, pelvic sphincter, and slow limb muscles – are relatively resistant to degeneration (Kaplan et al., 2014). In particular, ocular motor neurons and corresponding extraocular muscle sparing is a common feature of ALS and other MNDs, such as spinal muscular atrophy, spinobulbar muscular atrophy, and many forms of muscular dystrophy (Hedlund et al., 2010; Kallestad et al., 2011). In light of these premises, identifying intrinsic characteristics that confer motor units’ tolerance to degeneration could provide insight into the pathogenetic mechanisms underlying these pathologies and an important cue to prospect novel therapeutic intervention strategies.
Keywords: Neurodegeneration, ALS, Ocular motor neurons, Extraocular muscles, Tolerance
Highlights
Ocular motor units show relative resistance to degeneration in amyotrophic lateral sclerosis (ALS) as well as in other motor neuron diseases and aging. Ocular motor neurons and related extraocular muscles possess different molecular signatures compared to other motor units undergoing early degeneration in ALS. Unraveling determinants of selective susceptibility among different motor units could help change their responsiveness to the disease.
Introduction
ALS is a progressive, devastating, and fatal neurodegenerative disease that primarily affects motor neurons of the motor cortex, brainstem motor nuclei, and anterior horn of the spinal cord (Ragagnin et al., 2019). As a result, progressive degeneration and atrophy of voluntary skeletal muscles occur, ultimately resulting in paralysis (Ferrer et al., 2017). ALS worldwide prevalence and incidence are estimated to be 4.42 per 100,000 population and 1.59 per 100,000 person-years (Ilieva et al., 2023). ALS commonly begins in late- adulthood and it often has a focal onset: in fact, several clinical subsets can be distinguished by the anatomical site of the appearance of symptoms - bulbar or limb onset (Ragagnin et al., 2019). However, the disease subsequently spreads to different body regions, progressing relentlessly in most patients, and death occurs in a median of about three years, generally due to respiratory failure (Masrori and Van Damme, 2020). ALS appears sporadic in the majority of cases (sALS) but demonstrates inheritance (fALS) in 10% of cases due to mutations in, e.g., superoxide dismutase 1 (SOD1), TDP-43, FUS, and C90RF72 (Nijssen et al., 2017). The reason why motor neurons (MNs) are specifically targeted in ALS remains unclear, but remarkably, they are not all equally affected. Ocular motor neurons (OMNs) in the brainstem and Onuf’s nuclei MNs in the lumbosacral spinal cord are relatively resistant to degeneration, being spared until the disease's end stage. As a result, patients retain normal visual, sexual and bladder function throughout the disease course (Kaplan et al., 2014; Ragagnin et al., 2019). Additionally, there is a gradient of susceptibility also among spinal MNs. Fast fatigable (FF) MNs, the largest involved in short-lasting bouts of forceful contraction, are most severely affected and degenerate first, whereas slow (S) MNs, which are smaller and fire regularly to control postural muscles, are more resistant to degeneration (Kaplan et al., 2014; Nijssen et al., 2017; Ragagnin et al., 2019) (Figure 1).
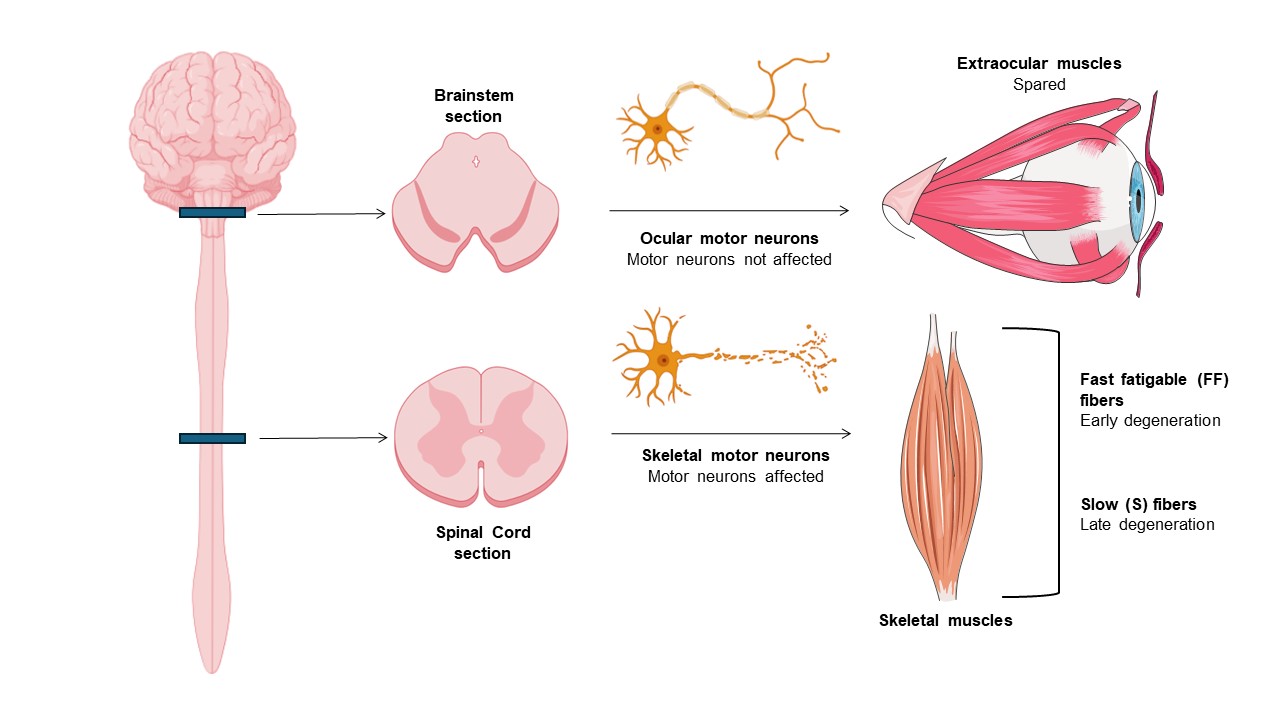
In a new window | Download PPT
Figure 1. Tolerance of oculomotor units in ALS. In ALS, ocular MNs and related extraocular muscles are spared until the disease’s end stage, whereas spinal MNs and associated muscles degenerate at different timepoints. Fast fatigable (FF) motor units degenerate first, while slow motor units (S) are affected later in the disease.
Ocular motor neurons
OMNs regulate eye movement thanks to neurons of the oculomotor (III), trochlear (IV), and abducens (VI) nuclei located in the midbrain and brainstem (Kaplan et al., 2014). They differ from other MNs in many respects, including their embryonic origin, anatomical location, structure, and function (Horn and Straka, 2021). Numerous studies have endeavored to define OMNs molecular signature to understand their known resilience to degeneration. With this purpose, microarray analysis and laser capture microdissection of OMNs compared to susceptible MNs have shown that they have distinct transcriptional profiles, revealing enriched transcripts for each population and, consequently, distinct protein signatures.
A pioneering study using SOD1G93A rats identified several genes as selectively expressed within motor neurons of oculomotor (III) and trochlear (IV) nuclei, with potential protective roles, such as Glucy1a3, early growth response protein 1, insulin growth factor 2 (IGF-2), and guanine deaminase (Hedlund et al., 2010). In particular, IGF-2 was shown to also be preferentially expressed in ocular motor neurons of SOD1G93A mice and end-stage ALS patients. More interestingly, viral delivery of IGF-2 into the hindlimb quadriceps and thoracic muscles of SOD1G93A mice promotes motor nerve regeneration and increases their lifespan (Allodi et al., 2016), confirming that differentially expressed genes between different MN subtypes may determine their responsiveness to disease, and their manipulation can protect vulnerable subpopulations from degeneration.
Microarray analysis from ocular and spinal cord MNs of post-mortem tissue from neurologically normal human controls revealed markedly different transcriptional profiles. The most significant differences were observed in genes involved in synaptic transmission, particularly those encoding γ-aminobutyric acid (GABA) and glutamate receptor subunits (Brockington et al., 2013). GluR2 and GABRA1 subunits were upregulated in ocular compared to spinal MNs, and these results showed concordance in mouse and rat studies (Brockington et al., 2013; Nijssen et al., 2017).
Similarly, other genes highly expressed in vulnerable MNs were linked to their susceptibility to degeneration. Of relevance, matrix metalloproteinase (MMP)-9 was shown to be strongly expressed only by fast MNs that undergo degeneration (but absent in ocular, Onuf’s nuclei, and S MNs) in ALS and that its ablation or partial reduction led to ~80%-delay in denervation and 25% increase in lifespan in SOD1G93A mice (Kaplan et al., 2014).
Altogether, these results show that MN subpopulations with different susceptibility to degeneration possess significant differences in their molecular signature, and they can be used to modify the fate of MNs destined to die early in the disease (Table 1).
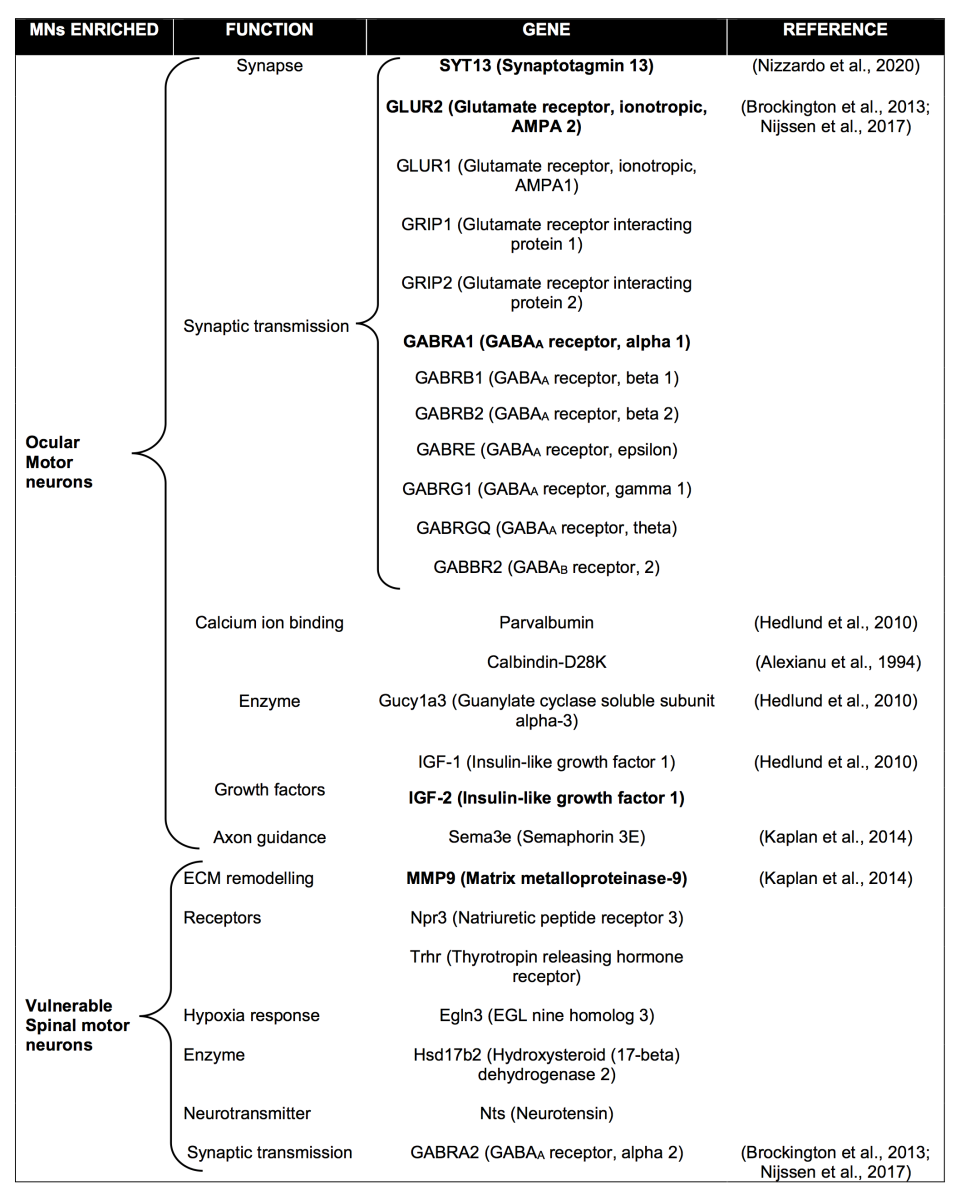
Extraocular muscles
As mentioned above, extraocular muscles are spared in ALS, so patients can use eye-tracking devices to communicate when muscles necessary for speech have degenerated (Comley et al., 2016). Extraocular muscles are responsible for the complex range of voluntary and reflexive eye movements, which includes smooth pursuit, vergence eye movements, saccades, gaze holding, optokinetic reflex, vestibulo-ocular and cervico-ocular reflexes, and spino-ocular motor coupling (Horn and Straka, 2021). All vertebrates, except for jawless vertebrates, have six extraocular muscles: four recti (superior, inferior, medial, and lateral) and two obliques (superior and inferior) (Spencer and Porter, 2006; Horn and Straka, 2021), while the levator palpebrae superior muscle is present in mammals (Spencer & Porter, 2006). The superior, medial, inferior recti and the inferior oblique are innervated by the oculomotor nucleus (III); the superior oblique is innervated by the trochlear nucleus (IV); the lateral rectus is innervated by the abducens (VI) (Nijssen et al., 2017).
EOMs have such unique properties that the term “allotype” has been attributed to their niche (Ketterer et al., 2010). They differ from other muscles in several aspects: embryologic origin, structural, and molecular characteristics (Nijssen et al., 2017; Bohlen et al., 2019; Horn and Straka, 2021; Kablar, 2023). In particular, EOMs have a specific embryologic origin from the prechordal and cranial paraxial mesoderm, while limb and trunk muscles originate from the somitic and lateral plate mesoderm (Spencer and Porter, 2006; Horn and Straka, 2021). Structurally, they are characterized by two layers, an external orbital and an internal global layer, both possessing individual characteristics. Unlike the four highly conserved muscle fiber types found in skeletal muscles (I, IIA, IIX, and IIB), EOMs are characterized by at least six fiber types, which are classified into three categories based on their pattern of innervation: singly innervated (twitch) fibers (SIFs) and two types of multiply innervated (nontwitch) fibers (MIFs), based on their location within the orbital or global layers (Spencer and Porter, 2006). SIFs are contacted at the center of the fiber by en-plaque endplates, and they respond to the typical “all-or-nothing” rapid twitch response. They account for 90% and 80% of fibers in the global and orbital layers, respectively. On the other hand, MIFs, which represent the remaining percentage of EOM fibers, have smaller endplates located in distinct, more distal bands of the muscle, forming the so-called “en-grappe” endplates and they undergo graded tonic contractions responsible for fine modulation of eye movement. MIFs of the orbital layer (20%) form typical “en-grappe” endplates but have additional en-plaque endings with corresponding contraction dynamics (Nijssen et al., 2017; Horn and Straka, 2021). Moreover, EOMs express 11 myosin heavy chain (MyHC) isoforms, including the four classical isoforms of limb muscles, three subtypes found in developing muscles, three EOM-specific subtypes, and one non-muscle MyHC (Horn and Straka, 2021). In contrast with skeletal muscle, they may co-express different MyHC isoforms in a single myofiber, accounting for the unique contractile properties of these muscles. Furthermore, unlike limb muscles, EOMs have higher proliferative capacity as they maintain a population of activated satellite cells even in aging, resulting in myofiber remodeling throughout life (Kallestad et al., 2011).
Not surprisingly, similar to OMNs in relation to other MNs, EOMs have a distinct molecular signature when compared to other skeletal muscles. This has been confirmed both in human and murine model studies. In particular, analysis of human EOM transcriptome using oligonucleotide-based expression profiling highlighted 338 differentially expressed genes (DEGs) in EOMs when compared to the quadriceps femoris (QF) limb muscle. These genes belonged to different functional groups, e.g., protein biosynthesis, intracellular signaling, sarcomeric elements, energy metabolism, and growth/development/regeneration (Fischer et al., 2005). On the other hand, an expression profile study of rat EOMs and tibialis anterior (TA) - a fast limb skeletal muscle - neuromuscular junctions (NMJs) revealed enriched genes for each synaptic region (Ketterer et al., 2010).
Taken together, all the features that distinguish EOMs from other muscle types may imply molecular, biochemical, and physiological characteristics, which are worth investigating and that could explain their singular resilience in different MN degenerative diseases and aging.
Conclusions and future prospectives
To date, no effective cure for ALS is available. Since it is well known that some motor unit subpopulations show high resistance to degeneration in this pathology, it has been hypothesized that intrinsic properties of specific subtypes of MNs and related muscles may somehow change their responsiveness to disease and so provide insight for novel therapeutic targets for its treatment.
An intense debate regarding ALS is due to the controversial question of where the disease begins: if it starts from cortical regions - a mechanism known as the dying forward hypothesis. - or if it starts from muscle cells and the NMJs - “dying back hypothesis”. Even if the answer remains debated, and there are many arguments in favor of each hypothesis, studies from murine models and patients have demonstrated that muscles and NMJs are targeted early in the disease (Ragagnin et al., 2019).
Consistent with this evidence and considering the singular and peculiar characteristics of EOMs, investigating and identifying molecular elements that can distinguish them from other muscles that undergo degeneration could give information about pathologic mechanisms in muscles and in relation to the specific classes of neurons they are matched with. Furthermore, this strategy could open the way to new potential therapeutic approaches for a wide range of MNDs.
Conflict of interests
The authors declare no conflict of interest. Giuseppe Pignataro is on the Editorial Board for Conditioning Medicine and he has not participated in the editorial review of this manuscript.
References
Xhesika Kolici1,2
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy. 2School of Advanced Studies, Center for Neuroscience, University of Camerino
Valeria Valsecchi1
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.
Giusy Laudati1
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.
Giuseppe Pignataro1
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.
Corresponding author:
Xhesika Kolici
Email: xhesikakolici@libero.it/xhesika.kolici@unicam.it
or
Giuseppe Pignataro
Email: giuseppe.pignataro@unina.it
In a new window | Download PPT
Figure 1. Tolerance of oculomotor units in ALS. In ALS, ocular MNs and related extraocular muscles are spared until the disease’s end stage, whereas spinal MNs and associated muscles degenerate at different timepoints. Fast fatigable (FF) motor units degenerate first, while slow motor units (S) are affected later in the disease.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 6107 | 46 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA