Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Is there a central role for the cerebral endothelium and the vasculature in the brain response to conditioning stimuli?
Time:2018-09-02
Number:12485
Author Affiliations

Conditioning Medicine, 2018. 1(5):220-232.
Abstract
A variety of conditioning stimuli (e.g. ischemia or hypoxia) can protect against stroke-induced brain injury. While most attention has focused on the effects of conditioning on parenchymal injury, there is considerable evidence that such stimuli also protect the cerebrovasculature, including the blood-brain barrier. This review summarizes the data on the cerebrovascular effects of ischemic/hypoxic pre-, per- and post-conditioning and the mechanisms involved in protection. It also addresses some important questions: Are the cerebrovascular effects of conditioning just secondary to reduced parenchymal injury? How central is endothelial conditioning to overall brain protection? For example, is endothelial conditioning sufficient or necessary for the induction of brain protection against stroke? Is the endothelium crucial as a sensor/transducer of conditioning stimuli?
Keywords: preconditioning, cerebral ischemia, blood-brain barrier, edema, permeability, cerebral blood flow.
Abstract
A variety of conditioning stimuli (e.g. ischemia or hypoxia) can protect against stroke-induced brain injury. While most attention has focused on the effects of conditioning on parenchymal injury, there is considerable evidence that such stimuli also protect the cerebrovasculature, including the blood-brain barrier. This review summarizes the data on the cerebrovascular effects of ischemic/hypoxic pre-, per- and post-conditioning and the mechanisms involved in protection. It also addresses some important questions: Are the cerebrovascular effects of conditioning just secondary to reduced parenchymal injury? How central is endothelial conditioning to overall brain protection? For example, is endothelial conditioning sufficient or necessary for the induction of brain protection against stroke? Is the endothelium crucial as a sensor/transducer of conditioning stimuli?
Keywords: preconditioning, cerebral ischemia, blood-brain barrier, edema, permeability, cerebral blood flow.
Introduction
Ischemic preconditioning was first described in heart where prior brief periods of ischemia were shown to protect against longer injurious durations (Murry et al., 1986). Ischemic preconditioning also occurs in brain (Kitagawa et al., 1991; Gidday, 2006; Li et al., 2017), and such brain protection can also be induced by ischemic events in distant tissues (remote ischemic preconditioning (Ren et al., 2008)); by other physiological stressors including hypoxia, hyperbaric oxygen, hyperoxia and exercise (Ding et al., 2004; Pan et al., 2016); and by certain pharmacological agents (Gidday, 2010). Although the effects of brain ischemic preconditioning were described first, intermittent reductions in cerebral blood flow during (perconditioning) and after (post-conditioning) an injurious ischemic event can also induce brain protection (Hess et al., 2013; Hess et al., 2015). These different conditioning stimuli induce protective adaptations in tissues that limit the effects of later injurious events (Gidday, 2006, 2010). Most of the attention on the effects of different conditioning stimuli has focused on the impact on neural function. However, in brain (e.g. Masada et al., 2001; Zhang et al., 2006; Stowe et al., 2011; Wacker et al., 2012; Zhang et al., 2017), as well as in other tissues (Rubino & Yellon, 2000; Aggarwal et al., 2016), such conditioning stimuli can also impact endothelial cell function, and this may play an important role in overall tissue protection (see below).
The cerebrovasculature is highly specialized, including forming the blood-brain barrier (BBB). Brain endothelial cells are linked by tight junctions (TJs) that limit the entry of many hydrophobic compounds from blood to brain (Abbott et al., 2010). They also possess a wide array of transporters involved in transporting nutrients into brain, preventing entry of potentially neurotoxic compounds from blood to brain and removing waste products from brain (Abbott et al., 2010). There is limited (although important) transcytosis at the brain endothelium compared to systemic capillaries, and there is limited leukocyte trafficking (Abbott et al., 2010). These brain endothelial properties are important for brain homeostasis. Although the endothelial cell is central to the BBB, those cells are part of a wider neurovascular unit (NVU) that is important for regulating endothelial and barrier function as well as cerebral blood flow (CBF) (Iadecola, 2017). The NVU includes astrocytes, pericytes, neurons, smooth muscle cells and their basement membranes.
Many neurological conditions impact cerebral endothelial function, e.g. ischemic and hemorrhagic stroke, traumatic brain injury, Alzheimer’s disease and multiple sclerosis. For example, ischemic stroke causes increased BBB permeability (with the potential entry of neurotoxic compounds), endothelial cell death, edema, leukocyte diapedesis and CBF dysregulation (Jiang et al., 2018). Such changes may contribute to brain injury (Jiang et al., 2018) and thus represent a target for conditioning-based therapies.
The aim of this review is to examine the evidence of the impact of conditioning stimuli on the cerebral endothelium, with a particular focus on ischemia/hypoxia-related stimuli protecting against stroke-induced BBB dysfunction. It also touches upon the effects of such stimuli on CBF, an area of disagreement in the field. This review raises several important issues: Are the effects of conditioning secondary to alterations in parenchymal injury? What is the importance of alterations in signaling within the NVU to the endothelial effects of conditioning stimuli? How central is endothelial conditioning to overall brain protection--e.g., is endothelial conditioning sufficient or necessary for the induction of brain protection against stroke? Is the endothelium a crucial sensor/transducer of conditioning stimuli?
Effects of conditioning stimuli on the blood-brain barrier and cerebral endothelial cell function (Figure 1)
Barrier permeability. Multiple studies have shown that a variety of conditioning stimuli can protect against later ischemia-induced BBB disruption in vivo (Table 1). For example, ischemic preconditioning (Masada et al., 2001; Zhang et al., 2006; Gesuete et al., 2011), ischemic post-conditioning (Han et al., 2014) and remote ischemic pre- and post-conditioning (limb) (Wei et al., 2012; Zhang et al., 2017) have all been reported to decrease ischemia-induced BBB permeability to tracers. Similar effects have been reported for hypoxia-induced preconditioning (Stowe et al., 2011; Wacker et al., 2012), although Chi et al. recently reported that hypoxia preconditioning with 2 hrs of exposure to 8% O2 24 hrs prior to a permanent middle cerebral artery occlusion (MCAO) in rats actually increased BBB permeability via a vascular endothelial growth factor (VEGF)-mediated mechanism (Chi et al., 2017). The protective effects of conditioning stimuli on stroke-induced BBB hyperpermeability are not limited to ischemia; similar effects have been reported for intracerebral hemorrhage (Geng et al., 2012; Lu et al., 2014).
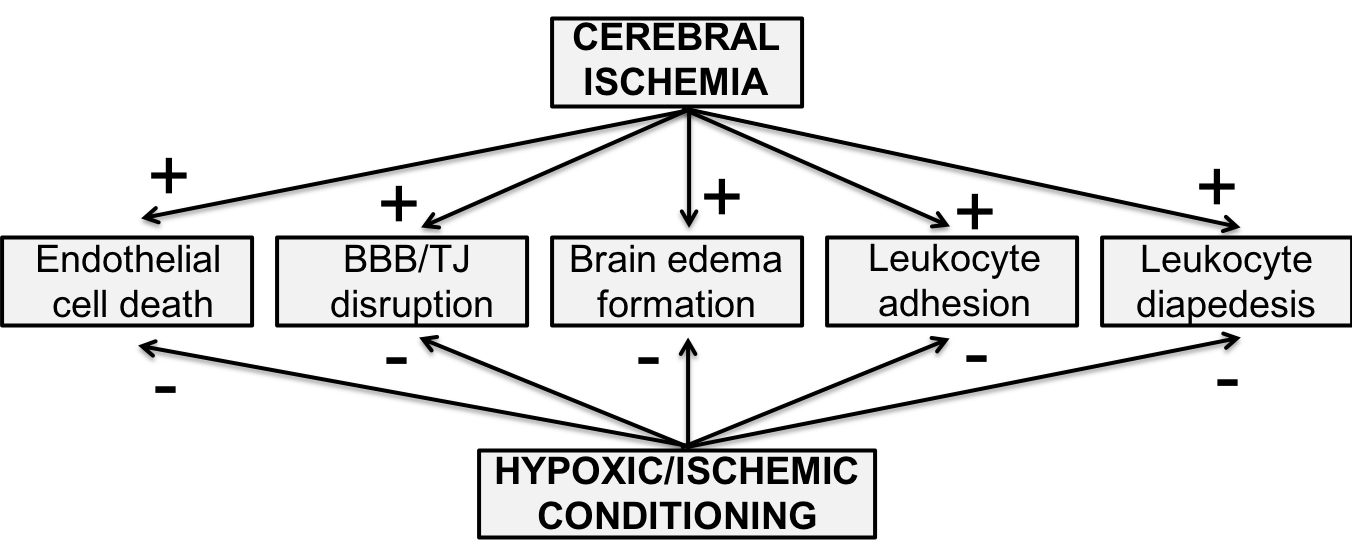
In a new window | Download PPT
Figure 1: Figure 1. Effects of cerebral ischemia on the cerebral endothelium and blood-brain barrier (BBB) are reduced by ischemic or hypoxic conditioning stimuli (pre-, per- and post-conditioning). TJ: tight junction.
In vitro (Table 1) exposure of brain endothelial cells to a brief period of oxygen glucose deprivation (OGD) or repetitive periods of hypoxia have been reported to reduce barrier hyperpermeability and cell death induced by a later prolonged period of OGD (in vitro ischemia model) (Andjelkovic et al., 2003; Zhang et al., 2007; An & Xue, 2009; Lee et al., 2009). Gesuete et al. also found that OGD preconditioning protected against OGD-induced BBB hyperpermeability in an in vitro BBB model involving co-culture of brain endothelial cells and astrocytes (Gesuete et al., 2011). However, they only found protection with co-culture, not with endothelial cells alone. In studies on non-brain endothelial cells in monoculture, Lin et al. (2013) found that OGD preconditioning reduced OGD-induced apoptosis in human microvascular endothelial cells-1 (HMEC-1) and Zhao et al. (2012) found that hypoxic preconditioning reduced OGD-induced cell damage and oxidative stress in rat arterial endothelial cells. Together these results suggest that endothelial cells can be directly preconditioned by exposure to ischemia/hypoxia, but that some effects of preconditioning may also occur via signals from other cells within the NVU (e.g. astrocytes--see below).
Table 1. Effects of hypoxia- or ischemia-related conditioning on BBB permeability and endothelial cell death after stroke in vivo or oxygen glucose deprivation in vitro.
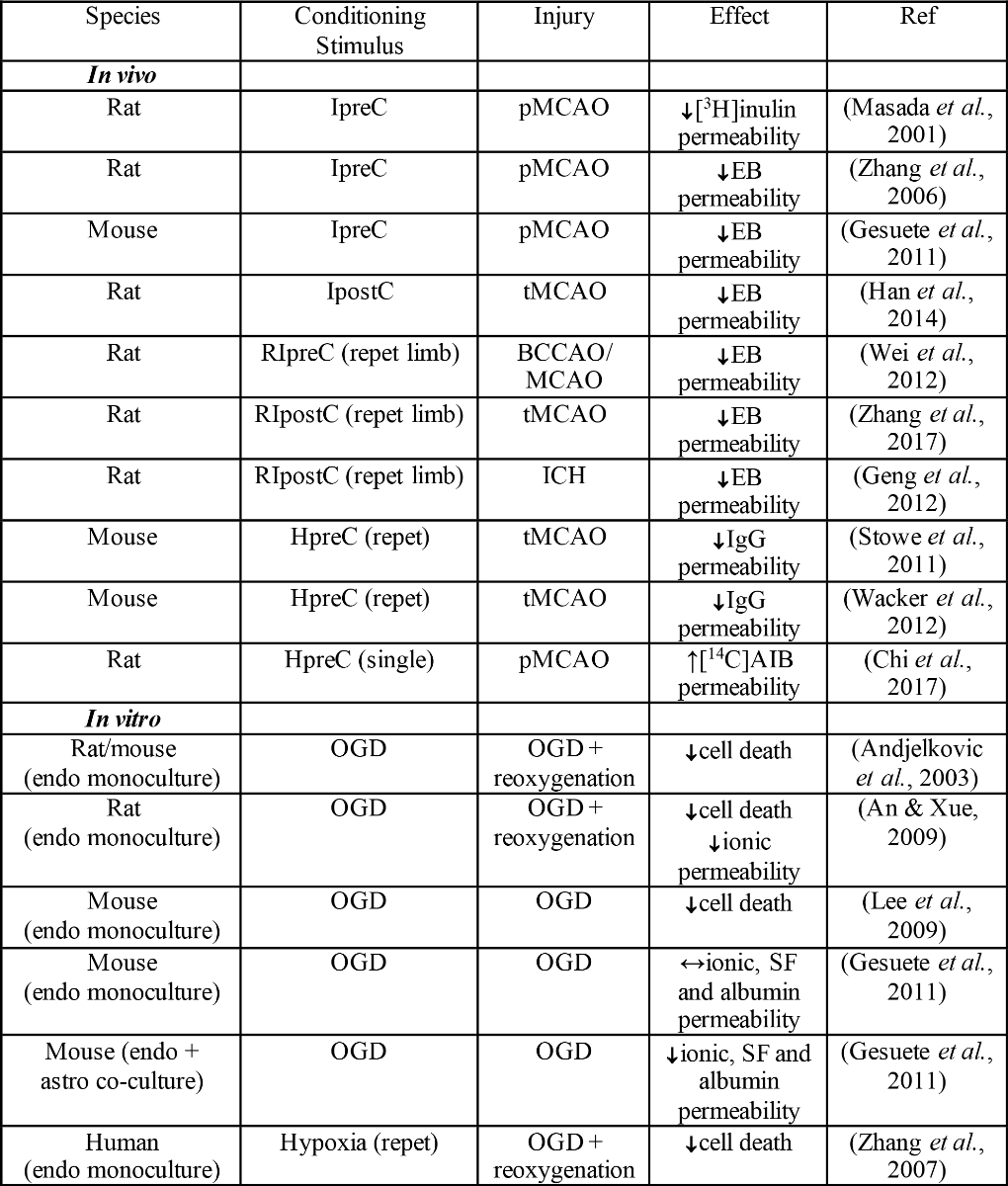
Key: ↓decrease compared to no conditioning; ↑increase compared to no conditioning; ↔ no change compared to no conditioning; AIB: alpha aminoisobutyric acid; astro: astrocyte; BCCAO: bilateral common carotid artery occlusion; endo: brain endothelial cells; EB: Evans blue; HpreC: hypoxic preconditioning; ICH: intracerebral hemorrhage; IpreC: ischemic preconditioning; IperC: ischemic perconditioning; IpostC: ischemic post-conditioning; pMCAO: permanent middle cerebral artery occlusion; tMCAO: transient middle cerebral artery occlusion OGD: oxygen glucose deprivation; SF: sodium fluorescein; repet: repetitive; RIpreC: remote ischemic preconditioning.
The TJs that link brain endothelial cells are an essential part of limiting BBB permeability. Cerebral ischemia causes a redistribution and/or loss of TJ proteins (e.g. claudin-5, occludin and ZO-1) leading to BBB hyperpermeability (Jiang et al., 2018). The TJs are linked to the cell actin cytoskeleton, and cytoskeletal changes after ischemia (e.g. stress fiber formation) contribute to TJ dysfunction (Shi et al., 2016). Several studies have shown conditioning stimuli can preserve TJ structure during subsequent ischemic events. In vitro, An et al. found that OGD-preconditioning prevented ZO-1 and F-actin redistribution during subsequent OGD + reoxygenation (An & Xue, 2009). Similarly, Gesuette et al. found that OGD preconditioning reduced redistribution of claudin-5 and ZO-1 induced by subsequent OGD (Gesuete et al., 2011). In vivo, ischemic post-conditioning after rat transient MCAO increased claudin-5 and occludin expression compared to non-conditioned animals (Han et al., 2014). Remote ischemic preconditioning (limb) in rat transient MCAO prevented claudin-5 redistribution and also increased occludin expression. It did not increase overall claudin-5 and ZO-1 expression (Ren et al., 2015). Wacker et al. (2012) found that a loss of ZO-1 and Ve-cadherin (an endothelial adherens junction protein) after transient MCAO in mice was prevented by hypoxic preconditioning. There was a similar trend (non-significant) for occludin. Preconditioning with hyperbaric oxygen protects against hypoxia- or ischemia-induced BBB disruption in vivo (Peng et al., 2008; Soejima et al., 2012) and in vitro (Hao et al., 2016). In vitro, the effects of hyperbaric oxygen preconditioning were associated with a reduced loss of occludin and ZO-1 at the cell membrane during subsequent hypoxia (Hao et al., 2016).
Insight into the effects of conditioning stimuli on the BBB can also be gained by examining the impact of such stimuli in the absence of a subsequent injurious event. Mark and Davis (2002) examined the effects of hypoxia (24 hours) with and without reoxygenation (2 hours) on brain endothelial TJ protein expression in vitro. They found that hypoxia + reoxygenation caused an upregulation in the expression of occludin, ZO-1 and ZO-2 compared to cells exposed to normoxic conditions. The direct effects of conditioning stimuli on TJ protein expression and organization, and how that interacts with the subsequent changes invoked by an injurious event, merit further investigation.
Brain edema. Classically, brain edema has been classified as vasogenic, associated with vascular disruption, and cytotoxic, linked to parenchymal cell injury (Klatzo, 1967), although many neurological conditions such as cerebral ischemia induce both. Evidence suggests that ischemic and hypoxic preconditioning reduces brain edema formation following later cerebral ischemia (Masada et al., 2001; Wacker et al., 2009; Shin et al., 2015), that ischemic post-conditioning also diminishes edema (Esmaeeli-Nadimi et al., 2015), and that remote (limb) ischemic pre- and post-conditioning have similar effects (Ren et al., 2015; Xia et al., 2017). It is currently uncertain whether these effects of conditioning stimuli on edema are due to reduced vasogenic or cytotoxic edema (due to smaller infarcts), or both. However, the effects of such stimuli on BBB permeability suggest at least some of the reduction is due to diminished vasogenic edema formation.
Leukocyte diapedesis into brain. Neuroinflammation has a major role in brain injury after stroke (Iadecola & Anrather, 2011). One component of stroke-induced neuroinflammation is an infiltration of circulating leukocytes (e.g. neutrophils, macrophages and lymphocytes) into brain. This infiltration is a multi-step process involving the production of cytokines and chemokines within brain, the expression of adhesion molecules on the cerebral endothelium, and the migration of leukocytes across the endothelium (Lopes Pinheiro et al., 2016). Evidence indicates that ischemic or hypoxic preconditioning alters cytokine/chemokine expression during subsequent injurious ischemia towards a more anti-inflammatory phenotype (Wang et al., 2014; Kim et al., 2015; McDonough & Weinstein, 2016). Similarly, ischemic or hypoxic preconditioning stimuli reduce expression of brain endothelial adhesion molecules (VCAM-1, P- and E-selectin) during subsequent stroke in vivo (Hoyte et al., 2010; Stowe et al., 2011). In vitro, brief exposure of brain endothelial cells to OGD also reduces the expression of ICAM-1 induced by prolonged OGD with reoxygenation (Andjelkovic et al., 2003). Stowe et al. found that preconditioning with repetitive bouts of hypoxia reduced the adherence of leukocytes to the cerebral endothelium induced by transient MCAO as well as leukocyte diapedesis (Stowe et al., 2011). Similarly, Selvaraj et al. reported that hypoxic preconditioning reduced total leukocyte infiltration into brain after transient MCAO (Selvaraj et al., 2017), and Doeppner et al. found that ischemic post-conditioning reduces the number of leukocytes in the brain after transient MCAO (Doeppner et al., 2017).
Blood flow regulation. The effects of ischemic/hypoxic preconditioning on CBF in ischemic stroke are controversial (Table 2). While multiple studies have indicated no effect of prior preconditioning on CBF during and after a subsequent ischemic event (e.g. Matsushima & Hakim, 1995; Chen et al., 1996; Cho et al., 2005; Stowe et al., 2011), some studies have suggested an important effect on blood flow, particularly in the ischemic penumbra (Hoyte et al., 2006; Zhao & Nowak, 2006; Cui et al., 2013). In line with the latter, some evidence exists of an impact of ischemic/hypoxic conditioning on the brain’s collateral circulation. Thus, Woitzik et al. found that hypoxic preconditioning in mice increased the diameter of the leptomeningeal anastomoses 72 hours later (Woitzik et al., 2006). The extent of the collateral circulation is an important determinant of stroke-induced brain injury in preclinical models and patients (Ginsberg, 2016).
Table 2. Effects of hypoxia- or ischemia-related preconditioning on cerebral blood flow.
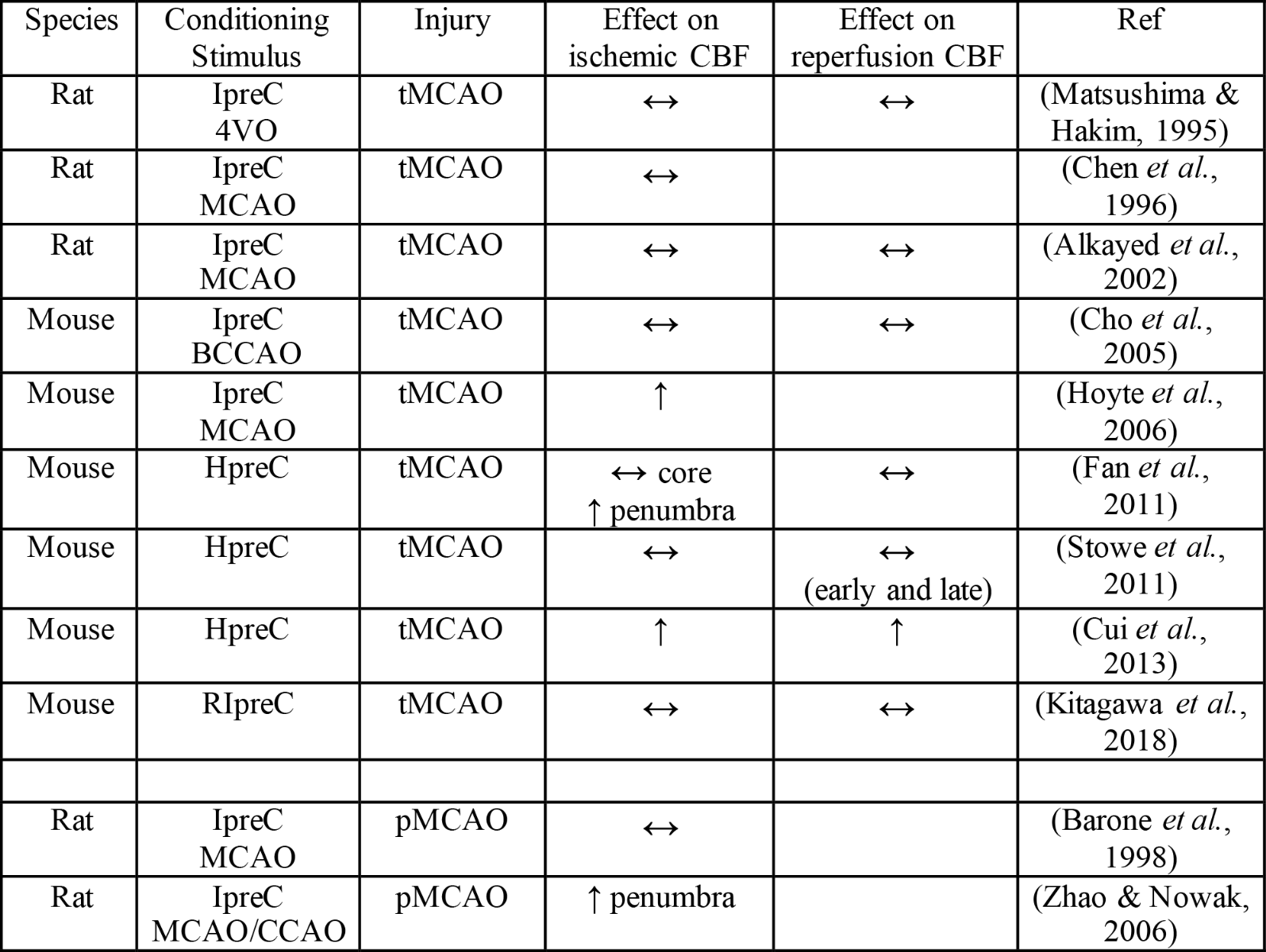
Key: ↑increase compared to no conditioning; ↔no change compared to no conditioning; 4VO: four-vessel occlusion; BCCAO: bilateral common carotid artery occlusion; CCAO: unilateral common carotid artery occlusion; HpreC: hypoxic preconditioning; IpreC: ischemic preconditioning; pMCAO: permanent middle cerebral artery occlusion; RIpreC: remote ischemic preconditioning; tMCAO: transient middle cerebral artery occlusion.
Experiments examining the effects of post-conditioning on CBF have been more consistent (Table 3) in showing reduced acute hyperemia and less delayed hypoperfusion (Zhao et al., 2006; Gao et al., 2008; Wang et al., 2008; Esmaeeli-Nadimi et al., 2015; Wu et al., 2015). Similarly, a number of studies have shown a beneficial effect of remote ischemic preconditioning (limb) on CBF. Hoda et al. (2012) found it increased CBF during embolic stroke in male mice as well as during tPA-induced reperfusion. That group also found similar effects in ovariectomized female mice (Hoda et al., 2014). Kitagawa et al. (Kitagawa et al., 1991) also found that remote ischemic perconditioning increased the diameter of the leptomeningeal anastomoses in mice undergoing transient MCAO, indicating that it likely improved collateral circulation. This was associated with an increase in CBF during the course of the MCAO. They did not report an effect of this conditioning stimulus on CBF upon reperfusion.
Table 3. Effects of hypoxia- or ischemia-related per- or post-conditioning on cerebral blood flow.
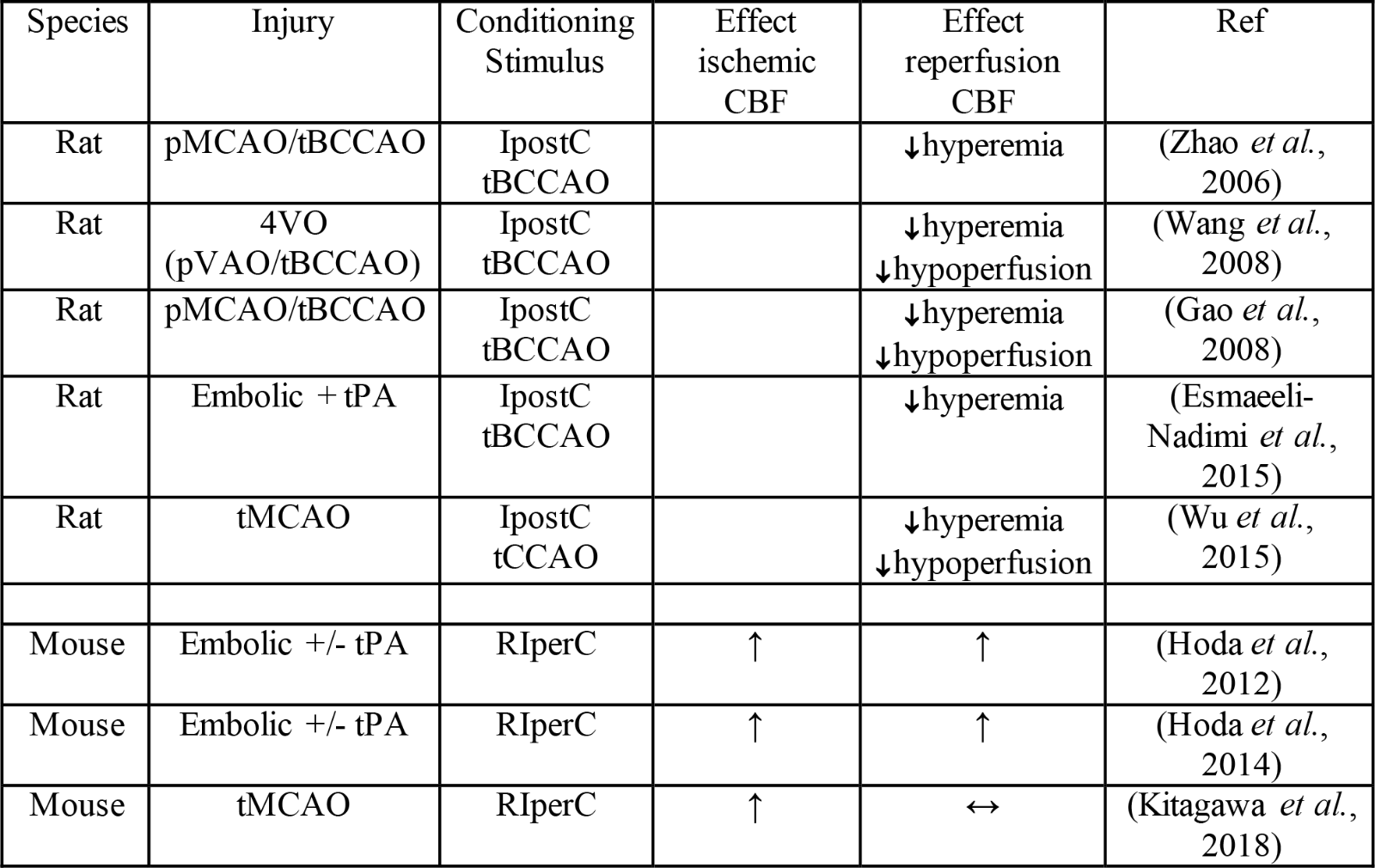
Key: ↓decrease compared to no conditioning; ↑increase compared to no conditioning; ↔no change compared to no conditioning; tBCCAO: bilateral common carotid artery occlusion; tCCAO: unilateral common carotid artery occlusion; IpostC: ischemic post-conditioning; pMCAO: permanent middle cerebral artery occlusion; pVAO: permanent vertebral artery occlusion; RIperC: remote ischemic perconditioning; tMCAO: transient middle cerebral artery occlusion; tPA: tissue plasminogen activator.
In patients with subarachnoid hemorrhage, Gonzalez et al. (2013) reported a transient cerebral vasodilation with remote ischemic preconditioning. In patients with intracranial arterial stenosis, Meng et al. (2012) found that remote ischemic preconditioning (limb, twice daily/300 days) and found improved cerebral perfusion as assessed by transcranial Doppler and SPECT. Similarly, Wang et al. (2017) and Mi et al. (2016) examined the effect of twice daily remote ischemic conditioning (limb) for one year in patients with cerebral small vessel disease. Wang et al. found a reduction in the pulsatility index of the middle cerebral artery, indicating that the conditioning improved cerebral perfusion and reduced the resistance of downstream vessels, while Mi et al. found an increase in the mean flow velocity in the middle cerebral artery.
Impact of comorbidities on conditioning
One potential impediment to the translation of pre-clinical conditioning data to the clinic is whether different diseases might lessen the beneficial effects of conditioning stimuli (Wang et al., 2013). For example, some evidence suggests that a variety of stroke comorbidities impact the ability of conditioning stimuli to protect against ischemic brain injury. Aging has been reported to diminish the efficacy of ischemic preconditioning in reducing ischemia-induced brain injury (Schaller, 2007; Della-Morte et al., 2013). Some evidence indicates that the effects of ischemic preconditioning are reduced in the spontaneously hypertensive rat (stroke prone (SHRSP)) compared to the less hypertensive SHR strain (Purcell et al., 2003). In heart, most but not all preclinical studies have shown reduced effectiveness of pre-and post-conditioning with diabetes, hypertension and aging, and some evidence indicates that this is the case in patients (reviewed in McCafferty et al. (2014)).
While aging, hypertension and diabetes/hyperglycemia all are risk factors for stroke and worsen stroke outcome, they all also impact BBB function after stroke (Jiang et al., 2018). Whether they also specifically affect the ability of conditioning stimuli to protect the cerebral endothelium and other elements of the NVU has not been specifically examined. This should be a priority for future research.
Are conditioning effects on the brain endothelium due to reduced parenchymal injury?
One mechanism by which conditioning stimuli might reduce stroke-induced BBB dysfunction is an indirect effect via reducing parenchymal injury. Ischemic and hypoxic pre-, per- and post-conditioning all have been shown to reduce infarct size/parenchymal injury (Hess et al., 2013; Stetler et al., 2014; Li et al., 2017). However, several pieces of evidence indicate that brain endothelial protection is not solely a result of reduced parenchymal injury. As noted above, several in vitro studies have shown that brain endothelial cells can be conditioned to reduce OGD-induced injury in the absence of other cell types (Andjelkovic et al., 2003; Zhang et al., 2007; An & Xue, 2009). In addition, Stowe et al. (2011) examined whether the effects of hypoxic preconditioning on reducing leukocyte adherence to endothelial cells after transient MCAO also occurred when mice were preconditioned prior to systemic administration of tumor necrosis factor (TNF)-α, an inflammatory cytokine. They found that hypoxic preconditioning reduced TNF-α-induced leukocyte adherence even though systemic TNF-α did not cause overt parenchymal injury.
Effects of conditioning stimuli on cerebral endothelial cells
There has been an extensive examination of the cellular mechanisms triggered by ischemic and hypoxic preconditioning in brain (Li et al., 2017). These mechanisms alter cell metabolism and reduce energy demand, protect against cell death and injury pathways, and produce responses that limit the severity of hypoxia/ischemia. The responses may occur early (e.g. classical preconditioning) and late (e.g. delayed preconditioning) after the conditioning stimulus. Examples of the former include phosphorylation of target proteins, while the latter often depend upon transcription and translation. Multiple cell signaling pathways are involved in the effects of conditioning including ERK, Akt and protein kinase C signaling (Li et al., 2017).
In general, the intracellular mechanisms involved in specific cell types are more easily studied in culture, but there is still a paucity of studies examining the mechanisms of hypoxia- or OGD-induced conditioning of brain endothelial cells in vitro. Both forms of conditioning do reduce OGD-induced endothelial cell death (Andjelkovic et al., 2003; Zhang et al., 2007; An & Xue, 2009), and Zhang et al. found that hypoxic preconditioning increased PI3-kinase/Akt signaling and activation of anti-apoptotic pathways including increased phosphorylated survivin (Zhang et al., 2007). Blocking the PI3-kinase/Akt pathway prevented hypoxic preconditioning from reducing OGD-induced cell death (Zhang et al., 2007). In the heart there is evidence of a crucial role of the mitochondrial changes (mitochondrial ATP-sensitive K+ channels, Bcl-2 family members and the mitochondrial permeability transition pore) in conditioning, including in the cardiac vasculature (Rubino & Yellon, 2000; Murphy, 2004). There is also evidence in the cerebrovasculature for a role of mitochondrial changes. Preconditioning with diazoxide, an ATP-sensitive K+ channel opener, protected against global ischemia-induced BBB disruption (Lenzser et al., 2005). Pre- or post-conditioning with BMS-191095, another mitochondrial ATP-sensitive channel opener, also protected rat brain endothelial cells from OGD-induced cell death. More in vitro studies are needed to fully elucidate the mechanisms involved in the effects of ischemic and hypoxic conditioning on the cerebral endothelium
Effects of conditioning stimuli on signaling within the neurovascular unit
Conditioning stimuli may also impact cell-to-cell or cell-to-extracellular matrix signaling within the NVU, indirectly affecting brain endothelial function. Abundant evidence suggests that in the absence of conditioning, endothelial cells receive signals from other components of the NVU. For example, at the levels of the cerebral capillaries, astrocytes release factors that enhance barrier tightness and co-culture with astrocytes, or astrocyte-conditioned media increases the transendothelial electrical resistance of brain endothelial monolayers in vitro (Abbott et al., 2010). Similarly, pericytes (Armulik et al., 2010; Daneman et al., 2010; Ben-Zvi et al., 2014) and endothelial-to-extracellular matrix interactions (Baeten & Akassoglou, 2011; Menezes et al., 2014) also regulate BBB function. It should also be noted the pericytes have been proposed to regulate CBF (Hall et al., 2014).
The effect of conditioning stimuli on intercellular signaling within the capillary NVU has generally been a neglected field. While individual cell types may react to a particular conditioning stimulus raising a protective response, those cell types may also send signals/mediators that affect other nearby (or distant) cells. Thus, for example, ischemic neurons may transmit “help-me” signals to other cell types to promote survival, including the cerebral endothelium (Xing & Lo, 2017). Some neuronal help-me signals include the chemokine CX3CL1, the cytokine IL-34, fibroblast growth factor-2, lipocalin-2 and IgG (Xing & Lo, 2017). Stowe et al. (2012) examined the role of the chemokine CCL2 in hypoxic preconditioning. They found that such preconditioning upregulated CCL2 mRNA and protein initially in neurons and then in endothelial cells. They also found an upregulation of the CCL2 receptor, CCR2, on endothelial cells. CCL2-null mice and wild-type animals treated with a CCL2 neutralizing antibody blocked the neuroprotective effects of hypoxic preconditioning, indicating the importance of CCL2-mediated signaling in the conditioning response.
There is also evidence for the importance of astrocyte-to-endothelial signaling in conditioning. Gesuette et al. (2011) found that OGD-induced preconditioning on co-cultures of brain endothelial cells and astrocytes reduced the barrier disruption caused by a later, more severe OGD exposure. Interestingly, they found this effect could be mimicked by preconditioning the astrocytes alone, but not the endothelial cells alone, and that inhibiting astrocyte metabolism with fluorocitrate also blocked OGD-induced preconditioning. These results indicate a crucial role of astrocyte-to-endothelial signaling in barrier protection.
Sphingosine-1-phosphate (S1P) is an important signaling molecule at the neurovascular unit with receptors on brain endothelial cells and astrocytes (Spampinato et al., 2015; Yanagida et al., 2017). Loss of the S1P receptor 1 specifically on endothelial cells causes BBB disruption (Yanagida et al., 2017). The phosphorylation of sphingosine to produce S1P is catalyzed by the sphingosine kinases. In vivo, Wacker et al. (2009; 2012) found that microvascular sphingosine kinase-2 (SphK-2) levels were increased by hypoxic preconditioning, and that inhibiting SphK or genetic knockout of SphK2 reduced the protective effects of hypoxic preconditioning on stroke infarct volume and on BBB disruption. The cellular location of SphK2 is still uncertain. In addition to effects on the BBB, evidence has shown that S1P receptor 1 activation increases the development of leptomeningeal collaterals improving stroke outcome in mice (Iwasawa et al., 2018).
VEGF-A is a crucial component of the brain response to hypoxia. It is upregulated in neurons, astrocytes and microglia after hypoxia (Ogunshola et al., 2000), and via receptors (VEGFR1 (Flt-1) and VEGFR2 (Flk-1)) on the cerebral endothelium it promotes angiogenesis, a chronic adaptation to a low-O2 environment (LaManna et al., 2004, and below). Laudenbach et al. (2007) found that a reduction in excitotoxic injury by hypoxic preconditioning in neonatal mice was prevented by a VEGFR2 antibody, and that mice lacking a hypoxia-responsive element on the VEGF-A gene actually had worse injury with hypoxic preconditioning rather than protection. Similarly, Lee et al. (2009) found that the cell death induced by hypoxia in neonatal rats was reduced by ischemic preconditioning, but that this neuroprotection was abrogated if VEGF-A or VEGFR2 (but not VEGFR1) were inhibited. Similarly, the protective effects of OGD preconditioning in limiting OGD-induced brain endothelial cell and neuronal death were blocked by antisense oligodeoxynucleotides targeting VEGF-A or VEGFR2, or a VEGF-A antibody (Lee et al., 2009). It should be noted that VEGF-A has non-endothelial (e.g. neuronal) effects that are important in brain injury responses and might contribute to hypoxic/ischemic conditioning (Li et al., 2017).
In addition to VEGF-A, there is evidence of a role of VEGF-C in ischemic preconditioning (Bhuiyan et al., 2015). Following ischemic preconditioning in mice, VEGF-C was upregulated in neurons. The VEGF-C receptor, VEGFR-3, is normally present in endothelial cells although it is also upregulated in neurons after preconditioning, and blocking that receptor prevents the neuroprotection induced by ischemic preconditioning (Bhuiyan et al., 2015).
Another type of signaling that has yet to receive attention with regards to the effects of conditioning at the NVU is that mediated b extracellular vesicles (exosomes or microvesicles depending on size (Ramirez et al., 2018)). Such vesicles are shed by almost all cell types and, via their microRNA and protein cargo, are involved cell-cell communication. At the NVU, the endothelium is both a source of such vesicles and a target (Ramirez et al., 2018). There is evidence in culture that endothelial cell-derived exosomes can protect neurons (Xiao et al., 2017). There is also evidence that circulating extracellular vesicles are involved in the cardioprotection induced by remote ischemic preconditioning (Barile et al., 2017; Giricz et al., 2014; Yamaguchi et al., 2015). Investigation is warranted on the impact of conditioning stimuli on the number and content of extracellular vesicles derived from brain endothelial cells, and on other components of the NVU as well as circulating extracellular vesicles, along with whether they affect the response of other cell types to ischemia and other forms of brain injury.
Cerebral blood flow is highly regulated at the level of the NVU (e.g. neurovascular coupling and autoregulation). One important regulator of CBF at the NVU is nitric oxide (NO). In brain, NO is produced by three nitric oxide synthase isoforms, eNOS (endothelial NOS), nNOS (neuronal NOS) and iNOS (inducible NOS). Evidence from knockout mice and inhibitor studies indicates that each isoform is required for preconditioning to protect against ischemic brain injury (Atochin et al., 2003; Cho et al., 2005; Iadecola et al., 2011). This may be via the actions of NO as a vasodilator (see discussion above on the CBF effects of hypoxic/ischemic conditioning), but NO has multiple other actions (Iadecola et al., 2011).
Important signaling events may also be invoked by conditioning stimuli outside the NVU. Circulating progenitor cells (e.g. endothelial progenitor cells (EPCs)) have an important role in angiogenesis and BBB repair after cerebral ischemia, and there has been considerable interest in the use of such cells in stroke therapy (Liu et al., 2014). Akita et al. (2003) found that hypoxic preconditioning induces the differentiation of peripheral blood mononuclear cells into EPC-like attaching cells, and that these cells could enhance neovascularization in ischemic hindlimb. Some evidence also indicates that remote ischemic preconditioning increases the number of EPCs in arteries of patients with heart disease (Liang et al., 2015).
Is endothelial preconditioning sufficient to induce brain tolerance? Endothelium as primary sensor/transducer of hypoxic/ischemic conditioning stimuli
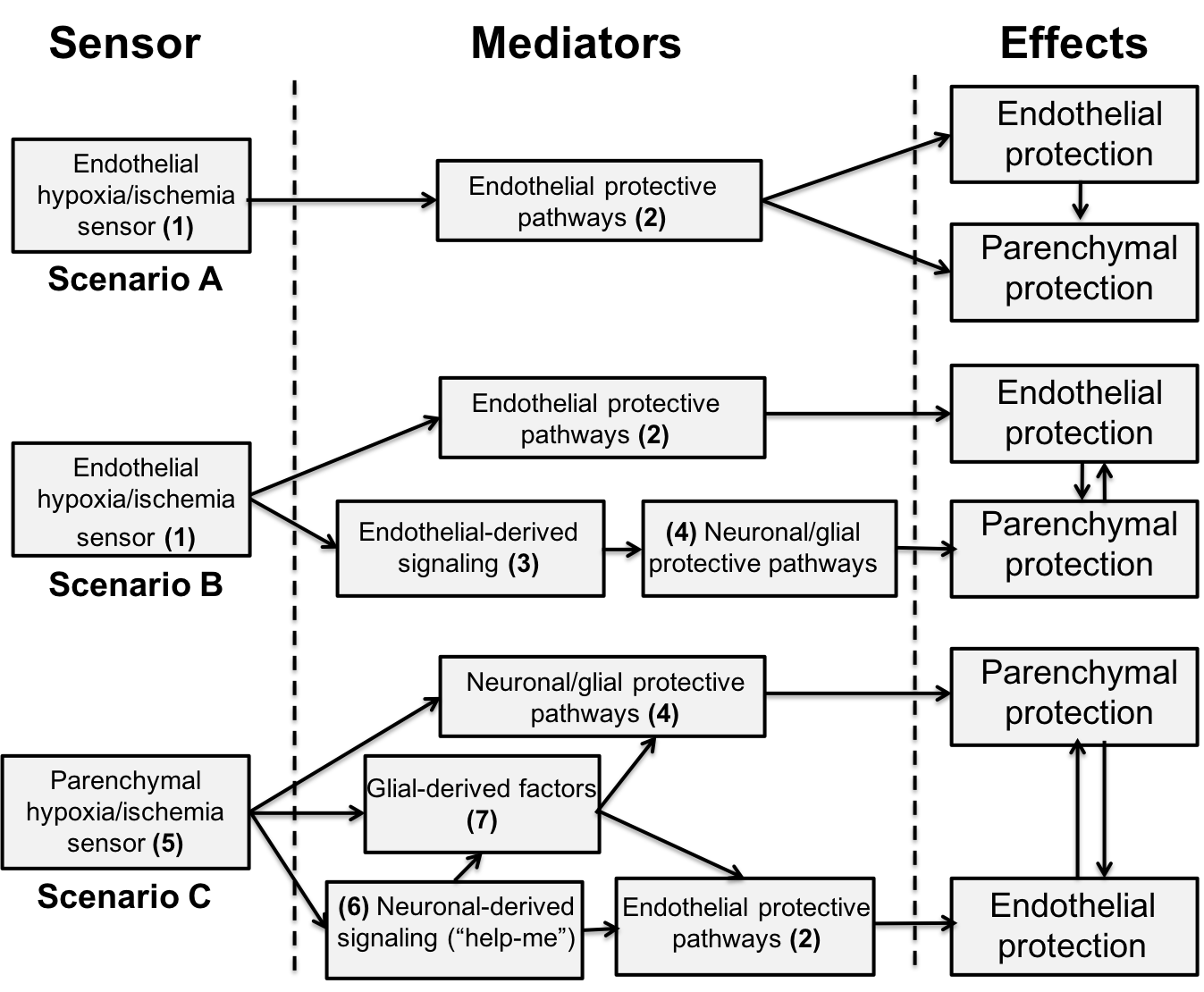
Growing evidence indicates that early BBB dysfunction plays a critical role in brain injury after ischemic stroke with specific manipulations to the cerebral endothelium not only protecting against BBB disruption but also reducing infarct size and behavioral deficits (Shi et al., 2016; Shi et al., 2017). Recent evidence also suggests that downregulating claudin-5 expression at the brain endothelium via adeno-associated virus-delivered shRNA causes behavioral deficits in rodents (Menard et al., 2017). Such experiments raise the question of whether the effects of conditioning stimuli on the cerebral endothelium can be a primary site of action that then leads to brain protection (Figure 2, scenarios A and B). There is some evidence supporting this hypothesis.
In a new window | Download PPT
Figure 2: Schematic showing alternate roles of the cerebral endothelium in the brain response to ischemic/hypoxic conditioning stimuli. The scenarios are not mutually exclusive. Scenario A: (1) The ischemic/hypoxic conditioning stimulus is sensed by the endothelium. This may involve HIF-1α. However, it may also involve response to humoral factors (e.g. adenosine or bradykinin) in remote ischemic conditioning, or shear stress-regulated ion channels. (2) The conditioning stimulus induces endothelial mechanisms that protect both the cerebral endothelium (e.g. anti-apoptotic, metabolic) and the brain (e.g. BBB protection, inhibition of leukocyte infiltration, production of nitric oxide) during subsequent ischemia. There is some evidence that reducing endothelial damage is sufficient to reduce parenchymal damage after stroke (Shi et al., 2016; Shi et al., 2017). Scenario B: The ischemic/hypoxic conditioning stimulus (1) is sensed by the cerebral endothelium and (2) elicits protective responses in those cells. In addition, (3) the stimulus causes the endothelium to release signaling molecules that (4) activate protective pathways in neurons and glial cells that limit subsequent ischemic brain damage. Scenario C: (5) The ischemic/hypoxic conditioning stimulus may be “sensed” by parenchymal cells. This may involve HIF-1α although there is evidence that neuronal HIF-1α is not critical for hypoxic preconditioning (Baranova et al., 2007; Zhu et al., 2013). Other non-neuronal cells might be involved, or a non-HIF-1α-dependent pathway could be important. (4) The conditioning stimulus may directly induce protective pathways in neurons/glia. (6) In addition, neurons can secrete “help-me” signals (e.g. CX3CL1, interleukin-34, lipocalin-2 and fibroblast growth factor-2) to endothelial and glial cells to elicit responses that will help protect the neurons (Xing & Lo, 2017). (7) Glial-derived factors may be particularly important in protecting neurons (e.g. by growth factor secretion) and BBB integrity (Gesuete et al., 2011).
Selvaraj et al. found that repetitive hypoxic preconditioning caused a long-term increase in the chemokine CXCL12 at the BBB (Selvaraj et al., 2017). An antagonist of the CXCL12 receptor, CXCR4, blocked the ability of hypoxic preconditioning to reduce transient MCAO-induced leukocyte infiltration into brain and limit infarct size. Interestingly, that study did not find a reduction in MCAO-induced brain edema or BBB disruption. These results suggest a critical role of both inflammation and the cerebral endothelium in hypoxic preconditioning.
Similarly, Ozaki et al. (2016) found that brain endothelial cells express high levels of the purinoreceptor P2X4. Inhibiting P2X4 prevented the ability of ischemic preconditioning to reduce infarct size and the behavioral deficits induced by transient MCAO. Importantly, conditional knockout of P2X4 receptor in Ve-cadherin-positive mice also prevented the protective effects of ischemic preconditioning. The P2X4 receptor is sensitive to shear stress, and the authors presented data showing that P2XR4 activation increases expression of osteopontin, a neuroprotectant, in endothelial cells. These results suggest a central role of the cerebral endothelium in ischemic preconditioning.
Another piece of evidence that may indicate the importance of the endothelium in conditioning is the effect of remote ischemic conditioning. The exact mechanisms underlying the effects of these stimuli are still not totally clear. Research has focused on the roles of the release of humoral factors from the ischemic tissue (e.g. limb) and a neurogenic component as well as modulation of the immune system (Hess et al., 2013; Hess et al., 2015). One proposed humoral factor is adenosine (Hu et al., 2012), which has receptors at the cerebral endothelium (A1/A2A receptors (Bynoe et al., 2015)) but only a low rate of blood-to-brain transport. Another is bradykinin (Hess et al., 2013); systemic administration can precondition the brain, causing reduced infarct size, brain edema and BBB permeability after transient MCAO in rats (Ping et al., 2005). However, bradykinin is an oligopeptide, and these, with some exceptions, have very low BBB permeabilities (Zlokovic, 1995). This suggests that the brain effects of these factors may be primarily at the level of the cerebral endothelium, which has abundant bradykinin receptors (primarily the kinin B2 receptor (Easton & Abbott, 2002; Dobrivojevic et al., 2015)).
Determining whether a pharmacological agent is acting as a conditioning stimulus at the level of the cerebral endothelium can be complex. Pre- and post-conditioning with systemic lipopolysaccharide (LPS) has been shown to reduce ischemic brain damage. While there is evidence that LPS has actions at the cerebral endothelium via endothelial nitric oxide synthase (Puisieux et al., 2000; Kunz et al., 2007; Orio et al., 2007) and that LPS does not cross the BBB, there is evidence that LPS can increase plasma ceramide concentrations, and that molecule can cross the BBB (Zimmermann et al., 2001). Ceramide can be found not only in brain vessels but also in perivascular cells and in the brain parenchyma after intravenous administration (Zimmermann et al., 2001).
Although most studies on the effects of hypoxic preconditioning have focused on acute or subacute scenarios, it should be realized that chronic hypoxic exposure has a profound effect of angiogenesis with increased capillary density (Pichiule & LaManna, 2002; LaManna et al., 2004; Benderro & LaManna, 2014). In addition, chronic cerebral hypoperfusion in rodents causes a delayed angiogenic response (Hai et al., 2003; Jing et al., 2015). These chronic angiogenic effects will help maintain cerebral oxygen delivery. Some evidence suggests that the adaptive response to chronic cerebral hypoperfusion may protect against later focal cerebral ischemia (Choi et al., 2007), although the relative importance of the angiogenic response in that protection is uncertain.
Studies using cell-specific knockout of signaling molecules can give insight into the relative roles of different cells in conditioning, e.g. examining the effect of cell-specific knockout hypoxia-inducible factor (HIF)-1α on hypoxic or ischemic preconditioning. Baranova et al. examined neuronal HIF-1α knockout mice and found no effect on hypoxic preconditioning (Baranova et al., 2007). Similarly, Zhu et al. found no effect of retinal ganglion cell-specific HIF-1α knockout on the effects of hypoxic preconditioning in the eye (Zhu et al., 2013). In heart, endothelial HIF-1α and β are both required for ischemic preconditioning (Sarkar et al., 2012). This suggests that the primary hypoxia/ischemia sensing in hypoxic/ischemic preconditioning in brain may be at the level of the cerebral endothelium. It should be noted that the cerebral endothelial response to conditioning stimuli might be critical in brain protection in two ways. Protection against endothelial damage itself may limit parenchymal damage during ischemia, e.g. by limiting the entry of potential neurotoxic compounds from blood that might exacerbate ischemic brain injury. However, an alternative is that the endothelial response is important in transducing the conditioning “signal” to the brain parenchyma. For example, the endothelium may respond to changes in blood oxygen or shear stress by emitting signaling molecules that elicit protective responses in parenchymal cells (i.e., may act as a transducer).
Future directions
While evidence is emerging that the brain endothelium response is a critical component in conditioning-induced protection against ischemic brain injury (Ozaki et al., 2016; Selvaraj et al., 2017), further studies are required. These could involve specifically blocking endothelial conditioning and examining whether protection against brain injury still occurs (e.g. using cell-specific knockouts) or examining whether a strategy to specifically induce a conditioning response in the cerebral endothelium (genetically or pharmacologically) is sufficient to protect against ischemic brain injury. In particular, blocking brain endothelial sensing of changes in blood flow or O2 could be very informative. A full elucidation of the signaling pathways involved in the brain endothelial conditioning response(s) would greatly assist in those goals.
One concern over the translation of any therapy from preclinical studies to patients is whether species differences might exist. Recently tremendous progress has been made in producing brain endothelial cells and other components of the NVU (e.g. astrocytes and pericytes) from human-induced pluripotent stem cells (iPSCs) (Lippmann et al., 2012; Lippmann et al., 2014). The iPSC-derived brain endothelial cells can be used in mono- or co-culture with astrocytes and/or pericytes and have permeability characteristics similar to those in vivo (Lippmann et al., 2012; Lippmann et al., 2014). These should provide an excellent resource for studying the effects of conditioning stimuli on the human brain endothelium and for examining the effects of such stimuli on signaling within the NVU.
A major goal of conditioning research is the identification of biomarkers that might identify whether a particular stimulus has been effective in inducing a conditioning response. For remote ischemic preconditioning, clinical studies have examined potential endothelial markers including serum VEGF, von Willebrand factor, nitric oxide metabolites and the number of endothelial progenitor cells, as well as brachial artery vasomotor function as a physiological marker (reviewed in Koch et al., 2014). It should be noted that the serum markers are produced by systemic as well as brain endothelial cells. If cerebral endothelial conditioning is critical for protecting the brain, examining biomarkers that are predominantly produced by that endothelium could be a major advance. For example, it has recently been suggested that extracellular vesicles specifically produced by the cerebral endothelium might be a marker for BBB status (Ramirez et al., 2018).
Conclusions
In stroke, cerebral endothelial/BBB dysfunction has often been considered a consequence of parenchymal cell injury. However, recent evidence has shown that specifically targeting the brain endothelium in stroke can not only ameliorate the endothelial dysfunction but also reduce parenchymal injury (e.g. infarct size and behavioral deficits) (Shi et al., 2016; Shi et al., 2017). Similarly, in the field of conditioning, some evidence indicates that conditioning of the cerebral endothelium is a critical component for overall brain protection. Further exploration of such findings may help elucidate new ways of protecting the brain and of identifying biomarkers that indicate whether conditioning strategies are impacting a target tissue.
Disclosures/conflicts
The authors declare they have no conflicts of interest.
Acknowledgements
This study was supported by grants NS-073595, NS-091545, NS-090925, NS093399, NS-096917, NS106746 and AG057928 from the National Institutes of Health (NIH) and grant 1-16-IBS-008 from the American Diabetes Association.
References
In a new window | Download PPT
Figure 1: Figure 1. Effects of cerebral ischemia on the cerebral endothelium and blood-brain barrier (BBB) are reduced by ischemic or hypoxic conditioning stimuli (pre-, per- and post-conditioning). TJ: tight junction.
In a new window | Download PPT
Figure 2: Schematic showing alternate roles of the cerebral endothelium in the brain response to ischemic/hypoxic conditioning stimuli. The scenarios are not mutually exclusive. Scenario A: (1) The ischemic/hypoxic conditioning stimulus is sensed by the endothelium. This may involve HIF-1α. However, it may also involve response to humoral factors (e.g. adenosine or bradykinin) in remote ischemic conditioning, or shear stress-regulated ion channels. (2) The conditioning stimulus induces endothelial mechanisms that protect both the cerebral endothelium (e.g. anti-apoptotic, metabolic) and the brain (e.g. BBB protection, inhibition of leukocyte infiltration, production of nitric oxide) during subsequent ischemia. There is some evidence that reducing endothelial damage is sufficient to reduce parenchymal damage after stroke (Shi et al., 2016; Shi et al., 2017). Scenario B: The ischemic/hypoxic conditioning stimulus (1) is sensed by the cerebral endothelium and (2) elicits protective responses in those cells. In addition, (3) the stimulus causes the endothelium to release signaling molecules that (4) activate protective pathways in neurons and glial cells that limit subsequent ischemic brain damage. Scenario C: (5) The ischemic/hypoxic conditioning stimulus may be “sensed” by parenchymal cells. This may involve HIF-1α although there is evidence that neuronal HIF-1α is not critical for hypoxic preconditioning (Baranova et al., 2007; Zhu et al., 2013). Other non-neuronal cells might be involved, or a non-HIF-1α-dependent pathway could be important. (4) The conditioning stimulus may directly induce protective pathways in neurons/glia. (6) In addition, neurons can secrete “help-me” signals (e.g. CX3CL1, interleukin-34, lipocalin-2 and fibroblast growth factor-2) to endothelial and glial cells to elicit responses that will help protect the neurons (Xing & Lo, 2017). (7) Glial-derived factors may be particularly important in protecting neurons (e.g. by growth factor secretion) and BBB integrity (Gesuete et al., 2011).
Table 1 - Effects of hypoxia- or ischemia-related conditioning on BBB permeability and endothelial cell death after stroke in vivo or oxygen glucose deprivation in vitro.
.png)
Key: ↓decrease compared to no conditioning; ↑increase compared to no conditioning; ↔ no change compared to no conditioning; AIB: alpha aminoisobutyric acid; astro: astrocyte; BCCAO: bilateral common carotid artery occlusion; endo: brain endothelial cells; EB: Evans blue; HpreC: hypoxic preconditioning; ICH: intracerebral hemorrhage; IpreC: ischemic preconditioning; IperC: ischemic perconditioning; IpostC: ischemic post-conditioning; pMCAO: permanent middle cerebral artery occlusion; tMCAO: transient middle cerebral artery occlusion OGD: oxygen glucose deprivation; SF: sodium fluorescein; repet: repetitive; RIpreC: remote ischemic preconditioning.
Table 2. Effects of hypoxia- or ischemia-related preconditioning on cerebral blood flow.
Key: ↑increase compared to no conditioning; ↔no change compared to no conditioning; 4VO: four-vessel occlusion; BCCAO: bilateral common carotid artery occlusion; CCAO: unilateral common carotid artery occlusion; HpreC: hypoxic preconditioning; IpreC: ischemic preconditioning; pMCAO: permanent middle cerebral artery occlusion; RIpreC: remote ischemic preconditioning; tMCAO: transient middle cerebral artery occlusion.
Table 3. Effects of hypoxia- or ischemia-related per- or post-conditioning on cerebral blood flow.
Key: ↓decrease compared to no conditioning; ↑increase compared to no conditioning; ↔no change compared to no conditioning; tBCCAO: bilateral common carotid artery occlusion; tCCAO: unilateral common carotid artery occlusion; IpostC: ischemic post-conditioning; pMCAO: permanent middle cerebral artery occlusion; pVAO: permanent vertebral artery occlusion; RIperC: remote ischemic perconditioning; tMCAO: transient middle cerebral artery occlusion; tPA: tissue plasminogen activator.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 12485 | 27 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA