Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The Role of Platelets in Ischemic Conditioning
Time:2018-11-08
Number:14323
Author Affiliations

Conditioning Medicine, 2018. 1(6):313-318.
Abstract
Abstract: Ischemic heart disease (IHD) is one of the leading causes of death and disability worldwide. Platelets, as the main regulators of hemostasis, are major players in acute myocardial ischemia/reperfusion injury (IRI). Additionally, platelets are modified by endogenous cardioprotective strategies such as ischemic preconditioning, postconditioning, and remote ischemic conditioning. In this article, we provide an overview of the functional role of platelets in acute myocardial IRI, and highlight their potential as targets for cardioprotection to improve health outcomes in patients with IHD.
Keywords: ischemia-reperfusion injury, platelets, ischemic conditioning, cardioprotection.
Abstract
Abstract: Ischemic heart disease (IHD) is one of the leading causes of death and disability worldwide. Platelets, as the main regulators of hemostasis, are major players in acute myocardial ischemia/reperfusion injury (IRI). Additionally, platelets are modified by endogenous cardioprotective strategies such as ischemic preconditioning, postconditioning, and remote ischemic conditioning. In this article, we provide an overview of the functional role of platelets in acute myocardial IRI, and highlight their potential as targets for cardioprotection to improve health outcomes in patients with IHD.
Keywords: ischemia-reperfusion injury, platelets, ischemic conditioning, cardioprotection.
Introduction
Cardiovascular disease (coronary heart disease and stroke) has been the leading cause of death worldwide over the last 15 years, accounting for 15.2 million deaths in 2016 (World Health Organization, 2018.; Cabrera-Fuentes et al., 2016a, 2016b). In addition to reducing quality of life, cardiovascular disease represents a major economic burden, with an estimated global cost by 2030 of US$1,044 billion (American Hearth Association and American Stroke Association, 2017).
In recent years, platelets have emerged as important mediators for various types of cardiovascular diseases (Malchow et al., 2017). Platelets are irregular anucleated cellular cytoplasmic fragments, derived from megakaryocytes in the bone marrow. They play key roles in thrombosis, hemostasis, inflammation, atherogenesis, and cancer metastasis (Nording et al., 2015; Leblanc and Peyruchaud, 2016; Papapanagiotou et al., 2016; Malchow et al., 2017; Ong et al., 2018).
Platelets in hemostasis and thrombosis
Platelet indices such as mean platelet volume (MPV) and platelet count, have been correlated with an increased risk of cardiovascular-related and non-cardiovascular-related deaths (Ranjith et al., 2009; Cortez-Espinosa et al., 2017; Malchow et al., 2017). Due to their diverse functions, platelets are highly regulated through membrane agonist receptors (adenosine diphosphate-ADP, thrombin, and thromboxane), adhesion proteins (fibrinogen, fibronectin, laminin, thrombospondin, vitronectin, von Willebrand factor-vWF), and ligands (collagen, fibrinogen). Upon vascular injury, platelets are captured on the endothelium via the integrins GPIIb/IIIa (CD41) and GP1b, which bind to different ligands (Splawińska et al., 1992). Subsequently, platelets take on a star-shaped form (Gawaz, 2004) after activation via respective surface receptors, and release granules containing ATP, Ca2+, serotonin, PF4, TGFB1, PDGF, fibronectin, and coagulation factors V and VIII (Smith et al., 1999; Papapanagiotou et al., 2016). In turn, the production of thromboxane A2 (TXA2) is induced, which is capable of activating additional platelets recruited to the growing hemostatic plug. Thrombus formation is initiated by the onset of the coagulation cascade. In circulating platelets, continuous exposure to inhibitory factors such as nitric oxide and prostacyclin ensures they maintain a resting, non-activated state (Köhler et al, 2009). A well-balanced equilibrium between these opposing processes is essential for normal platelet and vascular function (Figure 1) (Malchow et al., 2017).

In a new window | Download PPT
Figure 1: Platelets possess agonist receptors (adenosine diphosphate-ADP, thrombin, and thromboxane), adhesion proteins (fibrinogen, fibronectin, laminin, thrombospondin, vitronectin, von Willebrand factor-vWF), and ligands (collagen, fibrinogen). Upon vascular injury, platelets are captured at the endothelium via integrins GPIIb/IIIa, which initiates the activation of platelets through respective surface receptors, after which they take on a star-shaped form (Gawaz, 2004) and release granules containing ATP, Ca2+, serotonin, PF4, TGFB1, PDGF, fibronectin, and coagulation factors V and VIII. These granules induce additional platelet recruitment and thrombus formation. Platelets also can be activated by its agonist ADP via P2Y12 subsequently releasing PDVF, VEGF, adenosine, bradykinin, and endorphins into the bloodstream.
Platelets can also be activated by their agonist ADP via purinergic cellular receptors, namely P2Y1, P2Y12, and P2X1, or by collagen via the GPVI receptor. The P2Y1 receptor provokes calcium mobilization from internal stores that leads to conformational changes, and TXA2 and ADP-induced platelet aggregation (Hechler and Gachet, 2011). The P2Y12 receptor is a G-protein coupled receptor acting similar to P2Y1, but which also potentiates platelet activation via physiological agonists, inhibits adenylate cyclase, and turns off the cAMP-dependent inhibitory pathway (Koessler et al., 2018). The third receptor P2X1 is not related to G-protein activation, and facilitates the increase in Ca2+ concentration and activates ERK1/2 in the cell (Murugappa and Kunapuli, 2006). The GPVI receptor binds to collagen and stimulates 1) phospholipase C (PLC) mediated production of inositol triphosphate (I3P), which releases calcium from the dense tubular system, thereby increasing intracytoplasmic Ca2+ concentration and 2) the accumulation of diacylglycerol (DAG), which activates protein kinase C (PKC) that subsequently phosphorylates other proteins responsible for platelet aggregation. This latter process is shared with platelet activation induced by TXA2 and thrombin (Papapanagiotou et al., 2016).
Cellular metabolism in acute myocardial ischemia-reperfusion
During ischemia, oxygen levels decrease to critical levels, forcing cells to change from aerobic metabolism to anaerobic metabolism (Gourdin and Dubois, 2013). The duration of this ischemic-anoxic process can cause reversible damage (provided reperfusion is established in time), but can lead to cell death as damage spread to nearby cells if the anoxia is prolonged (Perrelli et al., 2011). Acute ischemia/reperfusion injury (IRI) results in organ dysfunction and is associated with serious clinical manifestations, including myocardial hibernation, acute heart failure, cerebral dysfunction, gastrointestinal dysfunction, systemic inflammatory response syndrome (Liehn and Hector A., 2015), and multiple organ dysfunction syndromes. The injury induced by IRI is divided into two parts: ischemic injury and reperfusion injury (Wu et al., 2018).
Due to low cellular oxygen concentrations, the mitochondrial electron transport chain decreases electron flux, affecting oxidative phosphorylation of ADP, thus reducing the production of ATP. The activation of Na+/K+-ATPase hydrolyzes ATP, increasing the concentration of ADP. High ADP levels lead to activation of anaerobic glycolysis, which generates lactate, cellular acidosis, and increased proton concentration (H+) causing dysfunction of intracellular organelles (Gourdin and Dubois, 2013). The cellular regulatory mechanisms of acidosis increase the Na+ concentration in the cell, which stimulates the Na+/K+-ATPase. This leads to an overload of Ca2+, which cannot be stored in the sarcoplasmic reticulum due to lack of ATP to activate its transporter. The high concentration of intracellular Ca2+ activates phosphatases, nucleases, and proteases, which in turn cause damage or total destruction of the membrane, and even cell death (Figure 2) (Perrelli et al., 2011; Gourdin and Dubois, 2013; Abad et al., 2018).
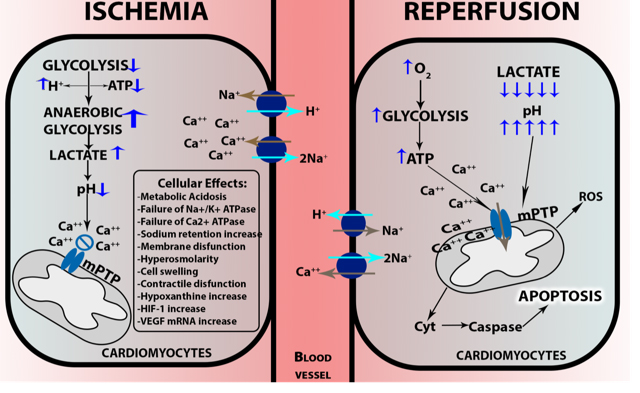
In a new window | Download PPT
Figure 2: Cellular damage in ischemia-reperfusion. During ischemia, oxygen levels become critically low, cells change from aerobic to anaerobic glycolysis, generating lactate, cellular acidosis, and increased proton concentrations. The regulatory mechanism of acidosis increases the intracellular Na+ concentration. This stimulates Na+/K+-ATPase leading to an overload of Ca2+, which cannot be stored in the sarcoplasmic reticulum due to lack of ATP to activate its transporter. During reperfusion, oxygen concentration rises and the recovery of ATP production in the presence of Ca2+ causes calcium oscillations, promoting mitochondrial mPTP opening, which leads the outer membrane to rupture, and the release of Cyt-C, which activates caspase-mediated apoptosis.
Platelets in ischemia and reperfusion
Ischemic heart disease (IHD) is mainly caused by atherosclerosis and its complications (Simsekyilmaz et al., 2014a, 2014b). Continuous activation and aggregation of platelets can release platelet-derived microparticles, which can at the least participate in the initiation of atherosclerotic plaques and coronary thrombus formation (Suades et al., 2012; Malchow et al., 2017). Significantly lower platelet count and higher platelet volume are associated with acute coronary syndrome in patients when compared to the normal population (Ranjith et al., 2009). Larger platelets are enzymatically and metabolically more active and have a higher potential thrombotic ability as compared with smaller platelets (Ranjith et al., 2009). Moreover, the MPV has been correlated with activation, aggregation, and release of granule content (Murugappa and Kunapuli, 2006). MPV is increased in several states of cardiovascular risk such as myocardial infarction and acute ischemic stroke, and is a convenient biomarker for risk and prognosis of heart diseases (Okoroiwu and Ogar, 2018).
Platelets and endothelial cells are the main regulators of hemostasis (Smith et al., 1999; Willoughby et al., 2002; Nording et al., 2015). A decrease in the flow of oxygen can cause or aggravate ischemic processes, which can lead to cell death. The damage caused by IRI in the vascular endothelium causes activation of circulating platelets, resulting in the release of the contents of alpha dense secretory granules and lysosomes (Golebiewska and Poole, 2015). The alpha granules are of utmost importance because they contain multiple cytokines, mitogens, and growth factors such as platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), and transforming growth factor beta (TGFβ), among others (Blair and Flaumenhaft, 2009). PDGF is a potent mitogen for mesenchymal cells and fibroblasts, as well as for smooth muscle and glial cells (Andrae et al., 2008). The PDGF receptor (PGDFR) is capable of modulatiing signal transduction through the PI3K-pathway or the reactive oxygen species (ROS)-Mediated STAT3 pathway, which then can regulate gene expression and the cell cycle, increasing cell proliferation and migration, and affecting angiogenesis (Figure 3) (Adair and Montani, 2010; Raica and Cimpean, 2010; Bodnar, 2013; Heldin and Lennartsson, 2013).
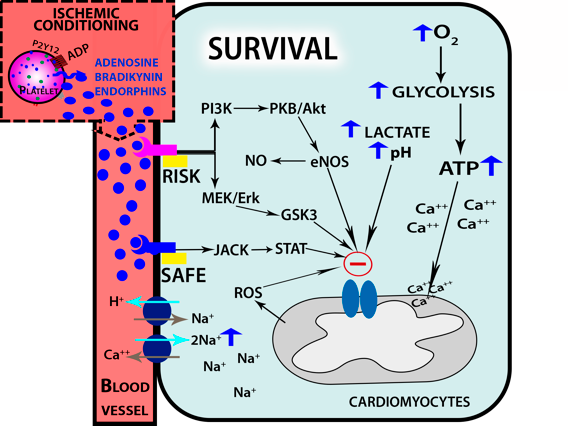
In a new window | Download PPT
Figure 3: Ischemic conditioning leads to endogenous platelet activation, which cause the release of its granular contents. During reperfusion, receptors on cardiomyocytes for adenosine, bradykinin, and endorphins activate protein signaling kinase cascades, which subsequently activate dowstream molecules such as PKC, ERK1/2, PI3K/AKT, RISK and SAFE. These molecules along with high concentrations of ROS produced by mitochondria block the opening of mPTP channel, thus protecting cells from apoptosis.
Examination of the granular factors released upon platelet activation was proposed 20 years ago by del Soppo (1998) who demonstrated increased plasma levels of PF4 and bTG upon platelet activation in patients suffering from stroke (del Zoppo, 1998). More recently, analysis of the differential proteomic profile of platelets from stroke patients identified 83 differentially expressed proteins out of 500 that are involved in various processes, such as the inflammatory response, cellular movement, immune cell trafficking, cell-to-cell signaling and interaction, hematological system development and function, and nucleic acid metabolism. Interestingly, in stroke patients 31 platelet proteins were involved in three pathways (inflammation, signaling and interaction, and the hematological system) (Cevik et al., 2016). More studies regarding proteomic profiling of platelet activation, aggregation, or degranulation need to be undertaken in order to better understand the complexity of platelet function in cardiovascular diseases.
Platelets in ischemic conditioning
During cardiac surgery, acute myocardial IRI is a crucial problem, and new techniques are needed to protect the heart (Bousselmi et al., 2014). Ischemic conditioning is a mechanical maneuver of repetitive controlled cycles of ischemia/reperfusion (I/R) (Hausenloy et al., 2016). It can be categorized as pre-, per- or post-conditioning, depending on the timing of the application. Ischemic preconditioning (IPC) was first described in 1986 by Murry et al. (1986), who found a 75% reduction in myocardial infarct size when the coronary artery was previously exposed to four cycles of five-minute of controlled I/R (Bousselmi et al., 2014). IPC protection can be divided into two phases. An early phase or classical ischemic preconditioning occurs within a few hours of the preconditioning stimulus and protects against myocardial necrosis. A delayed phase occurs at later time points after the preconditioning stimulus when the protection against myocardial stunning is mediated by de novo protein synthesis (Cohen et al., 2001; Yellon and Downey, 2003). IPC relies on maintaining mitochondrial ATP production by preventing the opening of the mitochondrial permeability transition pore (mPTP) induced by I/R, and activating the RISK (Reperfusion-Induced Salvage Kinase) and SAFE (Zhao and Vinten-Johansen, 2006) pathways via adenosine, bradykinin, and endorphins (Cohen et al., 2001). On the other hand, IPC late protection is mediated by iNOS and COX-2. Heusch et al. (2015) showed myocardial protection induced by IPC in a remote organ in which the remote ischemic pre-conditioning (RIPC) stimulus was repeated brief periods of ischemia in a limb at a distance from the target organ (Starlinger and Gruenberger, 2014; Heusch et al., 2015). The mechanisms of systemic tissue protection were studied by Starlinger and Gruenberger (2014) who proposed that immediately after RIPC, platelets are activated, and serotonin and VEGF are released from dense alpha granules. Serotonin acts on endothelial cells (EC) that release more VEGF, which in turn induces IL-10 and MMP-8 expression. EC also release HMG-B1, which decreases TNF-α tissue expression after reperfusion, causing decreased IRI (Starlinger and Gruenberger, 2014). Other studies found that upper arm intermittent ischemia induces protection of myocardial cells against injury caused by ischemia. It also reduces platelet activation and blunts platelet reactivity in response to ADP stimulation (Lanza et al., 2016). After RIPC, platelet activation is independent of collagen or arachidonic acid (Pryds et al., 2017). Moreover, the adenosine released by preconditioning favors thrombolysis (Przyklenk and Whittaker, 2017).
Platelets are also involved in IPC in patients with suspected acute myocardial infarction. When RIPC was delivered during ischemia and before reperfusion by primary percutaneous coronary intervention, this so-called ischemic per-conditioning (IPeC), resulted in potent cardioprotection (Hougaard et al., 2014). More recently, IPeC has been shown to be a clinically relevant neuroprotective strategy against acute ischemic stroke (Hahn et al., 2011). Although the mechanism underlying IPeC needs to be clarified, it is hypothesized that pathways similar to that seen in the early phase of IPC are involved.
During reperfusion, vascular cells are exposed to rapid changes in hydrostatic pressure, and the normal response to changes in osmotic gradients caused by these disturbances is edema formation (Friedrich, 2010). Capillary compression caused by more than 60-90 min of ischemia leads to membrane damage and leakage of water from the intravascular space into the interstitial space, causing interstitial edema (Friedrich, 2010). During this process, platelets can be activated by ligands or adhesion proteins expressed during capillary damage (Bath and Butterworth, 1996). During pulmonary hypertension that causes left ventricular myocardial interstitial edema (Davis et al., 1995; Bulluck et al., 2016) high MPV of platelets has been encountered (Mumpuni et al., 2016). This phenomenon contributes to and aggravates cell damage, which is characteristic of severe IRI.
Ischemic postconditioning (PoC) is a mechanical maneuver that transpires during the early phase of reperfusion. Similarly to IPC the stimulus is repetitive, controlled cycles of I/R (Staat, 2005); Vinten-Johansen, 2005; Zhao and Vinten-Johansen, 2006), which can be applied locally (LPoC) (Wang et al., 2018) or on the limbs (remote postconditioning, RPoC) (Wang et al., 2018; Zhao et al., 2018) to attenuate reperfusion injuries caused by endothelial activation and dysfunction at cellular and intracellular sites, infarction, or apoptosis (Zhao and Vinten-Johansen, 2006). Recent studies show that LPoC and/or RPoC not only improve myocardial survival and decrease myocardial edema in ST-elevation myocardial infarction patients without affecting final infarct size (Lou et al., 2018), but also improves neurological outcome after acute ischemic stroke (England et al., 2017).
Postconditioning triggers several mechanisms in the affected cells to attenuate oxidative damage induced by oxidants and nitric oxide. Those mechanisms can be activated by molecules in platelets after conditioning. Like in IPC, postconditioning induces cardioprotection by causing the release of adenosine from activated platelets, or by activated myocardial cells, preventing endothelium edema, and may also enhance endogenous cardioprotective mechanisms in the myocardium (Zhao and Vinten-Johansen, 2006). Less studied is the neurological protective mechanisms that may be mediated through HSP27 (Shimada et al., 2014; England et al., 2017).
During acute myocardial infarction, it has been proposed that ischemic preconditioning procedures (IPC, RIPC, IPeC, LPoC, or RPoC) commonly activate the release of adenosine, bradykinin, and opioid from platelets. In addition, ROS production activates the intracellular MEK1/2, PI3K, AKT, ERK1/2 signaling pathways. These pathways upregulate the RISK pathway, mediating the activation of downstream target molecules such as eNOS and GC in order to activate K+ ATP channels that ultimately cause inhibition of mPTP opening (Qian et al., 2018). Moreover, ADP release during IP may induce activation of signaling kinases in platelets. Soluble agonists such as ADP released from damaged endothelial cells and activated platelets acts on platelet P2Y1 and P2Y12 GPCR, causing further platelet activation and release of ADP. P2Y12 receptor sustains platelet activation in response to ADP, and therefore plays a central role in this process (Yun et al., 2016).
The fact that platelets possess ATP and ADP receptors signifies that platelets ensure responses are regulated according to the extent of the injury. More interestingly, CD39 and CD73 mediate ATP and ADP conversion to adenosine as another regulatory mechanism through which adenylate cyclase activates purinergic (P1) receptors (Koupenova and Ravid, 2018). While ATP activation of platelets via P2X1 receptors usually needs a co-stimulatory molecule, it regulates ADP-mediated P2Y12 receptor activation, increasing calcium influx into platelets. As reviewed above, when ADP is recognized by the P2Y12 receptor, the coupled G protein causes inhibition of the conversion of ATP to cAMP by deactivation of adenylate cyclase, and further increases in intracellular calcium via activation of IP3-receptor. The P2Y12 receptor is regulated via PI3K, inhibiting platelet activation after ADP-P2Y1 receptor activation, as a coordinated mechanism of regulation (Koupenova and Ravid, 2018). The blockade of P2Y12 receptors has been explored as an effective mechanism for regulating platelet responses during cardiovascular events. The ADP P2Y12 receptor antagonists clopidogrel, prasugrel, elinogrel, ticagrelor and cangrelor are clinically used in patients concomitantly with aspirin as antiplatelet therapy in percutaneous coronary intervention, peripheral angioplasty with stenting, and in acute coronary syndrome (). The drugs prescribed for patients with acute ischemic syndrome may have cardioprotective effects analogous to classic ischemic conditioning (Cortez-Espinosa et al., 2017; Pryds et al., 2017). However, those antiplatelet drugs can cause bleeding as a side effect (Serebruany et al., 2017; Looße et al., 2018).
Conclusion
Since cardiovascular diseases are a global health problem of enormous scale, any discovery in vascular or myocardial protection may prove to be very valuable for those at risk. Platelet function and activation upon vascular damage has been studied for many years, and still, the underlying molecular mechanisms have not been fully elucidated. Despite the fact that IPC was described 32 years ago, and has been demonstrated to be simple, low-cost, and very effective protective mechanism against injuries, the lack of extensive clinical studies prevents it from being adopted to the general population as a routine practice in vascular complications.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
Part of the original work presented by HCF was supported by the Russian Government Program for competitive growth of Kazan Federal University, Kazan (Russian Federation), by the SHF-Foundation Research Grant (SHF/FG657P/2017) and by the von Behring-Röntgen-Foundation (Marburg, Germany). This work was partially funded by Fondos del Programa de Fortalecimiento de la Calidad Educativa UABJO 2018 to JJAO. This project was funded by NIH grant HL139082 to JY.
References
Juan Alpuche1,2
1CONACyT-Facultad de Medicina, Centro de Investigación Facultad de Medicina, UNAM-UABJO. Universidad Autónoma Benito Juárez de Oaxaca. México.
2Centro de Investigación Facultad de Medicina, UNAM-UABJO. Universidad Autónoma Benito Juárez de Oaxaca.
Luz Quírino2,3
2Centro de Investigación Facultad de Medicina, UNAM-UABJO. Universidad Autónoma Benito Juárez de Oaxaca.
3Facultad de Odontología, Universidad Autónoma Benito Juárez de Oaxaca, México.
José T Sánchez-Vega4
4Parasitology Laboratory, Department of Microbiology and Parasitology, Faculty of Medicine, Universidad Nacional Autónoma de México, México City, México.
Jonathan Yap5
5Center for Cardiovascular Research, John A. Burns School of Medicine, University of Hawaii, Hawaii, USA.
Eduardo Pérez-Campos2,6
2Centro de Investigación Facultad de Medicina, UNAM-UABJO. Universidad Autónoma Benito Juárez de Oaxaca.
6Tecnológico Nacional de México/ IT Oaxaca. Oaxaca. México.
Hector A. Cabrera-Fuentes7-11
7Kazan Federal University, Department of Microbiology, Kazan, Russian Federation.
8Cardiovascular and Metabolic Disorders Program, Duke-NUS Medical School, Singapore, Singapore.
9National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
10Escuela de Ingeniería y Ciencias, Centro de Biotecnología-FEMSA, Tecnológico de Monterrey, Monterrey, NL, México.
11Institute of Biochemistry, Medical School, Justus-Liebig-University, Giessen, Germany.
Corresponding author:
Hector A. Cabrera-Fuentes
Email: cabrera.fuentes.h.a@nhcs.com.sg
or
Juan Alpuche
Email: Juan_Alpuche@hotmail.com
In a new window | Download PPT
Figure 1: Platelets possess agonist receptors (adenosine diphosphate-ADP, thrombin, and thromboxane), adhesion proteins (fibrinogen, fibronectin, laminin, thrombospondin, vitronectin, von Willebrand factor-vWF), and ligands (collagen, fibrinogen). Upon vascular injury, platelets are captured at the endothelium via integrins GPIIb/IIIa, which initiates the activation of platelets through respective surface receptors, after which they take on a star-shaped form (Gawaz, 2004) and release granules containing ATP, Ca2+, serotonin, PF4, TGFB1, PDGF, fibronectin, and coagulation factors V and VIII. These granules induce additional platelet recruitment and thrombus formation. Platelets also can be activated by its agonist ADP via P2Y12 subsequently releasing PDVF, VEGF, adenosine, bradykinin, and endorphins into the bloodstream.
In a new window | Download PPT
Figure 2: Cellular damage in ischemia-reperfusion. During ischemia, oxygen levels become critically low, cells change from aerobic to anaerobic glycolysis, generating lactate, cellular acidosis, and increased proton concentrations. The regulatory mechanism of acidosis increases the intracellular Na+ concentration. This stimulates Na+/K+-ATPase leading to an overload of Ca2+, which cannot be stored in the sarcoplasmic reticulum due to lack of ATP to activate its transporter. During reperfusion, oxygen concentration rises and the recovery of ATP production in the presence of Ca2+ causes calcium oscillations, promoting mitochondrial mPTP opening, which leads the outer membrane to rupture, and the release of Cyt-C, which activates caspase-mediated apoptosis.
In a new window | Download PPT
Figure 3: Ischemic conditioning leads to endogenous platelet activation, which cause the release of its granular contents. During reperfusion, receptors on cardiomyocytes for adenosine, bradykinin, and endorphins activate protein signaling kinase cascades, which subsequently activate dowstream molecules such as PKC, ERK1/2, PI3K/AKT, RISK and SAFE. These molecules along with high concentrations of ROS produced by mitochondria block the opening of mPTP channel, thus protecting cells from apoptosis.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14323 | 29 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA