Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Modified neural peptides as a potent therapy for neurodegenerative diseases
Time:2025-07-22
Number:16589
Author Affiliations

Conditioning Medicine 2024. 7(2): 41-47.
Abstract
Neurodegenerative diseases are a major health problem worldwide. Statistics suggest that in 2030, in America alone, there will be more than 12 million people who will present with a neurodegenerative pathology. Despite the multifactorial etiology of each neurodegenerative disease, all of them share highly marked immune and inflammatory components in their pathophysiology. Different immunomodulatory therapies have been suggested, particularly immunization with modified neural peptides, which have been introduced as a novel treatment over the last decade. Generally, this neural peptide therapy involves inoculating the central nervous system using natural constituents with minor biochemical modifications to boost protective autoimmunity, producing an autoreactive response that, instead of creating brain damage, induces a healing environment. Immunization with modified neural peptides has demonstrated encouraging results both in vitro and in vivo in multiple models of neurodegenerative diseases. In this review, we analyze the therapeutic effects of modified neural peptides in preclinical models of commonly diagnosed neurodegenerative disorders, namely Alzheimer’s disease, Parkinson’s disease, and stroke. Our goal is to highlight hallmark discoveries of modified neural peptides and identify gaps in knowledge that would allow safe and effective applications of these peptides to treat neurodegenerative disorders.
Keywords: Neurodegenerative diseases, Modified neural peptides, Alzheimer's disease, Parkinson's disease, Stroke
Abstract
Neurodegenerative diseases are a major health problem worldwide. Statistics suggest that in 2030, in America alone, there will be more than 12 million people who will present with a neurodegenerative pathology. Despite the multifactorial etiology of each neurodegenerative disease, all of them share highly marked immune and inflammatory components in their pathophysiology. Different immunomodulatory therapies have been suggested, particularly immunization with modified neural peptides, which have been introduced as a novel treatment over the last decade. Generally, this neural peptide therapy involves inoculating the central nervous system using natural constituents with minor biochemical modifications to boost protective autoimmunity, producing an autoreactive response that, instead of creating brain damage, induces a healing environment. Immunization with modified neural peptides has demonstrated encouraging results both in vitro and in vivo in multiple models of neurodegenerative diseases. In this review, we analyze the therapeutic effects of modified neural peptides in preclinical models of commonly diagnosed neurodegenerative disorders, namely Alzheimer’s disease, Parkinson’s disease, and stroke. Our goal is to highlight hallmark discoveries of modified neural peptides and identify gaps in knowledge that would allow safe and effective applications of these peptides to treat neurodegenerative disorders.
Keywords: Neurodegenerative diseases, Modified neural peptides, Alzheimer's disease, Parkinson's disease, Stroke
Highlights
We review the scientific evidence supporting modified neural peptide therapy as a promising interention for modulating immune responses towards an anti-inflammatory and self-repairing phenotype, potentially offering neuroprotection and regenerative effects in neurodegenerative diseases, specifically Parkinson's disease, Alzheimer's disease, and stroke. Immunization with modified neural peptides may represent a novel conditioning medicine approach to proactively strengthen the immune system against deleterious inflammatory responses associated with neurodegenerative diseases. Overall, this review highlights the urgent need for effective treatments against neurodegeneration and suggests modified neural peptide immunization as a promising conditioning medicine therapeutic intervention.
Introduction
Neurodegenerative disease (NDD) is a term commonly used to describe neurological disorders that compromise normal brain structure and physiology with progressive neurovascular damage following the onset of the disease. Public understanding of NDDs usually associates this term with chronic, non-curable, dementia-leading pathologies such as Alzheimer's Disease (AD). Nevertheless, the term neurodegeneration specifically alludes to brain damage that diminishes neurological function from a basal level of normal performance to a deficient condition (Kovacs, 2017). Thus, NDDs can be defined as brain disorders with impaired neurovascular units and reduced neurological capacity. This set of pathologies can be chronic, like AD or Parkinson's Disease (PD), as well as acute, such as stroke (Kovacs, 2017; Giri et al., 2024). NDDs are a health problem worldwide. In America alone, it is estimated that by the year 2030, there will be more than 12 million cases. As NDDs are closely related, their incidence is expected to rise accordingly as life expectancy increases (Kontis et al., 2017).
NDDs vary in etiology and clinical presentation. Nevertheless, neuroinflammation has been identified as a key element of every NDD pathophysiology. A fundamental inflammatory model applied to NDDs stipulates that the initial anti-inflammatory response to the brain injury helps to heal and limit the primary insult. Unfortunately, the anti-inflammatory response is usually overtaken by deleterious pro-inflammatory reactions (Iadecola, 2017; Cai et al., 2017), including microglial secretion of cytokines such as tumor necrosis factor-α, interleukin (IL)-1β, cluster of differentiation 1, IL-6, vascular adhesion proteins, and chemoattractants, among others (Subhramanyam et al., 2019), and activation of macrophages under an M1 phenotype that produces reactive lymphocyte infiltration, reactive oxygen species, oxidative stress, and a pro-apoptotic state (Subhramanyam et al., 2019; Hickman et al., 2018). Under these detrimental inflammatory conditions, the neurovascular unit begins to deteriorate, combined with other cell death mechanisms such as aberrant oxidative stress, mainly due to glutamate overproduction and NMDA receptor overstimulation (Yu et al., 2020). Similarly, astrocytes suffer from excitotoxicity that increases glutamate and Ca2+ release (Iadecola, 2017; Yu et al., 2020). Endothelial cell dysfunction leads to impaired nitric oxide-mediated vasodilation, and vascular cells release matrix metalloproteinases (MMPs) that lead to degradation of the extracellular matrix and loss of tight junction structure, altogether initiating blood-brain barrier (BBB) dysfunction and leakage (Thurgur and Pintaux, 2019).
Different NDD therapies have been investigated over the years, mainly because most current therapies purely produce symptomatic relief, with very few able to limit the expansion of tissue damage (Ridolfi and Abel, 2017). By recognizing neuroinflammation as a key element of NDD progression, finding a treatment that modulates inflammation may prove effective as a disease-modifying therapy. Immunization with modified neural peptides (MNPs) is a novel therapy targeting the inflammation associated with NDDs (Carniglia et al., 2017; Petrella et al., 2019). MNP therapy consists of inoculating with peptides that provide beneficial immune functions to the brain (Carniglia et al., 2017; Petrella et al., 2019). In this article, we present a brief review of MNPs and their potent therapeutic effects in preclinical models for AD, PD, and stroke.
Modified Neural Peptides
MNPs are short immunogenic amino acid sequences with modifications in their biochemical structure to confer efficient modulation of the immune response. The specific variation in the amino acid sequence of MNPs allows the peptides to bind with and trigger the T cell receptor to exhibit a state of anergy or even into an active anti-inflammatory phenotype (Rodríguez-Barrera et al., 2017; Monroy et al., 2023). MNPs produce their effects by modulating neuroinflammation rather than completely eradicating the immune response, which makes them unique compared to other immunomodulatory approaches. MNP therapy generates an autoreactive state in the protective anti-inflammatory phenotype. As noted above, the established cascade of inflammation-mediated cell death in NDDs consists of an initial acute anti-inflammatory response that is subsequently substituted by a chronic pro-inflammatory reaction, diminishing anti-inflammatory effects (Suárez-Meade et al., 2015; Wei et al., 2020). Accordingly, finding a strategy that sustains that therapeutic anti-inflammatory response over time and retards the onset of the harmful pro-inflammatory reaction will likely benefit NDDs. This sustained anti-inflammatory response appears to be achieved by MNP inoculation.
MNPs have shown promising effects against NDDs by inducing beneficial morphological and functional changes in in vitro and in vivo models of NDDs. Some MNPs exert their function using adaptive immunity, following a vaccine model that involves sensitizing the immune system against structural components of the central nervous system that manifest in NDDs. Thus, when the pathology actually takes place, the previous sensitization triggers an anti-inflammatory response characterized by elevated human type 2 helper (Th2) lymphocytes that promote a conducive microenvironment for brain regeneration. Furthermore, activated Th2 lymphocytes can create a neuroprotective microenvironment that may regulate secondary damage even days or weeks after the acute injury, thereby proactively preventing the persistent damage due to chronic neuroinflammation (García et al., 2019). By promoting a Th2 response, microglial cells go through T cell-mediated regulation, elevated IL-10 expression, and transforming growth-factor beta upregulation, while lipid peroxidation, free radical production, and apoptosis are halted (Parra et al., 2020). This MNP-sustained activation of the anti-inflammatory phenotype leads to the release of therapeutic molecules that promote neuroprotection and neuroregeneration, thereby affording a state of anergy that avoids the development of autoimmune diseases, i.e., aberrant chronic pro-inflammatory reactions.
Indeed, accumulating evidence demonstrates MNPs' capacity to modulate the immune response in NDDs and shift the pro-inflammatory state to an anti-inflammatory phenotype, which could lead to retardation of the initial injury and sequestration of the secondary injury or chronic progression, ultimately reducing the pathological symptoms of NDDs (Rodríguez-Barrera et al., 2017; Westwell and Berchere, 2019). The subsequent sections provide specific examples of successful MNP treatment in NDDs and identify remaining lab-to-clinic obstacles that warrant further investigations.
Alzheimer's disease
AD is a major health concern that affects people all over the world (Høgh, 2012). It is a progressive NDD that causes cognitive deterioration and is the leading cause of dementia (Lane et al., 2018). Understanding the role of two peptides that play a major part in the disease pathogenesis may allow key insights into AD etiology. Amyloid-β (Aβ) is an aberrant peptide of 39-43 amino acids that results from the misfolding of the α-helical domains into β-sheet structures (Hane et al., 2017). Aβ is produced by the breakdown of Aβ precursor protein (AβPP), which regulates cell survival, proliferation, motility, cholesterol binding, and metal ion homeostasis under physiological settings (Hane et al., 2017). The Aβ amyloidogenic pieces oligomerize and form aggregates after being cleaved by secretase enzymes, eventually depositing and accumulating to form plaques. This oligomerization activates several kinases (Tiwari et al., 2019). Aβ oligomers can cause synapse loss and dysfunction (Jeong, 2017). Alterations in ion channels, poor calcium homeostasis, increased mitochondrial oxidative stress, and decreased energy metabolism are other negative outcomes triggered by this oligomerization (Tiwari et al., 2019).
Tau protein, a microtubule-associated protein present in axons whose role is to promote cytoskeleton stability, is the other major molecule involved in AD pathogenesis (Hane et al., 2017; Naseri et al., 2019). Tau hyperphosphorylation is favored by the abundance of kinases released by Aβ, resulting in its oligomerization and production of insoluble neurofibrillary tangles (NFTs). NFT build-up within neural structures, disrupting neuronal transmission and cytotoxicity (Tiwari et al., 2019; Gao et al., 2018). NFT deposition and plaque formation, commonly known as the “M1-like phenotype” (Tiwari et al., 2019), are triggered by microglial activation and local inflammatory responses. This inflammatory state ultimately leads to neuronal and synaptic instability, which may specifically influence some behavioral functions, such as memory, learning, and emotion (Liu, 2019).
Pharmacological options for AD currently exist, including acetyl-cholinesterase inhibitors (AChEIs) like galantamine, rivastigmine, and donepezil (Cummings et al., 2019). These medications work by increasing the levels of acetylcholine by inhibiting its degradation, favoring its accumulation, and increasing neural function (Breijyeh et al., 2020). Memantine, combined with AChEIs or used by itself, is recommended for late stages of the disease and works by inhibiting the neurotransmission of glutamate, avoiding progressive neurodegeneration and continuous excitotoxicity (Kishi et al., 2020; Wang and Reddy, 2017).
The use of exogenous or synthetic neural peptides as a potential treatment for NDDs has been investigated for AD. Such is the case with glatiramer acetate (GA), an amino acid copolymer produced from alanine that has been shown to significantly slow the progression of AD (Koronyo-Hamaoui et al., 2011; Bakalash et al., 2011). Microglial activation and a considerable reduction of fibrillar myeloid presence in hippocampal areas are associated with reduced brain interferon expression after weekly GA administration (Frenkel et al., 2014). GA administration increases IL-10 levels that target the innate immune system and glial cells, assisting in the reduction of neuroinflammation (Koronyo et al., 2015) and macrophage infiltration, both of which are strongly linked to a shift from a proinflammatory to an anti-inflammatory profile (Butovsky et al., 2007). Long-term administration of GA is associated with improved cognitive performance, synaptic preservation, neurogenesis, and a reduction in astrogliosis (Koronyo et al., 2015; Janus et al., 2000).
MNP-based immunotherapy has progressed to the point where several vaccines can produce specific antibodies against the structural components of each illness. CAD106, a contemporary immunotherapy developed to produce antibodies against the Aβ neuropeptide fragments acting as a B-cell epitope, is among the most researched (Winblad et al., 2012). Its long-term administration can induce a specific antibody response against Aβ with a long decline time and a strong serological reaction in mild and severe stages of AD in patients without inducing a T cell immune response, reducing the risk of autoimmune reactivity (Farlow et al., 2015; Vanderberghe et al., 2016), and decreasing Aβ accumulation in the hippocampus, thalamus, and cerebral cortex (Vanderberghe et al., 2016). In addition, the antibodies produced by the vaccine aid in preventing neuroinflammation in high-risk locations by blocking the toxicity of Aβ aggregations. In transgenic mouse models, immunization with CAD106 has been shown to prevent up to 80% of the buildup of senile plaques (Wiessner et al., 2011).
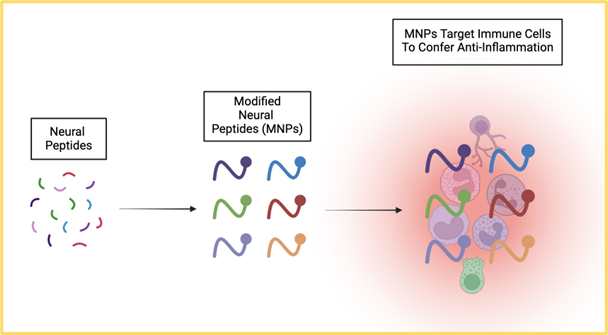
In a new window | Download PPT
Figure 1. Neural peptides can be modified to target immune cells in order to confer anti-inflammatory effects, thereby sequestering the inflammation-mediated secondary cell death associated with neurodegenerative diseases.
Parkinson’s disease
PD is the second most frequent NDD present in the elderly (Alexuidi et al., 2018). The incidence of PD incidence is constantly increasing and is expected to double by 2050, ultimately reaching 12 million cases worldwide (Tolosa et al., 2021; Dorsey et al., 2018). The pathophysiology of PD includes the loss of dopaminergic neurons in the substantia nigra pars compacta (Balestrino and Schapira, 2020). The most common clinical presentation of PD includes three key motor features: rigidity, bradykinesia, and resting tremors (Poewe et al., 2017; Cacabelos, 2017). The multifactorial etiology of PD results from a combination of biological and environmental factors (Cacabelos, 2017; Simon et al., 2020). Some genes that substantially enhance the development of PD have been identified, including Parkin (PARK) 2, PARK7, α-synuclein (SNCA), leucine-rich repeat kinase (LRRK2), and PTEN-induced and putative kinase 1 (PINK1) (Cacabelos, 2017).
α-Synuclein is a protein located in different areas of the brain, including the substantia nigra and the cerebellum. α-Synuclein has physiological roles in the fusion and trafficking of vesicles through the axon and its consequent transport (Simon et al., 2020; Jankovic and Tan, 2020). α-Synuclein has been identified as a pathognomonic marker of several NDDs because, when it is misfolded, it accumulates in intracytoplasmic inclusions, more commonly known as Lewy bodies. This accumulation induces the cellular expression of toxic cytokines, which consequently impairs synaptic function and leads to neuronal deterioration (Balestrino and Schapira, 2020). Lewy bodies have been found in the substantia nigra in autopsies of PD animal models and humans with PD (Dickson, 2020). It is recognized that PD pathophysiology presents with a significant pro-inflammatory component. Some neuroinflammatory processes that may enhance PD pathogenesis include the disproportionate amount of pro-inflammatory cytokines like tumor necrosis factor-α, IL-6, and apoptosis-related factors that promote neurotoxicity and cell death (Cacabelos, 2017). Pro-inflammatory environments may disrupt the BBB, allowing for cyclic peripheral inflammatory contributions to PD and magnification of the progressive dopaminergic loss (Ahmad et al., 2020). This dysfunction affects the L1 transporter localized in neuronal membranes, which, along with the metabolic precursor levodopa, are key for dopamine synthesis, suggesting that the alteration in BBB may lead to insufficient dopamine synthesis (Ahmad et al., 2020).
The current medications for PD include dopamine agonists, levodopa, monoamine oxidase inhibitors (MAO-i), and amantadine (Radhakrishnan, 2018). Non-motor symptoms, like autonomic symptoms, are treated with AChEIs like memantine (Gouda et al., 2020; Hayes, 2019). Pioneering evidence suggests that MNPs may play a neuroprotective role that can be implemented as a PD treatment. One MNP is Cop-1, which is shown to incite the switch from a Th1 to a Th2 phenotype, enabling a neuroprotective environment for dopaminergic neurons to survive and a modulating effect on microglial responses (Laurie et al., 2007; Valera and Masliah, 2013).
Due to α-synuclein misfolding and accumulation, there is an enhancement of toxic effects in the brain. Thus, a treatment that reduces α-synuclein accumulation must be implemented (Valera and Masliah, 2013). The AFFITOPE PD01 vaccine is a peptide carrier that induces antibodies. Cross-reactivity is prevented because the vaccine recognizes only α-synuclein, without interacting with β-synuclein, a protein with multiple neuroprotective properties (Schneeberger et al., 2012). For vaccination, PD01 is administered, which directly targets the site of α-synuclein accumulation, preventing further protein aggregation. This immunization has demonstrated abilities to decrease cerebral levels of α-synuclein, ameliorating neural alterations such as dendritic density loss and neuronal dysfunction. The vaccine also improves cognitive and motor functions. Furthermore, substantial memory changes were seen in several animal models after almost two years of the first PD01 vaccine administration (Masliah et al., 2011; Masliah et al., 2005). Taking this into consideration, MNPs represent a promising treatment for neurodegenerative diseases like PD. Further research is needed to assess its safety and other benefits of its usage.
Stroke
Cerebrovascular ischemia, better known as stroke, constitutes the second most common cause of death globally and the top cause of disability. Additionally, the increasing aging population has elevated the number of stroke cases (Campbellet al., 2019; Xu et al., 2020; Yang et al., 2019). Proinflammatory mediators are induced by ischemia, leading to cell death and dysfunction (Xu et al., 2020). Cerebrovascular ischemia originates from hypoperfusion, leading to impaired cerebral blood flow (CBF) characterized by embolism and thrombosis (Quin et al., 2020; Makris et al., 2018). Multiple hormonal, oxidative, and biochemical mechanisms constitute ischemic stroke’s pathophysiology, resulting in microvascular injury and BBB damage (Jayaraj et al., 2019).
Ischemic stroke manifests with impaired CBF and metabolic demand imbalance (Sekhar et al., 2020). Some regions of ischemia-affected tissue can be restored to normality with adequate treatment, while other areas have irreversible damage. Harmful and neuroprotective pathways are activated during an ischemic stroke (Goyal et al., 2020). Such pathways include alterations in the BBB, inflammation, apoptosis, reactive oxygen species, and hemostatic activation (Campbell et al., 2020). Another example is the deficient delivery of glucose and oxygen secondary to vessel obstruction and subsequent ATP production impairment. These metabolic changes generate cytotoxic edema (Makris et al., 2018). Another imperative aspect of stroke’s pathophysiology is excitotoxicity. When a significant reduction of phosphocreatine and ATP occurs, excitatory amino acids are discharged, causing an unrestricted depolarization within the penumbra, leading to a rise in calcium, glutamate, and sodium concentrations (Jayaraj et al., 2019). This increase can result in “reactive astrogliosis,” where neuronal death and astrocyte injury occur (Xu et al., 2020;).
Moreover, large amounts of reactive oxygen and reactive nitrogen species coupled with deficient cellular antioxidant production can damage cell components (Makris et al., 2018; Li et al., 2018). Yet another aspect of stroke’s pathophysiology is DNA damage, which triggers multiple pathways stimulating apoptosis and worsening the prognosis (Li et al., 2018). The anti-inflammatory responses to stroke are also important (Campbell et al., 2020). Tissue recovery and restoration of neurons are also triggered following anti-inflammatory responses in the pathology (Sakai and Shichita, 2019).
There are a variety of accepted treatments for ischemic strokes, such as thrombolytics like alteplase and tenecteplase, mechanical thrombolysis, stent retrievers, and endovascular therapy (Rabinstein and Alejandro, 2017; Catanese et al., 2017). The primary mechanism of these therapies is reperfusion of damaged areas, which has risks of hemorrhagic transformation and widespread cell death. Thus, recent studies have aimed to improve these treatments. For instance, immunization with NR2B9c (KLSSIESDV), a neuroprotective modified peptide, disrupts N-methyl-D-aspartate receptors, and PSD-95, the postsynaptic density protein-95, while not overtly blocking them (Meloni et al., 2020). Wheat germ agglutinin is used to deliver and preserve the therapeutic actions of NR2B9c (Aloizou et al., 2021; Arnon and Rina, 2019). Furthermore, regulatory and helper T cell actions are initiated by constitutive photomorphogenesis protein 1, which promotes neurogenesis. In tandem with neurogenesis, angiogenesis may be required for enhanced therapeutic outcomes in stroke. To this end, a hydrophilic protein called myelin basic protein may augment myelin sheath development of Schwann cells and oligodendrocytes, which is impaired during an ischemic stroke (Chen et al., 2019). In addition to myelin basic protein, myelin oligodendrocyte glycoprotein (MOG) has also been viewed as a potent contributor to vasculogenesis. MOG is a part of the immunoglobulin family that is presented on the surface of oligodendrocytes beside myelin sheaths (Peschl et al., 2019; Di Pauli and Berger, 2018). It is possible to modify MOG on its 35-55 amino acids. Thus MOG represents an attractive MNP therapy for cerebral ischemia due to its ability to reduce stroke area by stimulating IL-10 and T helper cell production (Wang and Reddy, 2017; Yang et al., 2017). In summary, MNPs offer a novel approach to stroke therapy by facilitating multiple neuroprotective and regenerative processes, such as neurogenesis, vasculogenesis, and anti-inflammatory mechanisms.
Conclusion
NDDs represent a detrimental health problem worldwide. Current treatments for these pathologies are mainly symptomatic, and they rarely modify disease progression or help in CNS functional recovery. Modulating immune responses is designed to reach a physiological state where the immune response is not suppressed but enhanced instead in an anti-inflammatory and self-repairing phenotype. MNPs are a promising therapy for achieving an autoreactive response in a protective phenotype using the patients' immune systems against NDDs. Various MNPs have been studied over the past decade with promising results suggesting that immunization with MNPs warrants serious pre-clinical evaluation toward clinical trials for NDDs.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
The authors thank the entire staff of Dr. Cesar Borlongan’s Neural Transplant Laboratory for critical discussions of this manuscript.
References
Germán Rivera Monroy1,2
1Centro de Investigación en Ciencias de la Salud (CICSA), FCS, Universidad Anáhuac México Campus Norte, Huixquilucan 52786, México. 2Center of Excellence for Aging and Brain Repair, Morsani College of Medicine, University of South Florida, 12901 Bruce B Downs Blvd, Tampa, FL 33612, USA.
Renata Murguiondo Pérez1
1Centro de Investigación en Ciencias de la Salud (CICSA), FCS, Universidad Anáhuac México Campus Norte, Huixquilucan 52786, México.
Ericka Cristina Loza López1
1Centro de Investigación en Ciencias de la Salud (CICSA), FCS, Universidad Anáhuac México Campus Norte, Huixquilucan 52786, México.
Enrique Blancarte Hernández1
1Centro de Investigación en Ciencias de la Salud (CICSA), FCS, Universidad Anáhuac México Campus Norte, Huixquilucan 52786, México.
Vanessa Castelli3
3Department of Life, Health and Environmental Sciences, University of L'Aquila, L'Aquila, Italy.
Corresponding author:
Vanessa Castelli
Email: vanessa.castelli@univaq.it
In a new window | Download PPT
Figure 1. Neural peptides can be modified to target immune cells in order to confer anti-inflammatory effects, thereby sequestering the inflammation-mediated secondary cell death associated with neurodegenerative diseases.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 16589 | 15 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA