Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Anesthetic Postconditioning in Acute Brain Injury—Anesthetics beyond Anesthesia
Time:2018-09-02
Number:13976
Author Affiliations

Conditioning Medicine, 2018. 1(5):243-258.
Abstract
Acute brain injuries like ischemic stroke, traumatic brain injury and intracerebral hemorrhage, if not treated properly, can leave behind sensory, motor and cognitive functional impairments, which can seriously affect the quality of life of patients. Considerable progress has been made in the treatment of these diseases, including endovascular thrombectomy, and decompressive craniotomy. However, due to their devastating impact on neurological outcomes, investigation into the management of acute brain injuries still warrants further attention. Anesthetic postconditioning is emerging as a promising therapeutic strategy for various forms of acute brain injury. The advantages of anesthetic postconditioning include (1) easy accessibility, (2) high controllability and its great effect on balancing metabolic demand, glutamatergic and GABAergic tone, controlling cortical spreading depression, neuroinflammation and ROS generation in the injured brain, which are all important pathological features of acute brain injuries. The advances in anesthesiology, including the development of new anesthetic agents and the innovation of anesthetic drug delivery system, could contribute to the clinical translation of anesthetic postconditioning in the early management of acute brain injury in an effort to further improve neurological outcomes.
Abstract
Acute brain injuries like ischemic stroke, traumatic brain injury and intracerebral hemorrhage, if not treated properly, can leave behind sensory, motor and cognitive functional impairments, which can seriously affect the quality of life of patients. Considerable progress has been made in the treatment of these diseases, including endovascular thrombectomy, and decompressive craniotomy. However, due to their devastating impact on neurological outcomes, investigation into the management of acute brain injuries still warrants further attention. Anesthetic postconditioning is emerging as a promising therapeutic strategy for various forms of acute brain injury. The advantages of anesthetic postconditioning include (1) easy accessibility, (2) high controllability and its great effect on balancing metabolic demand, glutamatergic and GABAergic tone, controlling cortical spreading depression, neuroinflammation and ROS generation in the injured brain, which are all important pathological features of acute brain injuries. The advances in anesthesiology, including the development of new anesthetic agents and the innovation of anesthetic drug delivery system, could contribute to the clinical translation of anesthetic postconditioning in the early management of acute brain injury in an effort to further improve neurological outcomes.
Introduction
Ischemic stroke, traumatic brain injury (TBI) and intracranial hemorrhage (ICH) are three common acute brain injuries that can be life-threatening and cause long-lasting or even permanent neurological symptoms, such as unconsciousness and sensory, motor and cognitive dysfunctions. Ischemic stroke is the fourth leading cause of death in the United States and second only to ischemic heart disease worldwide (Benjamin et al., 2017). TBI impacts 2.5 million people in the United States each year, leading to 5.3 million Americans with lifelong neurological deficits (Wilson et al., 2017). Primary ICH accounts for 85% of ICH and it affects 4 million people globally (Palm et al., 2013), with 30-day mortality as high as 40-50% (Kuramatsu et al., 2013). Although increased attention has been paid to these catastrophic brain injuries and new invasive procedures and surgical techniques have been developed, patient management remains a huge challenge in clinical practice.
Novel strategies that offer neuroprotection against acute brain injury are emerging that facilitate neurological recovery. Among these strategies, anesthetic postconditioning is a promising intervention with high clinical translation, as no prior knowledge of the ischemic event is required to provide effective protection. Postconditioning, typically referred to as ischemic postconditioning, was originally described by Zhao et al. (2003) to protect against prolonged myocardial ischemia by introducing several cycles of coronary occlusion/reperfusion after a sustained ischemic insult (Zhao et al., 2003; Kin et al., 2004). It can be elicited by brief episodes of ischemia or by administration of pharmacological agents, such as anesthetics. The protective effect of anesthetic postconditioning was first observed against cardiac ischemic and reperfusion injury in 2003 after administration of 1 minimum alveolar concentration (MAC) of sevoflurane during the first two minutes of reperfusion (Obal et al., 2003). Soon this effective protective strategy was used to treat acute brain injuries, including ischemic reperfusion injury, TBI and ICH. Here, we review the pathological changes shared by these acute brain injuries that are emerging as good therapeutic targets of anesthetic postconditioning, such as metabolic impairment, imbalanced glutamatergic and γ-aminobutyric acid (GABA)-ergic tone and cortical spreading depression. In addition, the advances in anesthetic agents and anesthetic delivery systems are also reviewed in an attempt to discuss the translational potential of anesthetic postconditioning in the clinical management of acute brain injury.
1. Acute brain injuries that could potentially benefit from anesthetic postconditioning
The management of acute ischemic stroke, especially mechanical thrombectomy, has evolved significantly during the past two decades (Berkhemer et al., 2015; Campbell et al., 2015; Saver et al., 2015). The thrombectomy procedure may require general anesthesia or conscious sedation, thus allowing early access of stroke patients to anesthetics. Both TBI and ICH have a common characteristic feature, cerebral edema, which can lead to increased intracranial pressure and cerebral ischemia, further promoting secondary injury and herniation of the brainstem down through the base of the skull. The cerebral edema may require surgical decompression, which would increase the likelihood that patients would be exposed to anesthetics early on, thereby receiving anesthetic postconditioning. Therefore, the three most common types of brain injury all have a high probability for surgical procedure intervention, with early access by patients to anesthetics and any accompanying protective postconditioning effects
With or without surgery, all acute brain injuries can potentially benefit from anesthetic postconditioning, which has the advantage of being highly effective and easily administered. The neuroprotective mechanisms underlying anesthetic postconditioning may target the aforementioned pathological changes observed in acute brain injuries, and will be discussed below (Figure 1).
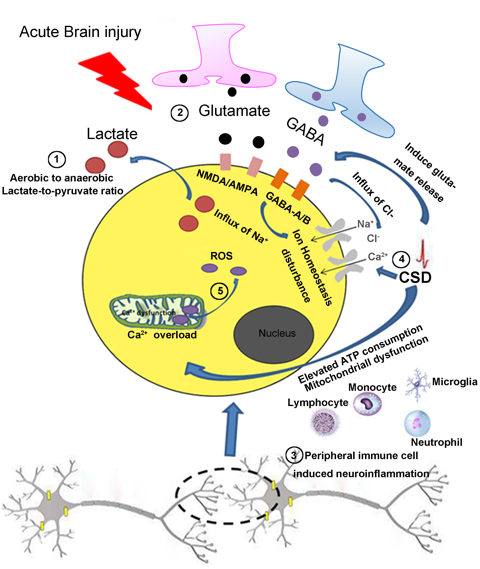
In a new window | Download PPT
Figure 1: Common features in the pathogenesis of acute brain injury. ①After acute brain injury, there is an increased lactate-to-pyruvate ratio, which indicates mitochondrial dysfunction and aerobic to anaerobic glycolysis of the cells in the injured brain. ②Glutamate, an excitatory neurotransmitter, induces excitotoxicity and an influx of Na+ by acting on NMDA/AMPA receptors. GABA an inhibitory neurotransmitter, induces the influx of Cl- by acting on GABA-A/B receptors. ③CSD induces the release of glutamate, promotes the influx of Ca2+ and Na+ then induces derangement of the ionic environment. CSD also elevates ATP consumption and causes mitochondrial dysfunction. ④ Resident glia cells and infiltrating peripheral immune cells can be activated after acute brain injury resulting in neuroinflammation. ⑤ After acute brain injury, voltage-gated Ca2+ channels are activated that accelerates the collapse of electron transport chain complexes in mitochondria, thus generating excessive ROS. NMDA, N-methyl-d-aspartate; AMPA, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; GABA, Gamma-aminobutyric acid; CSD, cortical spreading depression; ATP, adenosine triphosphate.
2. Therapeutic targets of anesthetics in acute brain injury
1) Metabolic impairments in the pathogenesis of acute brain injury
All acute brain injuries are associated with impairments in energy metabolism, resulting in disturbed ionic homeostasis, mitochondrial defects, hypoxia and inflammation (Carpenter et al., 2015; Guglielmetti et al., 2017; Simon et al., 2017). Microdialysis of brain metabolites, metabolomics or indirect measurement of arterio-venous metabolite concentrations demonstrated that injured brain tissue is characterized by high lactate levels, a high lactate/pyruvate ratio, altered glycolysis and purine metabolism (Gallagher et al., 2009; Jalloh et al., 2013; Carpenter et al., 2014; Lama et al., 2014; Koronowski et al., 2017; Koronowski et al., 2018). Studies using non-invasive proton magnetic resonance spectroscopy (H-MRS) revealed elevated lactate concentration in patients following brain injury and the level of lactate correlated with poor prognosis, cognitive decline and high mortality (Makoroff et al., 2005; Timofeev et al., 2011; Sala et al., 2013). With the development of in vivo real-time imaging, hyperpolarized 13C magnetic resonance spectroscopic imaging (HP 13C MRSI) has emerged as a clinically translatable neuroimaging method that notably improves the detection of metabolic conditions by monitoring the increased conversion of HP [1-13C] pyruvate to HP [1-13C] lactate under aerobic conditions. Using this new technique, it has been recently shown that there is an increased HP [1-13C] lactate-to-pyruvate conversion, which indicates mitochondrial dysfunction, and decreased activity of pyruvate dehydrogenase (PDH), the enzyme that converts pyruvate to Acetyl-coenzyme A (CoA), in the injured cortex at acute time points prior (4 hours) and also at acute (12 and 24 hours) and subacute (7 days) time points following TBI (DeVience et al., 2017; Guglielmetti et al., 2017). In ischemic stroke, the penumbra of stroke patients is a metabolically active region with reduced cerebral blood flow, characterized by an increased oxygen extraction fraction and preserved oxygen consumption (Marchal et al., 1993; Baron, 1999), thus metabolism switches from aerobic to anaerobic glycolysis, producing lactate. Now, using 1H magnetic resonance spectroscopy (MRS), changes in lactate levels in the ischemic region can be detected (Holmes et al., 2012). Using Xenon-enhanced computed tomography or computed tomography perfusion for cerebral blood flow (CBF), it was confirmed that the lactate/pyruvate ratio was persistently elevated in the perihemorrhagic zone compared to the seemingly normal cortical region (Tobieson et al., 2018). Further, increased acidosis is associated with increased cerebral ischemic brain injury, which can be observed in recurrent hypoglycemia exposed insulin treated diabetic rats (Rehni et al., 2018; Shukla et al., 2018). Thus, metabolic disturbances characterized by increased lactate/pyruvate ratio represent one of the important pathological changes of acute brain injuries.
There are several ways to combat the metabolic changes following acute brain injury. First, continued maintenance of aerobic metabolism is key to the survival of tissue since it is significantly more efficient at producing adenosine triphosphate (ATP) than anaerobic metabolism. Hence, it is critical to decrease the metabolism of the injured brain to salvage the tissue at risk. Notably anesthetic postconditioning is a good therapeutic choice for acute brain injury because it could effectively decrease brain metabolism (Alkire et al., 1995; Alkire, 1998; Bonhomme et al., 2001; Kaisti et al., 2002; Kaisti et al., 2003; Jeong et al., 2006; Schlunzen et al., 2012). Different anesthetics produce different characteristic effects on brain metabolism and CBF. Sevoflurane, a widely used inhalation anesthetic, decreases cerebral metabolism more than CBF (Kaisti et al., 2002; Kaisti et al., 2003; Jeong et al., 2006), while propofol induces a proportional decrease in both brain metabolism and CBF (Alkire et al., 1995; Alkire, 1998; Bonhomme et al., 2001; Schlunzen et al., 2012). Dexmedetomidine, an α2-adrenoceptor agonist, which induces sufficient sedation while allowing patient arousal, can significantly reduce the glucose metabolism rate. Furthermore, the suppressive effect of dexmedetomidine is even stronger than propofol (Laaksonen et al., 2018). It can also produce a 33% global decrease of CBF from baseline (Zornow et al., 1993; Prielipp et al., 2002). It was suggested that the cerebral metabolic rate and CBF coupling is preserved during dexmedetomidine administration in healthy volunteers (Drummond et al., 2008).
Second, direct suppression of the metabolic rate by inducing a “hibernation-like state” is also an intriguing choice to achieve neuroprotection against acute brain injury. Hibernation, also termed torpor, is a state of inactivity and metabolic depression in endotherms during winter seasons to conserve energy when sufficient food is unavailable (Dave et al., 2012; Geiser, 2013). During hibernation, metabolic rate can be reduced as low as 1%, CBF dropped to 10% and glucose utilization to 2% of active state (Storey and Storey, 2010; Geiser, 2013). This hypometabolism allows animals in deep torpor to withstand drastic fluctuations in CBF without causing damage to the brain in case of stroke and TBI (Zhou et al., 2001; Dave et al., 2012). Brains of torpor animals, such as arctic ground squirrels are remarkably tolerant to global cerebral ischemia during euthermia (Zhou et al., 2001; Dave et al., 2012). Therefore, an artificially induced “hibernation-like” state is a promising strategy to protect against acute brain injuries, such as stroke and TBI. There are several methods that induce a neuroprotective hibernation-like state, including therapeutic hypothermia, anesthetics, ethanol, ischemic preconditioning (Stenzel-Poore et al., 2003; Thompson et al., 2013a) and resveratrol preconditioning (Koronowski et al., 2015; Khoury et al., 2016). Resveratrol preconditioning offers a wide therapeutic intervention window through activation of nuclear factor erythroid-2 related factor 2 (Nrf2), an astrocyte-enriched transcription factor that has previously been shown to upregulate cellular antioxidant systems in response to ischemia (Narayanan et al., 2015; Narayanan et al., 2018). Notably, a number of anesthetic agents are excellent candidates to induce artificial hibernation, including isoflurane, sevoflurane and propofol. Each of these anesthetic agents has been confirmed to reduce cerebral metabolic rates (Alkire et al., 1995; Alkire, 1998; Bonhomme et al., 2001; Kaisti et al., 2002; Kaisti et al., 2003; Jeong et al., 2006; Schlunzen et al., 2012).
Finally, sirtuins are emerging as important targets for neuroprotection after cerebral ischemia (Thompson et al., 2013b; Koronowski and Perez-Pinzon, 2015; Morris-Blanco et al., 2016; Koronowski et al., 2017; Koronowski et al., 2018). Sirtuins are a family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases with homology to the yeast silent information regulator 2 (Sir2). There are seven mammalian members of the sirtuin family (SIRT1–7), which localize to different subcellular compartments. Among these sirtuins, mitochondrial SIRT1 has been suggested to modulate mitochondrial function and to regulate glycolysis after global cerebral ischemia (Thompson et al., 2013b; Koronowski and Perez-Pinzon, 2015). SIRT5 regulates oxygen consumption in the mitochondria and modulates purine metabolism (urea, adenosine, adenine, xanthine), nitrogen metabolism (glutamic acid, glycine), and malate-aspartate shuttle (malic acid, glutamic acid) after focal ischemia (Morris-Blanco et al., 2016; Koronowski et al., 2018). Therefore, both SIRT1 and SIRT5 are promising therapeutic targets through which neuroprotection can be achieved via metabolic regulation. Previous studies have demonstrated that SIRT1 and SIRT5 play important roles in the metabolic regulation and neuroprotection afforded by ischemic preconditioning and resveratrol preconditioning in ischemic brain injury (Thompson et al., 2013b; Koronowski and Perez-Pinzon, 2015; Morris-Blanco et al., 2016; Koronowski et al., 2017; Koronowski et al., 2018). However, currently, there is no evidence that anesthetic agents influence the activity of SIRT1 and SIRT5, making this an intriguing direction for future investigation.
2) Imbalanced glutamatergic and GABAergic tone in the brain after acute brain injury
In the brain, there are excitatory and inhibitory neurotransmitters that balance each other to maintain normal neurologic function. Glutamate is the principle excitatory neurotransmitter and GABA is the primary inhibitory neurotransmitter. Glutamate is synthesized from glutamine in presynaptic glutamatergic neurons, after which it is stored in presynaptic vesicles, and released into the synaptic cleft by a depolarizing current. It acts on ionotropic or metabotropic G-protein coupled receptors to induce an intracellular signaling cascade. The ionotropic receptors include N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), and kainate receptors.
In the early phase of acute brain injury, such as TBI, the immediate glutamate release sets off a series of metabolic cascades, subsequently leading to excitotoxicity and eventually to cell death (Zlotnik et al., 2012; Goodrich et al., 2013). This cell death leads to functional impairments, including hypoperfusion due to failure in cerebrovascular autoregulation, edema via accumulation of intracellular Na+, glutamate-induced excitotoxicity, epileptogenesis, and cognitive decline (Smith et al., 1993; Obrenovitch and Urenjak, 1997; Zlotnik et al., 2012). Decreasing acute glutamate excitotoxicity has been widely accepted to provide neuroprotection against acute brain injury, including stroke and TBI. Indeed, removing glutamate from the synapse has been suggested to provide neuroprotection (Zlotnik et al., 2012; Goodrich et al., 2013). In contrast, high levels of glutamate release have been demonstrated in acute ischemic stroke (Davalos et al., 1997) or ICH (Liu and Sharp, 2012). Inhibiting glutamate release by using antioxidants, or granulocyte-colony stimulating factor provides neuroprotection against stroke (Hurtado et al., 2003; Han et al., 2008; Dohare et al., 2014). Decreasing the glutamate receptor AMPAR currents by protein kinase C epsilon (PKCɛ) activation confers neuroprotection against global cerebral ischemia (Cohan et al., 2017).
GABA is the principle inhibitory neurotransmitter in the central nervous system and is synthesized from glutamate by glutamate decarboxylase (GAD). It is also stored in presynaptic vesicles and is released onto postsynaptic terminals and acts on GABA receptors located on dendritic projections, axons or axon terminals. There are two types of GABA receptors, GABAA and GABAB. GABAA are ionotropic receptors that cause Cl- channel opening and GABAB are metabotropic G-protein coupled receptors. GABAA receptors contain at least 16 subunits among which, the α1, γ2, α4 and δ1 receive the most attention in TBI research. After TBI, down-regulation of the ε- and θ-subunits of the GABAA receptor correlates with susceptibility to post-traumatic epileptogenesis (Huusko and Pitkanen, 2014). Immunoreactivity of the GABAA receptor α1 subunit was significantly decreased in the ipsilateral thalamus 5-6 days after TBI, and it was further decreased in the cortical lesion area and thalamus at 40-42 days after TBI. These data correlate with the reduction in the GABAA signal measured using ex vivo autoradiography with 18F-GE-194 (Lopez-Picon et al., 2016). There are differential alterations in tonic and phasic GABAA receptor currents in hippocampal dentate granule cells 90 days after controlled cortical impact, which was associated with the development of posttraumatic epilepsy after TBI (Mtchedlishvili et al., 2010). Activation of GABAA receptors by ischemic preconditioning confers neuroprotection in rat organotypic hippocampal slices early after oxygen-glucose deprivation (OGD) injury (DeFazio et al., 2009). However, in the late phase of ischemic injury, the role of GABAA receptor differs from that in the early phase, as excessive tonic GABA activity in hippocampal interneurons late after stroke leads to impaired synaptic plasticity and memory deficits (Orfila et al., 2017). Selective inhibition of extra-synaptic α5-GABAA receptors by S44819 enhanced hippocampal synaptic plasticity and thus improved cognitive function in neurodegenerative disorders and facilitated long-term post-stroke recovery (Etherington et al., 2017). Therefore, regulating GABA activity in the acute phase after ischemia is an important means by which damage incurred from acute brain injury can be minimized and also to enhance long-term functional outcome of patients with TBI and ischemic stroke. However, an association between GABA activity and ICH is still lacking and thus, warrants further investigation.
There are a number of anesthetic agents, such as midazolam, ketamine, volatile and intravenous anesthetics that have been found to decrease glutamate release or increase local concentrations of GABA after acute brain injury (Table 1) (Bickler et al., 1995; Sakai and Amaha, 2000; Lehner et al., 2010). Midazolam (0.3-30 μM) inhibits glutamate release via the glial glutamate transporter (GLT-1) (Sakai and Amaha, 2000), significantly increases the local concentration of GABA (0.75 mg/kg, i.p), and inhibits the expression of contextual fear in anxious rats (Lehner et al., 2010). Midazolam acts on the GABAA receptor and leads to spontaneous Ca2+ oscillation in cultured hippocampal neurons (Sinner et al., 2006). Propofol (0.5 µM) yields analogous potentiation of GABA-mediated currents, prevents the rise in reactive oxygen species (ROS), and inhibits intracellular release of apoptosis-inducing factor (AIF), thus preventing neuronal cell loss in rat brainstem slices (Ghezzi et al., 2017). By decreasing glutamate release and increasing local GABA concentrations, a variety of anesthetic agents, as described above, alter the glutamatergic and GABAergic tone in the brain after acute brain injury. Thus, regulation of glutamatergic and GABAergic tone may be important mechanisms by which anesthetic postconditioning exerts its neuroprotection against acute brain injury.
Table 1. Excitatory and inhibitory receptors targeted by anesthetic agents in acute brain injury.
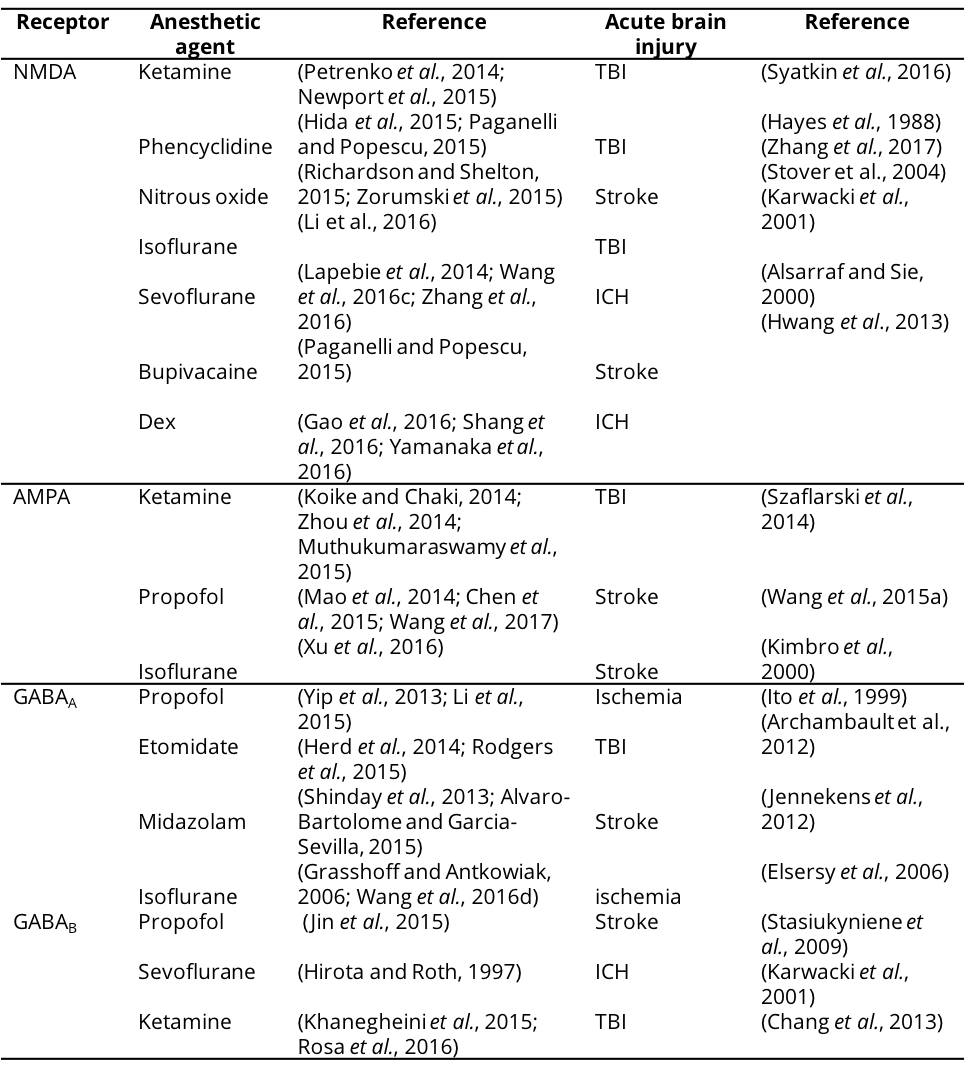
NMDA, N-methyl-D-aspatic acid; AMPA, α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; GABA, γ-aminobutyric acid; Dex, Dexmedetomidine; TBI, Traumatic brain injury; ICH, Intracranial hemorrhage.
3) Cortical spreading depression deteriorates neurological outcomes of acute brain injury
Cortical spreading depression (CSD) is a slowly propagating (2-5 mm/min) wave of rapid, near-complete depolarization of neurons and astrocytes followed by a period of electrical suppression of a distinct population of cortical neurons (Dreier et al., 2017; Hartings et al., 2017). These events contribute to the progression of brain injury in humans. More specifically, CSD refers to the electrical silence of brain electrical activity following spreading depolarization (Lauritzen et al., 2011). Unlike a normal neuronal action potential, CSD induces a greater magnitude of extracellular potential shift, which continues for at least several minutes (Ayata and Lauritzen, 2015). It commonly occurs in individuals with migraine, aneurismal subarachnoid hemorrhage (SAH), delayed ischemic stroke after SAH, malignant hemispheric stroke, spontaneous intracerebral hemorrhage, or TBI (Hadjikhani et al., 2001; Dohmen et al., 2008; Hartings et al., 2011; Lauritzen et al., 2011; Dreier and Reiffurth, 2015).
The occurrence of CSD is usually followed by several distinct phases of secondary changes in CBF, an initial brief hypoperfusion, a marked but transient hyperemia, a later and smaller hyperemia, and a long-lasting oligemia. CSD can induce the release of glutamate, which later binds to NMDA receptors to further promote the influx of Ca2+ and Na+. CSD not only induces derangement of the ionic environment but it also elevates ATP consumption and imposes a considerable energetic burden on brain tissue (Shinohara et al., 1979; Kocher, 1990; Shibata and Suzuki, 2017), exacerbating tissue acidosis (Menyhart et al., 2017). In addition to neurons and astrocytes, it has been recently suggested that microglia also exhibit dramatic changes in activity in response to CSD, such as increased NMDA-dependent inward rectifying potassium conductance (Wendt et al., 2016), increased expression of major histocompatibility complex (MHC) class II antigen (Gehrmann et al., 1993), increased ROS production (Grinberg et al., 2012), and increased secretion of IL-1β (Jander et al., 2001) and TNFα (Grinberg et al., 2013). With limited ATP supply, increased excitotoxicity and increased inflammation, CSD could contribute to the expansion of infarct volume and brain injury after cerebral ischemic stroke or TBI (Risher et al., 2010; Hinzman et al., 2015). After ischemic stroke, depolarization occurs spontaneously in the tissue surrounding the freshly developing ischemic infarct (i.e. ischemic penumbra), which is functionally and metabolically compromised. Peri-infarct depolarization (PID) refers to slow potential changes in the absence of electrocorticography (ECoG) background activity in ischemic stroke. PID usually occurs in brain tissue that is not yet irreversibly damaged, but this tissue damage may be augmented when PIDs appear in clusters after cerebral artery occlusion (Saito et al., 1997; Ohta et al., 2001). In contrast, negative slow voltage variations with simultaneous transient ECoG suppressions are signatures of CSD. The CSD/PID events can be detected over the course of 7 days after stroke. Lesion progression has recently been demonstrated by sequential MR imaging in a malignant stroke patient specifically in the zone experiencing 100 PIDs over a period of 5 days (Nakamura et al., 2010). Similar deterioration has recently been observed in subarachnoid hemorrhagic patients. Measurement of tissue oxygen partial pressure revealed progressive stepwise decline with subsequent CSDs within clusters (Bosche et al., 2010). These clusters of CSDs are associated with delayed ischemic neurological deficits (Dreier et al., 2006). Thus, it is well established that CSDs occur after the three most common acute brain injuries, with potentially devastating impact on neurological outcomes, and thus CSDs may well turn out to be an important mechanistic target for neuroprotection.
Fortunately, multiple anesthetic agents have been shown to effectively suppress CSDs. Some analgesics and sedative drugs, such as benzodiazepines and barbiturates (GABA receptor agonists), and ketamine (NMDA receptor blocker) have been shown to alter the susceptibility to and the course of spreading depolarization by regulating neuronal activity and synaptic transmission (Somjen, 2001; Sakowitz et al., 2009). The administration of ketamine reduces the occurrence of isoelectric spreading depolarization in patients suffering from TBI and malignant hemispheric stroke (Hertle et al., 2012). Inhalation anesthetics, such as isoflurane, are also suggested to suppress CSDs and improve recovery from the metabolically demanding CSD waves in neurocritical care patients (Takagaki et al., 2014). Although GABAA receptor (GABAAR) antagonists can be sufficient to generate spreading depression, (Kohling et al., 2003), propofol, a GABAAR agonist, did not effectively prevent the CSD event or the propagation rate of CSDs, which may likely be due to a ceiling effect (Aiba and Shuttleworth, 2014; Takagaki et al., 2014). Therefore, attenuation of CSD may be an important underlying mechanism through which anesthetic postconditioning exerts its neuroprotective effect against acute brain injury.
4) Neuroinflammation
Mounting evidence suggests that ischemic stroke, TBI, and ICH all inevitably cause neuroinflammation, which can be caused by the activation of resident glia cells or by infiltrating peripheral immune cells (An et al., 2014; Corps et al., 2015). Notably, unlike the primary brain injury itself, neuroinflammation can often cause extensive and lasting damage through a complex cascade of events, thus it is usually referred to as “secondary injury.” Importantly, the neuroinflammation, which can be detected during the subacute phase (≤ 3 weeks post-injury) by positron emission tomography imaging with the translocator protein radioligand [18F]PBR111 and diffusion tensor imaging, is suggested to correlate with chronic TBI deficits (Missault et al., 2018). The damage and pathogen-associated molecular patterns (e.g.alarmins) released from the injured brain interact with receptors on inflammatory cells, such as toll-like receptors (TLRs), nucleotide oligomerization domain-like receptors and scavenger receptors to initiate the inflammatory cascade after TBI, stroke, SAH and ICH (Jassam et al., 2017; PrabhuDas et al., 2017; Lu et al., 2018). It has been recently suggested that the inflammation that occurs in injured brains after acute brain injury may not always be detrimental to neurological recovery. There are various types of immune cells that can alleviate inflammation and promote the clearance of brain debris after acute brain injury, such as regulatory T cells, regulatory B cells, regulatory dendritic cells, M2-phenotype microglia, macrophage, and monocytes (Liesz et al., 2009; Ren et al., 2011; Li et al., 2013; Hu et al., 2015; Jassam et al., 2017; Zhou et al., 2017a; Zhou et al., 2017b).
Although the network of neuroinflammation is complex, sevoflurane and propofol, two commonly used general anesthetics, have been suggested to affect neuroinflammation after ischemic stroke and TBI. However the evidence regarding sevoflurane is conflicting. Propofol attenuates microglia-mediated proinflammatory responses after ischemic stroke and TBI (Luo et al., 2013; Zhou et al., 2013; Zheng et al., 2018). However, sevoflurane may either change microglia/macrophage dynamics to promote brain repair after stroke or aggravate microglia-mediated neuroinflammation and exacerbate cognitive decline (Zhu et al., 2017a; Dang et al., 2018; Dong et al., 2018). The direct effect of anesthetics on the inflammatory response of the brain is conflicting; however, these conflicting results may derive from the use of different treatment paradigms across studies. It still remains unknown how anesthetic postconditioning impacts the neuroinflammation after acute brain injury. Further investigation into the effect that anesthetic postconditioning have on neuroinflammation after acute brain injury are therefore warranted.
5) Free radical generation and mitochondria dysfunction
Acute brain injury produces high levels of ROS, which can be lethal to neurons. In ischemic stroke, rapid depletion of energy leads to a loss of membrane potential and depolarization, activating voltage-gated Ca2+ channels, which release excitatory amino acids, such as glutamate, into the extracellular space. The binding of glutamate to NMDA and AMPA receptors further deteriorates Ca2+ homeostasis, which accelerates the collapse of electron transport chain complexes in mitochondria, thus generating excessive ROS (Doyle et al., 2008). Excessive ROS and Ca2+ activate calpain protease, which promotes apoptosis and other forms of programmed cell death, primarily through modification of proteins and lipids present at the outer membrane of the mitochondria in a caspase-dependent or caspase-independent manner (Perez-Pinzon et al., 2012). Ischemia induces NAD+ depletion and compromises several NAD+-dependent processes that may ultimately lead to cell death (Khoury et al., 2018). Inhibiting the activation of protein kinase C in the mitochondria by ischemic preconditioning can induce ischemic tolerance by preserving mitochondrial pools of NAD+ and nicotinamide phosphoribosyltransferase, an enzyme involved in NAD+ production (Dave et al., 2011; Morris-Blanco et al., 2014; Thompson et al., 2015). Increased ROS production is also associated with axonal alterations after TBI, which is characterized by diffuse axonal injury (Frati et al., 2017). Similar to the ROS generation in ischemic neurons, increased intracellular Ca2+ in the mitochondria produces excessive ROS in the axons, causing swellings or varicosities along the axons and terminal bulbs (Siedler et al., 2014). In intracerebral hemorrhage, the initial hematoma induces glutamate release and leads to mitochondrial dysfunction and ROS production (Brunswick et al., 2012). Mitochondrial dysfunction leads to insufficient ATP generation resulting in the failure of cellular pumps causing cytotoxic edema and neuronal apoptosis (Kim-Han et al., 2006). Thus, ROS is also an important common pathological change that could be targeted to improve neurological outcomes after acute brain injury.
Mounting evidence suggests that ROS generation can also be targeted by several anesthetics (Yu et al., 2015a; Bellanti et al., 2016; Wang et al., 2016e; Ghezzi et al., 2017; Shinjo et al., 2018). Propofol has been proposed to protect cardiomyocytes and hepatic cells from oxidative stress presumably by reducing ROS production (Bellanti et al., 2016; Ghezzi et al., 2017; Shinjo et al., 2018). However, there are conflicting reports as to whether propofol increases ROS production and oxidative stress in developing neurons (Liang et al., 2018). Likewise, evidence regarding the role of sevoflurane in ROS production is also conflicting. However, sevoflurane postconditioning is consistently protective against oxidative stress in multiple cell types (Yu et al., 2015a; Wendt et al., 2016). The discrepancy of the role of anesthetics in ROS production may due to differences in the experimental parameters, such as the dose and duration of anesthesia, as well as the disease models being investigated. How postconditioning of propofol or sevoflurane affects the underlying pathology of acute brain injury therefore warrants further investigation. Besides the aforementioned common features that are shared by different acute brain injuries, there are also some signaling cascades that may mediate the neuroprotective effects of anesthetic postconditioning after acute brain injury (Table 2).
Table 2 Anesthetic postconditioning protocols and related signaling pathways.
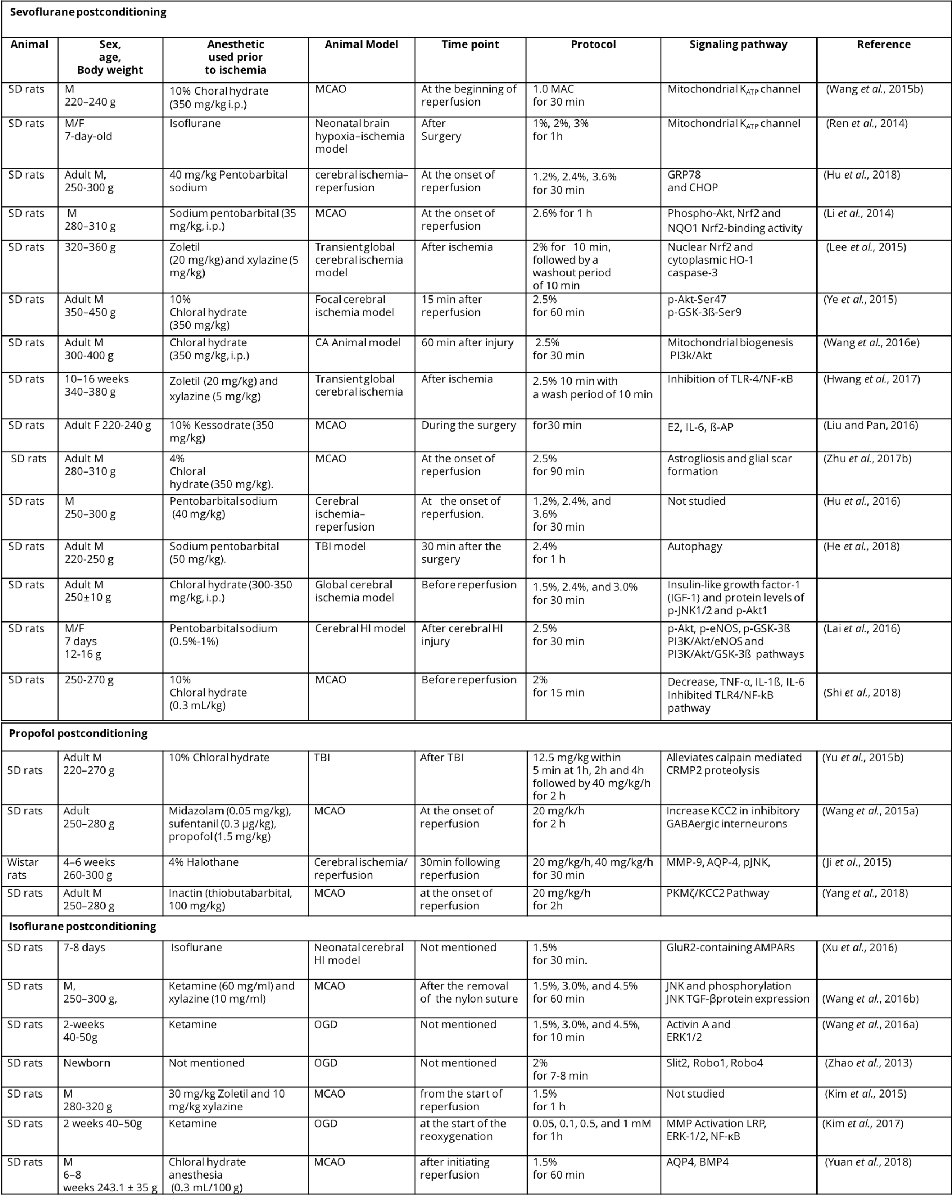
M, male; F, female; SD rats, Sprague-Dawley rats ;TBI, traumatic brain injury; MCAO, middle cerebral artery occlusion; MAC, minimum alveolar concentration; CA, cardiac arrest; HI, hypoxia-ischemia; CRMP2, Collapsin response mediator protein-2; DIOA,[(dihydroindenyl)oxy] alkanoic acid; OGD, oxygen-glucose deprivation; GRP78, glucose-regulated protein-78; CHOP, C/EBP-homologous protein; Phospho-Nrf2, Phosphorylated nuclear factor-erythroid 2-related factor 2; Akt, protein kinase B; NQO1, quinine oxidoreductase1; HO-1, hemoxygenase-1; PI3k, phosphatidylinositol-3-kinase; TLR-4, toll-like receptor-4; NF-κB, nuclear factor kappa B; E2, serum estradiol-2; IL-6, interleukin-6; β-AP, beta-amyloid protein; TNF-α, tumor necrosis factor-α; eNOS, endothelial nitric oxide synthase; MMP-9, matrix metalloproteinase-9; AQP-4, aquaporin-4; pJNK, phosphorylated c-Jun N-terminal kinase; KCC2; potassium chloride co-transporter 2; PKMζ, protein kinase Mζ; TGF-β, transforming growth factor-β; ERK1/2, extracellular signal regulated kinase1/2; LRP, lipoprotein receptor-related protein ; AQP4,Aquaporins4; BMP4, bone morphogenetic protein 4.
3. Paradigms of anesthetic postconditioning that may combat acute brain injuries
Although the formal term anesthetic postconditioning is relatively new, the idea of using anesthetics to sedate patients with acute brain injury has a long history, and has been practiced in clinical settings for many years. Anesthesia-induced pharmacological coma is a common approach used in the intensive care unit to treat refractory intracranial hypertension (Vutskits, 2014). A variety of anesthetics have been shown to have neuroprotective effects against TBI, such as propofol, pentobarbital, and sevoflurane. Pentobarbital is a commonly used sedatives used to decrease cerebral oxygen consumption in the acute phase of TBI (Carney et al., 2017). Pentobarbital infusion changes the metabolomics of TBI patients compared to TBI patients who did not receive pentobarbital (Wolahan et al., 2018). Pentobarbital coma used in TBI patients could help to maintain higher cerebral perfusion pressure and is associated with a higher 1 year-survival rate in patients with severe TBI and refractory intracranial hypertension (Marshall et al., 2010). However, it is important to be cautious of the fact that arterial hypotension could be observed with barbiturates (Roberts and Sydenham, 2012), bolus of midazolam (Papazian et al., 1993) or bolus of opioids (Albanese et al., 1999). Control of system hemodynamics is of great importance for acute brain injuries, such as TBI and ICH, especially for those whose intracranial compliance is compromised (Helbok et al., 2012). It was shown that the daily interruption of sedation or neurological wake-up test might cause cerebral hypoperfusion and raise intracranial pressure. This can lead to an imbalance of energy supply and demand, especially for the injured brain, and can increase the risk for metabolic distress and brain tissue hypoxia (Helbok et al., 2011; Helbok et al., 2012). Overall, the current guidelines on sedation and analgesia in the intensive care unit should be extended to stabilize brain injury patients (Barr et al., 2013). Using clinical scores and protocols to manage sedation and analgesia may provide neurological benefits and reduce the risk of over sedation and hemodynamic instability (Egerod et al., 2010; Yu et al., 2013).
Since 2003, when the idea of anesthetic postconditioning was first introduced, a lot of studies on inhalation anesthetic postconditioning and propofol-induced postconditioning have been published involving a variety of conditioning paradigms.
1) Inhalation anesthetic postconditioning
Anesthetic postconditioning can be achieved by administration of 1 minimum alveolar concentration of sevoflurane for only 2 minutes or 2% of isoflurane for 15 minutes at the onset of ischemic reperfusion in a rat myocardial ischemia model (Obal et al., 2003; Feng et al., 2005; Lucchinetti et al., 2005; Obal et al., 2005). In the brain, anesthetic postconditioning was first introduced in 2008 in a study in which 2% isoflurane was applied for 30 minutes starting 10 minutes after OGD in brain slices or maintained under anesthesia with 2% isoflurane via an endotracheal tube for 60 min in a middle cerebral artery occlusion (MCAO) rat model (Lee et al., 2008).
2) Propofol-induced postconditioning
Propofol postconditioning at doses of 10 or 20 mg/kg/h infusion rate at the onset of reperfusion for 30 minutes could provide not only early neuroprotection but also long term (28 days after reperfusion) neuroprotection in transient MCAO rats (Wang et al., 2009; Wang et al., 2011). Variations in the protocols of anesthetic postconditioning are listed in Table 2.
4. Advances in anesthesiology brings anesthetic postconditioning to a new era
1) Development of anesthetic agents
In the past two decades, both anesthetic agents and anesthetic delivery systems have experienced evolutional changes. New formulations of existing agents and new chemical entities have been developed to improve safety, predictability, efficacy, onset and recovery profile and to minimize side effects. Currently, some general anesthetic agents that are used in routine clinical practice have been tested in anesthetic postconditioning, including propofol, sevoflurane, and isoflurane. In addition, some newly developed anesthetic agents are proving to be good candidates for anesthetic postconditioning because of their unique pharmacological properties.
Remimazolam is a new, fast- and short-acting, ester-based anesthetic agent targeted for procedural use in the United States. It was initially developed for the sedation of adult colonoscopy patients (Borkett et al., 2015). It combines the properties of two unique drugs already established in anesthesia, midazolam and remifentanil, acting on both GABA receptors, as does midazolam, and exhibiting pharmacokinetic properties common to the ester-based opioid remifentanil, thus achieving a more rapid onset and faster recovery. Because it is primarily cleared by tissue esterase enzymes, accumulation should not occur after infusion as it has a relatively short context-sensitive half-life of 7-8 minutes (Wiltshire et al., 2012).
Xenon, which was first administered to humans in 1951 (Cullen and Gross, 1951), has been suggested as an important neuroprotective agent. It offers the lowest blood gas partition coefficient of any anesthetic, is non-flammable, non-teratogenic, and has a minimal effect on the cardiovascular system. Importantly, it has no deleterious effects on neurocognition in non-human primate models (Derwall et al., 2008; Chakkarapani et al., 2012). With the above advantages and a faster emergence than both inhalation and propofol anesthesia (Law et al., 2016), it may have great translational potential as an anesthetic postconditioning agent.
2) Advances in anesthetic drug delivery system
Innovations in computer technologies have fostered the development of new anesthetic drug delivery systems, such as target-controlled infusions (TCI) (Figure 2), closed-loop drug delivery systems, and computer-assisted personalized sedation systems. With TCI, intravenous drugs are delivered based on computer models, with the goal of achieving a defined or target drug concentration at a specific site (Absalom et al., 2016). TCI is now widely used in clinical practice to administer propofol and opioids for sedation and general anesthesia to millions of patients each year (except in the USA). It was designed to overcome the anesthetic accumulation in the tissue during drug delivery by adjusting the infusion rate based on patient characteristics (weight, height, age, sex, and additional biomarkers) to achieve and maintain a steady-state drug concentration in the plasma or target site (Struys et al., 2016). There are now two types of TCI systems, open loop system and closed-loop system. The open loop system infuses drugs based on the drug library incorporated in the device but lacks real-time feedback from the patient. The closed-loop system monitors hypnosis to determine a new input to the system (Hemmerling et al., 2013). Compared to the open-loop system, the closed-loop system offers advantages of precise dosing, less workload, better control of sedation or anesthesia depth, less drug consumption and improved hemodynamic stability. These new innovations in anesthetic agents and drug delivery have the potential to advance the field of anesthetic postconditioning to more widespread use to treat acute brain injury.
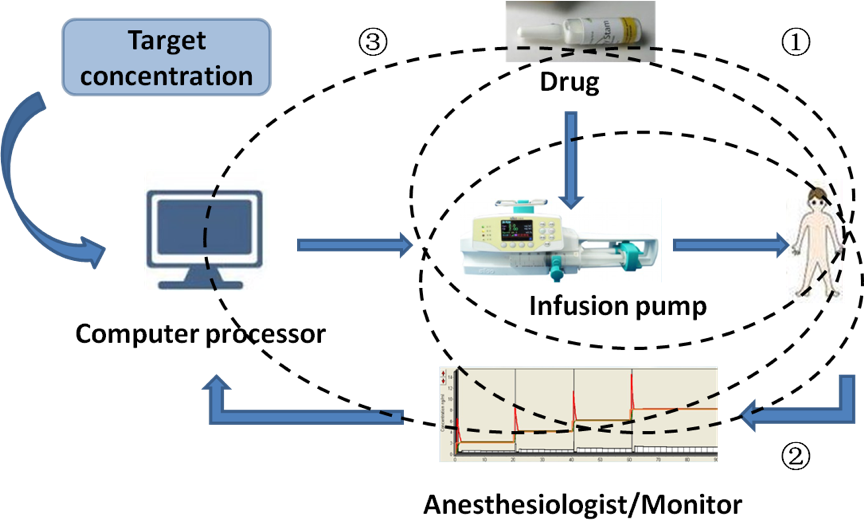
In a new window | Download PPT
Figure 2: Advances in anesthetic drug delivery system. ①TCI refers to the maintenance of appropriate anesthesia depth by adjusting the drug concentration at the target site based on the principles of pharmacokinetics and pharmacodynamics when injecting intravenous anesthetics. ②Closed-loop drug delivery system is an automatic control technological device with feedback signals to control TCI data and pumping speed. ③Computer-assisted personalized sedation system is an intravenous drug delivery device that uses computer models, with the goal of achieving a defined or target drug concentration at a specific site. TCI, target controlled infusion.
Concluding remarks
Finally, we would like to concede that most of our knowledge on anesthetic postconditioning in acute brain injury is derived from animal models. However, considering that anesthetic agents are already being widely used for general anesthesia and sedation, the clinical translation of anesthetic postconditioning is particularly promising. This is especially true with the advances in anesthesiology, which brings safer and easier controllable anesthetic agents and drug delivery systems to patients’ bedside. Large-scale clinical trials will be needed to verify the effectiveness of anesthetic postconditioning in different types of acute brain injury.
Disclosures/conflicts
The authors declare they have no conflicts of interest.
Acknowledgements
P.L. is supported by the National Natural Science Foundation of China (NSFC) grant (81722017) and the Shanghai Rising-Star Program (16QA1402600).
References
Tingting Huang1*
1Department of Anesthesiology, Renji Hospital, School of Medicine, Shanghai Jiaotong University.
Yan Li1*
1Department of Anesthesiology, Renji Hospital, School of Medicine, Shanghai Jiaotong University.
Peiying Li1
1Department of Anesthesiology, Renji Hospital, School of Medicine, Shanghai Jiaotong University.
Corresponding author:
Dr. Peiying Li
Email: peiyingli.md@gmail.com
Tingting Huang and Yan Li contributed equally to this article.
In a new window | Download PPT
Figure 1: Common features in the pathogenesis of acute brain injury. ①After acute brain injury, there is an increased lactate-to-pyruvate ratio, which indicates mitochondrial dysfunction and aerobic to anaerobic glycolysis of the cells in the injured brain. ②Glutamate, an excitatory neurotransmitter, induces excitotoxicity and an influx of Na+ by acting on NMDA/AMPA receptors. GABA an inhibitory neurotransmitter, induces the influx of Cl- by acting on GABA-A/B receptors. ③CSD induces the release of glutamate, promotes the influx of Ca2+ and Na+ then induces derangement of the ionic environment. CSD also elevates ATP consumption and causes mitochondrial dysfunction. ④ Resident glia cells and infiltrating peripheral immune cells can be activated after acute brain injury resulting in neuroinflammation. ⑤ After acute brain injury, voltage-gated Ca2+ channels are activated that accelerates the collapse of electron transport chain complexes in mitochondria, thus generating excessive ROS. NMDA, N-methyl-d-aspartate; AMPA, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; GABA, Gamma-aminobutyric acid; CSD, cortical spreading depression; ATP, adenosine triphosphate.
In a new window | Download PPT
Figure 2: Advances in anesthetic drug delivery system. ↘TCI refers to the maintenance of appropriate anesthesia depth by adjusting the drug concentration at the target site based on the principles of pharmacokinetics and pharmacodynamics when injecting intravenous anesthetics.♭Closed-loop drug delivery system is an automatic control technological device with feedback signals to control TCI data and pumping speed.♩Computer-assisted personalized sedation system is an intravenous drug delivery device that uses computer models, with the goal of achieving a defined or target drug concentration at a specific site. TCI, target controlled infusion.
Table 1. Excitatory and inhibitory receptors targeted by anesthetic agents in acute brain injury.
NMDA, N-methyl-D-aspatic acid; AMPA, α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; GABA, γ-aminobutyric acid; Dex, Dexmedetomidine; TBI, Traumatic brain injury; ICH, Intracranial hemorrhage.
Table 2 Anesthetic postconditioning protocols and related signaling pathways.
M, male; F, female; SD rats, Sprague-Dawley rats ;TBI, traumatic brain injury; MCAO, middle cerebral artery occlusion; MAC, minimum alveolar concentration; CA, cardiac arrest; HI, hypoxia-ischemia; CRMP2, Collapsin response mediator protein-2; DIOA,[(dihydroindenyl)oxy] alkanoic acid; OGD, oxygen-glucose deprivation; GRP78, glucose-regulated protein-78; CHOP, C/EBP-homologous protein; Phospho-Nrf2, Phosphorylated nuclear factor-erythroid 2-related factor 2; Akt, protein kinase B; NQO1, quinine oxidoreductase1; HO-1, hemoxygenase-1; PI3k, phosphatidylinositol-3-kinase; TLR-4, toll-like receptor-4; NF-κB, nuclear factor kappa B; E2, serum estradiol-2; IL-6, interleukin-6; β-AP, beta-amyloid protein; TNF-α, tumor necrosis factor-α; eNOS, endothelial nitric oxide synthase; MMP-9, matrix metalloproteinase-9; AQP-4, aquaporin-4; pJNK, phosphorylated c-Jun N-terminal kinase; KCC2; potassium chloride co-transporter 2; PKMζ, protein kinase Mζ; TGF-β, transforming growth factor-β; ERK1/2, extracellular signal regulated kinase1/2; LRP, lipoprotein receptor-related protein ; AQP4,Aquaporins4; BMP4, bone morphogenetic protein 4.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13976 | 27 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA