Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Responses of Endothelial Cells Towards Ischemic Conditioning Following Acute Myocardial Infarction
Time:2018-09-02
Number:15669
Author Affiliations

Conditioning Medicine, 2018. 1(5):267-278.
Abstract
One of the primary therapeutic goals of modern cardiology is to design strategies aimed at minimizing myocardial infarct size and optimizing cardiac function following acute myocardial infarction (AMI). Patients with AMI who undergo reperfusion therapy display dysfunction of the coronary endothelium. Consequently, ischemic endothelial cells become more permeable, thereby weakening their natural anti-thrombotic and anti-inflammatory potential. Ischemia-reperfusion injury (IRI) is associated with activation of humoral and cellular components of the hemostatic and innate immune system. It is also associated with excessive production of reactive oxygen species (ROS), the inhibition of nitric oxide synthase, and with inflammatory processes. Given its essential role in the regulation of vascular homeostasis, including the regulation of platelets and leukocytes among others, dysfunctional endothelium can lead to increased risk of coronary vasospasm and thrombosis. Endothelial dysfunction can be prevented by ischemic conditioning based on limited intermittent periods of ischemia and reperfusion. The molecular mechanisms and signal transduction pathways underlying conditioning phenomena in the coronary endothelium have been described as involving less ROS production, reduced adhesion of neutrophils to endothelial cells and diminished inflammatory reactions. This review summarizes our current understanding of the cellular and molecular mechanisms regulating IRI-affected and -damaged coronary endothelium, and how ischemic conditioning may preserve its function.
Keywords: ischemia reperfusion injury, endothelium, inflammation, ischemic conditioning, cardioprotection
Abstract
One of the primary therapeutic goals of modern cardiology is to design strategies aimed at minimizing myocardial infarct size and optimizing cardiac function following acute myocardial infarction (AMI). Patients with AMI who undergo reperfusion therapy display dysfunction of the coronary endothelium. Consequently, ischemic endothelial cells become more permeable, thereby weakening their natural anti-thrombotic and anti-inflammatory potential. Ischemia-reperfusion injury (IRI) is associated with activation of humoral and cellular components of the hemostatic and innate immune system. It is also associated with excessive production of reactive oxygen species (ROS), the inhibition of nitric oxide synthase, and with inflammatory processes. Given its essential role in the regulation of vascular homeostasis, including the regulation of platelets and leukocytes among others, dysfunctional endothelium can lead to increased risk of coronary vasospasm and thrombosis. Endothelial dysfunction can be prevented by ischemic conditioning based on limited intermittent periods of ischemia and reperfusion. The molecular mechanisms and signal transduction pathways underlying conditioning phenomena in the coronary endothelium have been described as involving less ROS production, reduced adhesion of neutrophils to endothelial cells and diminished inflammatory reactions. This review summarizes our current understanding of the cellular and molecular mechanisms regulating IRI-affected and -damaged coronary endothelium, and how ischemic conditioning may preserve its function.
Keywords: ischemia reperfusion injury, endothelium, inflammation, ischemic conditioning, cardioprotection
1. Introduction
The endothelium is defined as a cell monolayer covering the innermost apical surface of all blood and lymphatic vessels. Beyond its role as a physical and permeability barrier, endothelial cells (EC) can be anatomically divided into different phenotypes such as continuous, fenestrated and discontinuous endothelium, which are all functionally involved in a variety of tissue- and organ-specific functions contributing to vascular homeostasis (Kladakis and Nerem, 2004). Due to the specific expression of anti-inflammatory and anti-aggregatory molecules, quiescent EC provide a non-thrombogenic luminal surface and also actively contribute to the regulation of vascular tone, inflammation, and metabolism (de Agostini et al., 1990; Preissner, 1990; van Hinsbergh, 1997; Kladakis and Nerem, 2004; Urbich and Dimmeler, 2004). At particular sites in the vascular tree, the endothelium also plays a central role in innate immunity, allowing monocyte and neutrophil rolling and diapedesis to occur (de Agostini et al., 1990; van Hinsbergh, 1997; Urbich and Dimmeler, 2004; Rajendran et al., 2013). Stress-, damage- or metabolite-related dysfunctions of EC (Chavakis and Preissner, 2005) contribute to a variety of diseases in a wide range of organs such as heart, lung, liver or kidney (Rajendran et al., 2013). The most prominent naturally occurring endothelial dysfunction is the life-long exposure of non-laminar fluid shear stress to blood vessels at predestined sites (such as bifurcations) in the circulatory system. As a consequence, young individuals have early signs of atherogenesis, such as fatty streaks, that may develop into atherosclerotic plaques as they get older, depending on the person´s genetic make-up and lifestyle (Chatterjee, 2018).
Following the treatment of such atherosclerosis-related arterial occlusions in the context of myocardial infarction, the underlying process of ischemia-reperfusion (IR) induces deleterious effects not only on large vessels but also in the microcirculation of the heart. Several studies indicate that EC are more sensitive to IR than cardiomyocytes and are critical mediators of cardiac IR-injury (IRI) (Tsao et al., 1990; Lefer et al., 1991; Richard et al., 1994; Singhal et al., 2010; Hausenloy and Yellon, 2016). In this context, endothelial dysfunction implies diminished production or availability of nitric oxide (NO), an imbalance in the contribution of endothelium-derived relaxing molecules (Michiels, 2003) and a deficiency of essential vasodilators to provide control of vascular tone and blood pressure, such as prostacyclin (PGI2) (Pearson, 2000). After summarizing some of the risk and initiation factors of endothelial dysfunction, this review will focus on the protective endogenous strategy of ischemic conditioning and its potential beneficial effects on vascular homeostasis.
2. Endothelial dysfunction upon ischemia-reperfusion (IR)
Multiple mechanisms, including inflammation, increased levels of ROS, cellular apoptosis, and raised vasoconstriction, but decreased vasodilator production and vascular remodeling are involved in the damage of EC during IR (Richard et al., 1994). Ischemia is characterized in general by the interruption of oxygen supply, either in a specific tissue or in a whole organ. Importantly, cardiovascular diseases are often initiated by ischemic episodes, contributing to the main cause of death in developed countries (Remme, 2000; Celermajer et al., 2012).
The endothelium seems to be particularly sensitive to conditions of ischemia and posterior reperfusion, because these conditions promote e.g. hypoxia-dependent changes in gene expression and permeability properties of EC, contributing to endothelial dysfunction (Harrison, 1997) and the pathogenesis of cardiovascular disorders, including myocardial ischemia (Shimokawa and Yasuda, 2008). Other responses of dysfunctional EC include the chronic imbalance between ROS versus NO, the continued expression of adhesion receptors such as E-selectin, intercellular cell adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and receptor for advanced glycation end products (RAGE), as well as the reduction of acetylcholine production (Gaboury et al., 1993; Chavakis et al., 2003; Quagliaro et al., 2005). Consequently, the imbalance in favor of adhesive and thrombogenic properties of the endothelium makes it more prone to dysregulated leukocyte attachment, increased oxidative stress, thereby triggering inflammatory, thrombotic, and necrotic processes (Heitzer et al., 2001; Galle et al., 2003). As already indicated, a major irreversible outcome and consequence of dysfunctional endothelium is associated with atherosclerosis and vascular diseases (Poredos, 2002b, a; Widmer and Lerman, 2014).
2.1. Cytotoxicity caused by Ca2+ ions. The control of cellular Ca2+ levels in arterial EC and smooth muscle cells is necessary for the control of vascular tone, including EC-dependent vasodilation activities (Lefer et al., 1991; Sandow et al., 2012). Several studies have shown that IRI leads to EC damage in coronary arteries by disturbance of Ca2+ homeostasis (Karasawa et al., 1991; Lefer et al., 1991; Lucchesi, 1993; Vinten-Johansen et al., 1999; Kimura et al., 2000; Ladilov et al., 2000; Symons and Schaefer, 2001; Kumar et al., 2007). A combination of anoxia with extracellular acidosis (pH 6.4) results in excessive accumulation of Ca2+ in the cytosol of EC mainly due to the leak of Ca2+ from the endoplasmic reticulum (ER). The ischemic factors (low pH and lactate) leading to cytosolic acidosis cause intracellular Ca2+ overload in EC (Ladilov et al., 2000), while anoxia and glucose deprivation play only a minor role in this context (Lefer et al., 1991). Uncontrolled depletion of Ca2+ in the ER may lead to ER-stress, followed by cleavage of ER-bound caspase-12 (Kumar et al., 2007) and caspase-3, and the activation of apoptosis through cytochrome-c (Ladilov et al., 2000; Borutaite et al., 2003; Kumar et al., 2007). By increasing intracellular Ca2+ stores, store-operated Ca2+ entry (SOCE) mainly regulates the cytosolic Ca2+ concentration of EC (Putney, 1986). Following ischemia, extracellular, but not intracellular, acidification induces EC dysfunction by suppressing SOCE (Asai et al., 2009). Additionally, unregulated influxes of Ca2+, secondary to plasma membrane damage and disruption homeostasis. It also activates intracellular lipases, proteases and endonucleases that are important in the apoptotic signaling pathway. During reperfusion, EC increase their permeability, and an inflammatory signal transduction pathway, initiated by the oxidation of specific cellular modulators, alters the EC cytoskeleton and down-regulates natural anti-thrombotic and anti-inflammatory processes, thus increasing membrane permeability (Higginson et al., 1982; Mullane et al., 1984; Hinshaw et al., 1989; Mertens et al., 1990; Geeraerts et al., 1991; Patel et al., 1991; Natarajan et al., 1993; Hastie et al., 1997; Ladilov et al., 2000; Kumar et al., 2007). Moreover, in in vitro studies with coronary EC, acidic preconditioning conferred protection against ischemia-apoptosis induction, associated with over-expression of the anti-apoptotic protein Bcl-XL (Kumar et al., 2008). Further investigation of Ca2+ fluxes in coronary EC may lead to the characterization of new therapeutic approaches for the treatment of chronic inflammation and coronary artery disease (Steppich et al., 2009; Stepien et al., 2012; Suades et al., 2015).
2.2 Oxidative stress. Increased ROS levels have been generally attributed to IRI (Hinshaw et al., 1989; Lucchesi, 1993; Gaboury et al., 1994; Szocs, 2004; Therade-Matharan et al., 2005; Hernandez-Resendiz et al., 2018), whereby xanthine oxidase, NADPH oxidase, and the mitochondrial electron transport chain are the most frequently implicated sources of ROS in myocardium exposed to IR. The rapid burst of oxygen-derived free radicals during reperfusion coincides with the time course of progression in endothelial dysfunction (Karasawa et al., 1991). For example, exposure of EC for 6h to hypoxia followed by 45 min of re-oxygenation increased the generation of superoxide anion (O2•-) in EC (Tsao et al., 1990; Kimura et al., 2000), whereby cytosolic xanthine oxidase and the mitochondrial electron transport chain (complexes I and III) provide the primary sources of endothelial superoxide anion. Since NADPH oxidase (NOX) 4, one of the NOX isoforms, is present in EC and co-localizes with mitochondria, it may be considered as a source of ROS as well (Szocs, 2004; Dymkowska et al., 2014).
Moreover, as a result of an excess of superoxide anion, hydroxyl (OH-) and peroxynitrite anions (ONOO-) are also produced by EC, provoking NO inactivation. As a consequence, decreased vasorelaxation and eventually vasoconstriction are observed. Peroxynitrite is a free radical that mediates lipid peroxidation and uncoupling of NO synthase (NOS). Usually, O2•- can be dismutated to H2O2, either spontaneously or by superoxide dismutase (SOD). The exposure of EC to high levels of H2O2 (< 500 M) increases the intracellular Ca2+ concentration, the expression of the aforementioned adhesion receptors (Szocs, 2004; Therade-Matharan et al., 2005), as well as complement activation (Patel et al., 1991; Gaboury et al., 1994) that collectively induces neutrophil attachment to the dysfunctional vascular endothelium (Lucchesi, 1993). Needless to say, damage of EC by oxidative stress is reduced or prevented by ROS scavengers and antioxidants.
Activation of endothelial cells by oxidative stress thereby promotes inflammation: Following IRI, activated EC express P-Selectin, E-Selectin and intercellular adhesion molecules (ICAMs), boosting the recruitment of neutrophils and contributing to cytokine production as major inflammatory reactions (Winn et al., 1997). Oxidative stress also induces the activation of NF-kB, which plays a key role in endothelial apoptosis via the down-regulation of Bcl-2, Bax translocation and p53 upregulation (Aoki et al., 2001).
2.3. Inhibition of nitric oxide synthase (NOS). Both, decomposition of NO during reperfusion and inhibition of NO synthesis, can increase leukocyte adherence to the venular wall. Tetrahydrobiopterine (BH4) is a cofactor, which is critical for NO production by regulating endothelial NOS (eNOS) activity. During IR, free radicals can oxidize BH4, causing a fall in the BH4/eNOS ratio, thereby uncoupling eNOS. In such a scenario, uncoupled eNOS behaves like NADPH oxidase, increasing the production of O2•-, H2O2 and OONO- to induce cytotoxicity and inflammation (Figure 1). In EC, eNOS competes with arginase for the substrate L-arginine, whereby the activity of arginase, the primary L-arginine-consuming enzyme, has been reported to become elevated following IRI (Hein et al., 2003). Since NO is a potent inhibitor of neutrophil activation and adhesion, the decreased levels of NO may lead to the development of an acute inflammatory response (Laude et al., 2001; Ong et al., 2018).

In a new window | Download PPT
Figure 1: Overview of endothelial cell response towards acute myocardial infarction. This scheme depicts the reperfusion-induced damage of endothelial cells following acute myocardial infarction.
3. Endothelial dysfunction and inflammation
The stages of acute inflammation (as part of the innate immunity response) in a blood vessel are vasodilation, increased permeability of the microvasculature, and vascular stasis (Szmitko et al., 2003). As EC undergo cytoskeletal changes that disrupt junctions in venules and capillaries, a delayed cellular response is observed in this process, starting about 6h after the initial stimulus and lasting for days. In response to risk factors, such as IR, the endogenous protection mechanisms (anti-oxidant, anti-inflammatory, etc.) of the coronary endothelium begin to break down. The already indicated production of ROS and the reduction of NO provoke the expression of inflammatory cytokines such as interleukin-6 (IL-6) (Verma et al., 2002) or monocyte chemoattractant protein-1 (MCP-1) as well as the upregulation of VCAM-1 on EC. Upon EC activation, the first luminally expressed adhesion molecule is P-selectin (Vinten-Johansen et al., 1999), which is derived from degranulation of Weibel-Palade bodies, mediating the initial rolling of leukocytes along the vessel wall surface. After 4-6h of reperfusion, de novo expressed E-selectin, ICAM-1 and RAGE appear to induce the firm adhesion of leukocytes via there activated ß2-integrins, preparing these inflammatory cells for their transmigration across the dilated endothelium into the myocardium (Ma et al., 1992; Ampofo et al., 2017). Expression of endothelial ICAM-1, VCAM-1 and RAGE are considered essential processes for providing firm adhesion and transmigration of neutrophils and monocytes to the subendothelial space in blood vessels, contributing to the initial steps of atherosclerotic lesion development as well (Ziche et al., 1994; Deem and Cook-Mills, 2004; Nawroth et al., 2005; Lopez-Diez et al., 2016).
The expression of the aforementioned adhesion molecules is further induced by proinflammatory cytokines such as interleukin-1 (IL-1) or tumor necrosis factor-α (TNF-α), which have been reported to be expressed in IR within minutes (Cardozo et al., 2005; Schmidt et al., 2006). Once adhered to the inflamed endothelium, monocytes cross the arterial wall between EC and transmigrate into the neo-intimal space (Vinten-Johansen et al., 1999).
It is well established that after reperfusion, IR induces the accumulation of platelets via P-selectin - ligand interactions on the dysfunctional (activated), but intact endothelium in the post-ischemic microvasculature (Coulter et al., 2000; Mirabet et al., 2002). The initial expression of P-selectin by the activated endothelium can be considered an early biomarker for the IR-induced platelet-EC interactions (Massberg et al., 1998). The first attachment of platelets to the still intact but activated endothelium during IR is mediated by platelet glycoprotein Ibα as a ligand for endothelial P-selectin and a receptor for von Willebrand factor, the latter being released from endothelial Weibel-Palade bodies as well (Massberg et al., 1998; Romo et al., 1999). Following adhesion, platelet activation is potentiated by the release of soluble platelet agonists at sites of EC injury, finally resulting in prolonged granule release and the formation of platelet aggregates (Maiocchi et al., 2018). Among the platelet agonists are ROS, as well as platelet granule products, including proinflammatory cytokines, chemokines, thromboxane A2, leukotrienes or proinflammatory lysolipids (Marcus, 1979; Leo et al., 1997). These events, combined with the release of vasoconstricting molecules from platelets, further exacerbate the physical obstruction of the microvasculature.
As an interventional strategy, NO was found to mediate a cardio-protective effect in a number of common clinical strategies, such as preconditioning, postconditioning and remote ischemic preconditioning, further supporting the concept that targeting of ROS, NOS and inflammatory response mechanisms may provide myocardial protection under conditions of IRI.
4. Influence of ischemic conditioning on the coronary endothelium
Brief non-lethal episodes of IR are known to protect against the deleterious effects of a sustained lethal time span of IR. This phenomenon is known as “ischemic conditioning” and has been observed in the heart and other organs (Thielmann et al., 2013; Meybohm et al., 2015; Cabrera-Fuentes et al., 2016a; Cabrera-Fuentes et al., 2016b; Hausenloy et al., 2016). Although several interrelated molecular mechanisms are operative here, ischemic conditioning protects the heart against IRI via a culminating “reperfusion injury signaling kinase” (RISK) pathway. The RISK pathway has been extensively studied in cardiomyocytes (Frohlich et al., 2013) and hepatocytes (Carini et al., 2001; Teoh et al., 2003; Hausenloy and Yellon, 2004).
4.1. Ischemic preconditioning. One of the most promising avenues of research into potential therapeutic treatments for IRI is ischemic preconditioning (IPC). Several studies have demonstrated that IPC has direct effects on tissue cells, making them resistant to ischemic damage but also preventing the characteristic vascular dysfunction at the arteriolar, venular and capillary levels (DeFily and Chilian, 1993; Bouchard and Lamontagne, 1996; Kaeffer et al., 1996; Thourani et al., 1999; Kharbanda et al., 2001). Upon application of IPC, surgery studies in pediatric and adult patients revealed a significant reduction of tissue damage, indicated by decreased serum levels of troponin I and creatinine kinase KB (Botker et al., 2010). Likewise, other reports demonstrated a decrease of angina episodes, reduced ST deviation on the electrocardiogram and less severe arrhythmias following IPC (Jenkins et al., 1997; Sloth et al., 2014). However, specific mechanisms of IPC at the cellular and molecular level are not well understood, and most of the studies prior to or after preconditioning using a clinical approach are focused on cardiomyocyte pathophysiology.
Acute IP is operative if the time interval between a brief stimulus of preconditioning and the posterior ischemic insult lasts less than 2h (Korthuis and Gute, 1997). The second phase of protection termed “delayed preconditioning” or “ischemic tolerance” becomes important if the time period between insults is longer than 24h (Korthuis and Gute, 1997). There are differences between both types of protection; the acute preconditioning is too short to be dependent on gene expression and cellular protein synthesis, while delayed preconditioning necessarily includes phenomena of de novo protein synthesis (Downey et al., 1994; Korthuis and Gute, 1997).
4.2. Cellular mechanisms in the acute preconditioning phase. In the acute phase of IP, activation of PKC in cardiomyocytes induces phosphorylation of effector molecules that may contribute to cellular protection. These factors are associated with adenosine receptors via their pertussis-sensitive G proteins like ATP-sensitive potassium channels (KATP) and can contribute to protective effects of preconditioning by triggering the mKATP/ROS pathway (Heurteaux et al., 1995; Korthuis and Gute, 1997). The importance of KATP channels in this context is supported by experimental studies using respective agonists and antagonists. Although the protective effects involve KATP channels in both plasma and mitochondrial membranes, the underlying mechanisms are still unclear (Auchampach and Gross, 1993; Jerome et al., 1995). In addition to its influence on KATP channels within the context of protective preconditioning, PKC can induce 5’ nucleotidase to increase adenosine levels during sustained ischemia. This process mobilizes cellular depots of EC metabolic energy and prevents leukocyte adherence and subsequent inflammatory reactions (Kitakaze et al., 1994).
4.3. Cellular responses upon delayed IP. Cardiac protection can also be achieved if the ischemic episode occurs within 24h after the initial brief stimulus of ischemia or IP. This delayed response is more an adaptive response and a consequence of the induction of gene expression and de novo protein synthesis of metabolic enzymes and heat shock proteins following the first brief ischemic insult (Korthuis and Gute, 1997). In post-ischemic phases, induction of gene and protein expression in leukocytes and EC involves activation of PKC and other kinases that become translocated to the nucleus to regulate nuclear transcription factor activities (Kitakaze et al., 1994; Korthuis and Gute, 1997). Here, the nuclear factor κB (NF-κB) is a central transcription factor in delayed responses of IP at least in EC and it is considered to be the key factor for the transcription of post-ischemic inflammatory mediators. Importantly, if NF-κB translocation is blocked, the delayed phase of IP is also inhibited (Yellon and Baxter, 1995).
When EC and cardiomyocytes are exposed towards short ischemic episodes, they respond with an increased production of NO (Lu et al., 1993; Yamashita et al., 1994). NO also plays a central role during delayed responses due to a decrease in oxidative stress and cell adhesion (Korthuis and Gute, 1997). In an autocrine fashion, eNOS-produced NO is a mediator of EC protection upon delayed IP. Administration of the non-selective NOS inhibitor L-NAME prior to prolonged myocardial IR did abolish the protective effect of IPC on the coronary endothelium, indicating that NO corresponds with the EC-dependent reactions of delayed IPC (Laude et al., 2003). In addition to the influence of NO during oxidative stress, the delayed response of IP may enhance the activity of antioxidant enzymes such as SOD, catalase or glutathione peroxidase, thereby potentiating the initial protective effect of NO (Yellon and Baxter, 1995).
When arteries are subjected to IR, IPC has been shown to prevent endothelium-dependent relaxing responses to acetylcholine (Kaeffer et al., 1996). In most studies, the protection by IPC occurs during the reperfusion period, when arterioles are thought to be most vulnerable to dysfunction. IPC reduces the expression of EC adhesion molecules, resulting in reduced adhesion of neutrophils to the endothelium and hence, lowering the inflammatory reactions. In response to myocardial IR, circulating and heart-derived levels of TNF-α increase within minutes, mainly from macrophages, monocytes and mast cells. Moreover, IPC has been shown to decrease cardiac as well as circulating TNF-α levels during sustained ischemia, thereby reducing myocardial infarct size in rabbits (Meldrum et al., 1998; Belosjorow et al., 2003). In another report, IPC reduced the expression of ß2-integrins on neutrophils in preconditioned human subjects, and this observation could be linked to decreased EC injury (Kharbanda et al., 2001).
Another adaptive mechanism upon IPC is the increased biosynthesis and liberation of heat shock proteins, whereby this family of intracellular proteins may normally control cell metabolism and maintain structure and function of important proteins during stress. Nevertheless, the underlying mechanisms and signaling pathways are poorly understood (Marber and Yellon, 1996; Gray et al., 1999). The possibility to reproduce this phenomenon in human subjects may allow the identification of related (circulating) biomarkers and pathophysiological parameters by testing pharmacological approaches to prevent IRI in cardiac patients.
4.4. Ischemic Postconditioning. Ischemic postconditioning (IPost) was first reported in dogs, and it is performed after the onset of reperfusion. In anesthetized open-chest animals, the left anterior descending (LAD) artery was occluded for 1h and reperfused for 3h. In controls, there was no intervention. In pre-conditioning (pre-con), the LAD was occluded for 5 min and reperfused for 10 min before the prolonged occlusion. In post-conditioning (post-con), at the start of reperfusion, three cycles of 30-s reperfusion and 30-s LAD re-occlusion preceded the 3h period of reperfusion. Infarct size was significantly less in the pre-con and post-con groups compared with controls (Zhao et al., 2003), however, hardly any differences were reported in the reduction of infarct sizes between animals treated with pre-con or post-con protocols, respectively (Donato et al., 2007). These results were confirmed in other vertebrate models such as rabbits, rats, pigs and mice (Schwartz and Lagranha, 2006; Gomez et al., 2007; Skyschally et al., 2009). Since the model algorithm is based on the delay at the first re-occlusion time point and the duration of the ischemic-reperfusion stimulus (Skyschally et al., 2009), caution should be taken for interpretation of the model algorithm used in IPost due to its high variability. Protection conferred by IPost is associated with the improvement of EC function, reduction of inflammation and necrosis (Engelman et al., 1995; Kin et al., 2004).
IPost can also protect EC from IRI via phosphatidylinositol 3-kinase (PI3K)/ protein kinase B (Akt) activation (Zhang et al., 2007). Respective investigations of IRI in EC of human subjects confirmed that IPost inhibits the opening of the mitochondrial permeability transition pore (mPTP) by preventing KATP channel activation, leading to cellular protection from reperfusion injury (Okorie et al., 2011; Ong et al., 2015). The perturbations of coronary EC can be analyzed by brachial artery ultrasound (Lind et al., 2005), demonstrating that IPost may protect the coronary endothelium. Moreover, it has been shown that the vasodilation function of endothelium was improved in patients by IPost (Ma et al., 2006).
An important phenomenon for explaining IPost-associated protection is the increase in NO production and the reduction of the “no-reflow” phenomenon (Schwartz and Kloner, 2012). The no-reflow phenomenon is defined as the reperfusion failure at the microvascular level after primary percutaneos coronary intervention (PCI) in patients with ST-segment elevation myocardial infarction (STEMI) (Ramjane et al., 2008; Bouleti et al., 2015; Gupta and Gupta, 2016). This mechanism is a partial limitation of microcirculatory blood flow after the onset of reperfusion, despite the elimination of vascular occlusion. No-reflow is caused by vascular spasms, EC damage, sarcolemmal bubbles in the endothelium and by smooth muscle migration (Rajendran et al., 2013). Histologic analysis of the no-reflow microvasculature area shows endothelial swelling with a general loss of pinocytotic vesicles, an evidence for endothelial disruption (Kloner, 2011). After long periods of preconditioning, capillary no-reflow is attenuated by a mechanism involving KATP channels (Jerome et al., 1995). Some studies showed that IPost reduces no-reflow, thereby improving the blood flow in the microcirculation. Interestingly, hypercholesterolemia may significantly reduce this beneficial effect (Reffelmann and Kloner, 2006; Zhao et al., 2007).
4.5. Remote ischemic preconditioning (RIP). After the discovery and the medical development of the indicated conditioning procedures to directly protect the target organ, it was demonstrated that the ischemic conditioning stimulus could be applied in a non-invasive way (such as by a blood pressure cuff) and distant from the organ to be targeted (Oxman et al., 1997). Against this background, in the first human study, endothelial IRI of the forearm was induced by 20 min of upper limb ischemia followed by reperfusion. Remote preconditioning was induced by three 5 min cycles of ischemia of the contralateral limb, whereby venous occlusion plethysmography was used to assess forearm blood flow in response to acetylcholine at baseline and 15 min after reperfusion. Interestingly, the response to acetylcholine was significantly attenuated in the control group after 15 min reperfusion, but remote preconditioning prevented this reduction (Kharbanda et al., 2002). This procedure is designated as remote ischemic conditioning (RIC), whereby brief episodes of ischemia and reperfusion applied in a particular organ or vascular bed may confer protection in a remote tissue and organ exposed to IRI (Heusch et al., 2015).
According to two recent randomized trials, RIP improves long-term clinical prognosis after primary percutaneous coronary intervention (PPCI) (Davies et al., 2013; Sloth et al., 2014). Nevertheless, a prospective randomized controlled clinical multi-centric trial named “Effect of Remote Ischemic Conditioning on Clinical Outcomes in STEMI Patients Undergoing PPCI (CONDI2/ERIC-PPCI)” is currently investigating whether RIC can reduce cardiac death and hospitalization for heart failure at 1 year in 5,200 patients, presenting with a ST-elevation myocardial infarction (STEMI) and treated by PPCI. The trial is scheduled to be completed in December 2019 (online reference https://clinicaltrials.gov/ct2/show/NCT02342522).
RIC prior to PPCI significantly improves EC function in patients with AMI (Manchurov et al., 2014). Moreover, it has been demonstrated in animal studies that whole blood transfusion and transfer of coronary effluent from one organism to another is sufficient to transfer cardio-protection from one RIP-treated animal to a naïve one. This phenomenon has been replicated in several species, supporting the role of humoral factors in RIC-mediated cardio-protection (Dickson et al., 1999). In particular, increased levels of adenosine in coronary effluent after RIC, as well as neuro-humoral factors and other biochemical and electrical responses caused the RIP cardio-protective signaling system (Patel et al., 2002; Weinbrenner et al., 2004; Gross et al., 2011; Donato et al., 2013). In RIP, the activation of peripheral sensory fibers appear to activate PKCγ as observed in other ischemic conditioning techniques (Gross et al., 2013).
Besides the non-invasive procedure used for inducing RIC, it is able to confer cardio-protection when applied at different time points in relation to the induction of the main IR episode. This characteristic is also beneficial in terms of clinical outcomes (Sivaraman et al., 2015). RIC could be used immediately or 12- 24h before a long ischemic episode (RIP), after ischemia but before reperfusion, at the moment of reperfusion (RIP) or 15 min following reperfusion (delayed IPC) (Sivaraman et al., 2015).
Some recent studies in the context of RIC revealed the existence of an as yet uncharacterized humoral factor involved in cardio-protection (Serejo et al., 2007; Shimizu et al., 2009). This humoral factor could have properties of a protein with a size between 3.5 and 30 kDa, whereby the vasodilator peptide bradykinin could be excluded (Serejo et al., 2007; Shimizu et al., 2009). Some proteomic studies indicated differentially expressed proteins in plasma derived from patients exposed to RIC as compared to controls. Such putative proteins appear to be associated with inflammatory responses, hemostasis and lipid transport, supporting the hypothesis that complex interactions of molecular pathways contribute to RIC in cardio-protection (Hepponstall et al., 2012). In particular, as in other types of conditioning procedures, an increase in adenosine levels as well as the contribution of the NOS system, of stromal cell-derived factor-1α (SMC-1α), interleukin 10, or microRNA-144 were found (Cai et al., 2012; Davidson et al., 2013; Li et al., 2014), whereby such molecules possibly play a role as local triggers of KATP channels and as NO effectors (Schmidt et al., 2007). Nevertheless, each of these factors alone is insufficient to promote the entire phenomenon of cardio-protection.
Recently, the role of the extracellular RNA (exRNA)/RNase1 system in cardiac I/R injury has received increased attention (Cabrera-Fuentes et al., 2014; Cabrera-Fuentes et al., 2016a; Cabrera-Fuentes et al., 2016b). ExRNA induces vascular hyper-permeability by increasing intracellular calcium ions in EC (Balint et al., 2014), and interacts with VEGF to initiate VEGF-mediated signal transduction through neuropilin-1, VEGF-R2 phosphorylation, activation of phospholipase C (PLC) and intracellular release of Ca2+ (Fischer et al., 2009). Vascular EC constitutively produces extracellular circulating RNase1, which has different functions in EC responses (Fischer et al., 2011). In a recent clinical study, plasma levels of RNase1 increased after cardiac surgery subjected to RIP, while there was a significant reduction of exRNA and the pro-inflammatory mediator TNF-α, suggesting a role of vascular RNase1 as a potential mediator of RIP for cardio-protection (Cabrera-Fuentes et al., 2015).
4.6. Methodology used to measure coronary EC function. The methodologies used to measure coronary EC dysfunction were designed to be non-invasive to achieve higher security margin and better acceptance from the patients. At the same time, determinations must reflect the actual state of EC function based on our knowledge of endothelial physiology and activation, described before.
Indirect assessment: Some studies have used the assessment of vasoconstriction in response to acetylcholine that consists in flow-mediated vasodilation in the brachial artery by ultrasound to detect coronary artery EC dysfunction and coronary artery disease as a surrogate measure (Anderson et al., 1995; Corretti et al., 2002). One advantage of this technique is that an observer can measure EC function in response to a stimulus. However, such an effect is indirect and brachial condition is assumed to be the same as in the coronary artery, which may not be accurate. Another disadvantage is the operator-dependent variation and the wide range of reported normal values (Berry et al., 2000). However, brachial function is proposed as a strong predictor of coronary function in a statistically significant manner, showing that a positive predictive value of abnormal brachial dilation (<3%) in predicting coronary EC dysfunction is 95% (Anderson et al., 1995). Both approaches of indirect assessment are based on endothelium-dependent relaxation as a result mainly of NO bioavailability. Nevertheless, other vascular mediators can be involved such as bradykinin.
Direct assessment: Direct assessment is considered the "gold standard" and it determines the change in coronary artery diameter, coronary blood flow and coronary vascular resistance to an intracoronary infusion of acetylcholine (Hasdai and Lerman, 1999). In coronary arteries with a quiescent endothelium coverage, the response to intracoronary acetylcholine is epicardial and microvascular dilation results in an increase in coronary blood flow (Bonetti et al., 2003). When the EC lining is disrupted, activated or dysfunctional, intracoronary acetylcholine induces paradoxical responses, for example, vasoconstriction and a decrease in coronary blood flow. The response to acetylcholine serves as a marker for the bioavailability of NO; an abnormal response suggests a lack of NO bioavailability such as for endothelial dysfunction.
Magnetic resonance imaging (MRI) has been used for the direct assessment of coronary endothelial dysfunction (Bulluck et al., 2017). It assesses coronary endothelium responses to pharmacological stimuli. Patients need to be prepared for the imaging session through the administration of vasoactive medications, selected according to the respective study and the individual patient (Hays et al., 2012). Some authors have reported that MRI is performed after an angiography to localize vascular structures more accurately (Hays et al., 2012; Bulluck et al., 2018).
5. Conclusions
A particular stimulus to mediate conditioning of organs such as the heart can act at different biological levels to engage molecular programs that produce durable (> 24h) tolerance against ischemic injury. The cardio-protection conferred by ischemic conditioning is a complex system of successive events, and the mechanisms still remain to be resolved. However, we can enumerate three necessary steps to explain cellular responses after ischemic conditioning (Figure 2): (1) Generation of a cardio-protective signal in conditioned tissue with an anticipated role for EC signaling; (2) transport pathways via circulatory and nervous systems that confer the cardio-protective signal from the conditioned tissue to the heart; (3) activation of cellular signaling pathways in the heart to initiate protection. (1) and (2) involve interactions between neural and circulating factors, whereby the endothelium appears to play a crucial role in both steps (Sivaraman et al., 2015). The central role played by dysfunctional EC in promoting thrombus formation and inflammatory responses during IRI is well recognized. There is considerable progress in understanding the role of dysfunctional EC as a potential therapeutic target for myocardial protection as well. The development of approaches to control EC activation, focussing on the RISK pathway in EC may contribute to a better understanding of IRI and provide new opportunities for its treatment or prevention.
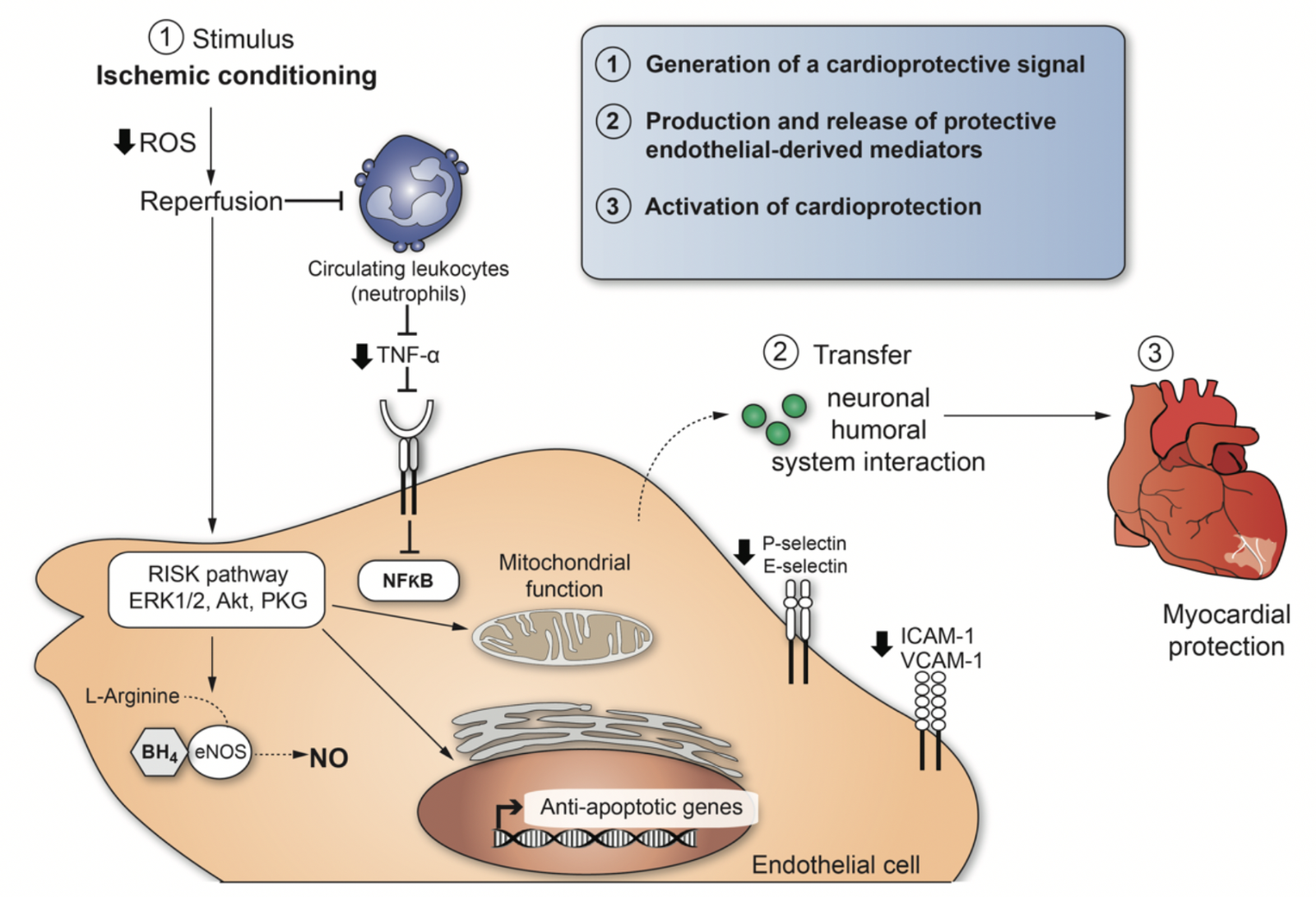
In a new window | Download PPT
Figure 2: Cardio-protective mediators and molecular mechanisms that are activated by ischemic conditioning in endothelial cells. The illustration enumerates three necessary steps to explain cellular responses after ischemic conditioning-induced cardioprotection.
Disclosures/conflicts
The authors declare that they have no conflicts of interest.
Acknowledgements
SHR is funded by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund – Young Individual Research Grant (OF-YIRG) – 0078/2018. Part of the original work presented by HCF and KTP was supported by the Russian Government Program for competitive growth of Kazan Federal University, Kazan (Russian Federation), by the SHF-Foundation Research Grant (SHF/FG657P/2017) and by the von Behring-Röntgen-Foundation (Marburg, Germany). This work was supported by the European Cooperation in Science and Technology (COST Action CA16225 / EU-CARDIOPROTECTION). This project was funded by NIH grant R01HL81863 to WAB.
References
Sauri Hernández-Reséndiz1,2,3
1Cardiovascular and Metabolic Disorders Program, Duke-NUS Medical School, Singapore, Singapore.
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
3Escuela de Ingeniería y Ciencias, Centro de Biotecnología-FEMSA, Tecnológico de Monterrey, Monterrey, NL, México.
Mónica Muñoz-Vega7
7Molecular Biology Department, Instituto Nacional de Cardiología “Ignacio Chávez”, C.D de México, México.
Whendy E. Contreras1,2
1Cardiovascular and Metabolic Disorders Program, Duke-NUS Medical School, Singapore, Singapore.
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
Gustavo E. Crespo-Avilan1,2
1Cardiovascular and Metabolic Disorders Program, Duke-NUS Medical School, Singapore, Singapore.
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
Julian Rodriguez-Montesinos5
5Institute of Biochemistry, Medical School, Justus-Liebig-University, Giessen, Germany.
Oscar Arias-Carrión6
6Unidad de Trastornos del Movimiento y Sueño, Hospital General Dr. Manuel Gea González. Ciudad de México, México.
Oscar Pérez-Méndez7
7Molecular Biology Department, Instituto Nacional de Cardiología “Ignacio Chávez”, C.D de México, México.
William A. Boisvert8
8Center for Cardiovascular Research, John A. Burns School of Medicine, University of Hawaii, Hawaii, USA.
Klaus T. Preissner4,5
4Kazan Federal University, Department of Microbiology, Kazan, Russian Federation.
5Institute of Biochemistry, Medical School, Justus-Liebig-University, Giessen, Germany.
Hector A. Cabrera-Fuentes1-5
1Cardiovascular and Metabolic Disorders Program, Duke-NUS Medical School, Singapore, Singapore.
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
3Escuela de Ingeniería y Ciencias, Centro de Biotecnología-FEMSA, Tecnológico de Monterrey, Monterrey, NL, México.
4Kazan Federal University, Department of Microbiology, Kazan, Russian Federation.
5Institute of Biochemistry, Medical School, Justus-Liebig-University, Giessen, Germany.
Corresponding author:
Hector A. Cabrera-Fuentes
Email: cabrera.fuentes.h.a@nhcs.com.sg
In a new window | Download PPT
Figure 1: Overview of endothelial cell response towards acute myocardial infarction. This scheme depicts the reperfusion-induced damage of endothelial cells following acute myocardial infarction.
In a new window | Download PPT
Figure 2: Cardio-protective mediators and molecular mechanisms that are activated by ischemic conditioning in endothelial cells. The illustration enumerates three necessary steps to explain cellular responses after ischemic conditioning-induced cardioprotection.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 15669 | 64 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA