Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Positive and negative conditioning in the neonatal brain
Time:2018-11-08
Number:12444
Author Affiliations

Conditioning Medicine, 2018. 1(6):279-293.
Abstract
Brain injury in the perinatal period occurs in many clinical settings, e.g. hypoxic-ischemic encephalopathy (HIE) in term infants, neonatal stroke, encephalopathy of prematurity, and infections. These insults often result in life-long disabilities including cerebral palsy, cognitive deficits, visual dysfunction, hearing impairments, and epilepsy. However, the success of clinical implementation of a broad array of potential neuroprotective strategies tested experimentally has been limited with the exception of therapeutic hypothermia (TH) used within hours of birth in term human babies with mild to moderate HIE. There is an extensive search for adjuvant therapeutic approaches to enhance the outcomes. One strategy is to modify susceptibility in the developing CNS by means of preconditioning or postconditioning using sublethal stress. The pre-clinical and clinical literature has shown that CNS immaturity at the time of ischemic insult plays a central role in the response to injury. Thus, better understanding of the molecular regulation of the endogenous vulnerability of the immature brain is needed. Further, the use of sublethal stressors of different origin may help shed light on mechanistic similarities and distinctions beween conditioning strategies. In this review we discuss the mechanisms of protection that are achieved by an interplay of changes on the systemic level and brain level, and via changes of intracellular and mitochondrial signaling. We also discuss the barriers to improving our understanding of how brain immaturity and the type of insult—hypoxic, ischemic or inflammatory—affect the efficacy of conditioning efforts in the immature brain.
Keywords: preconditioning, postconditining, hypoxia-ischemia, stroke, immune system, mitochondria, blood-brain barrier.
Abstract
Brain injury in the perinatal period occurs in many clinical settings, e.g. hypoxic-ischemic encephalopathy (HIE) in term infants, neonatal stroke, encephalopathy of prematurity, and infections. These insults often result in life-long disabilities including cerebral palsy, cognitive deficits, visual dysfunction, hearing impairments, and epilepsy. However, the success of clinical implementation of a broad array of potential neuroprotective strategies tested experimentally has been limited with the exception of therapeutic hypothermia (TH) used within hours of birth in term human babies with mild to moderate HIE. There is an extensive search for adjuvant therapeutic approaches to enhance the outcomes. One strategy is to modify susceptibility in the developing CNS by means of preconditioning or postconditioning using sublethal stress. The pre-clinical and clinical literature has shown that CNS immaturity at the time of ischemic insult plays a central role in the response to injury. Thus, better understanding of the molecular regulation of the endogenous vulnerability of the immature brain is needed. Further, the use of sublethal stressors of different origin may help shed light on mechanistic similarities and distinctions beween conditioning strategies. In this review we discuss the mechanisms of protection that are achieved by an interplay of changes on the systemic level and brain level, and via changes of intracellular and mitochondrial signaling. We also discuss the barriers to improving our understanding of how brain immaturity and the type of insult—hypoxic, ischemic or inflammatory—affect the efficacy of conditioning efforts in the immature brain.
Keywords: preconditioning, postconditining, hypoxia-ischemia, stroke, immune system, mitochondria, blood-brain barrier.
Introduction
Brain injury in the perinatal period occurs in many clinical settings, e.g. hypoxic-ischemic encephalopathy (HIE) in term infants, neonatal stroke, encephalopathy of prematurity, and infections (Hagberg et al., 2015). These insults often result in life-long disabilities including cerebral palsy, cognitive deficits, visual dysfunction, hearing impairments and epilepsy (Wood et al., 2000; Delobel-Ayoub et al., 2009; Fernandez-Lopez et al., 2014; Hagberg et al., 2016). A broad array of potential neuroprotective strategies has been tested pre-clinically, targeting various signaling pathways to attenuate neuronal cell death and/or limit toxic aspects of oxidative stress and inflammation caused by injury. However, the success of clinical implementation has been limited. Therapeutic hypothermia (TH) used within hours of birth in term human babies with HIE has been the only successful treatment thus far (Edwards et al., 2010). TH has been approved for treating HIE since 2010 in at-term human neonates and it has become the standard of care in many countries. However, beneficial effects of TH are limited to only mild-to-moderate brain injury, and many infants still develop significant adverse outcomes. There is an extensive search for adjuvant therapeutic approaches to improve the outcomes. One strategy is to better understand the molecular regulation of the endogenous vulnerability of the immature brain by using sublethal stressors of different origin. The present review will focus on various ways to modify susceptibility in the developing CNS by means of preconditioning (pre-C) or postconditioning (post-C) stressors.
Conditioning
A sublethal stress (e.g., hypoxia, ischemia) or drug administration can induce resistance to a subsequent potentially lethal insult, a phenomenon known as pre-C (Janoff, 1964). Pre-C was first described in the heart (Murry et al., 1986) but can be induced in most organs, including the brain (Chen and Simon, 1997; Dirnagl et al., 2003; Gidday, 2006; Thompson et al., 2015). In the adult, the application of pre-C and post-C stressors have been extensively studied as protective strategies in models of cerebral ischemia. Multiple lines of evidence have suggested that protection is achieved by an array of mechanisms at the systemic level, at the neurovascular unit interface, and by modifying cell-cell interactions and intracellular signalling and, in particular, mitochondrial function. Altered temporo-spatial patterns of cerebral blood flow (CBF) upon reperfusion, immune-neurovascular interactions, suppression of neuroinflammation, and changes in mitochondrial function in injured adult brain are among the most important changes caused by pre-C and post-C stressors.
Pre-C seems to be particularly robust in the immature brain (Gidday et al., 1994). In most studies, conditioning is induced by exposure to the stressor or drug some time prior to the severe insult (usually > 6-12 h), but tolerance can also be triggered by exposure to the stressor after the main insult, referred to as post-C (Zhao et al., 2006a; Teo et al., 2015). Conditioning stimuli can both reduce immature brain vulnerability (positive conditioning; Gidday et al., 1994) or increase vulnerability (negative conditioning; Eklind et al., 2001). Furthermore, subthreshold stress in a peripheral organ (e.g. repeated limb ischemia) can induce conditioning in the developing brain, a phenomenon called remote pre-C or post-C (R-pre-C or R-post-C) (Tropak et al., 2011; Khan et al., 2015; Ezzati et al., 2016). Conditioning can be induced by sublethal hypoxia, ischemia, epileptic seizures, hyperbaric oxygenation, hyperthermia, toll-like receptor (TLR) agonists, or drugs such as magnesium sulphate, glucocorticoids or anesthetics (e.g. xenon) (Barks et al., 1991; Gidday et al., 1994; Chen and Simon, 1997; Eklind et al., 2001; Dirnagl et al., 2003; Gidday, 2006; Koning et al., 2017). As in the adult brain, several pre-C and post-C mechanisms have been proposed to alter vulnerability in the developing brain. Some of the different regimens of conditioning applied in the immature brain are summarized in Table 1.
Table 1. Examples of conditioning paradigms in the developing brain.
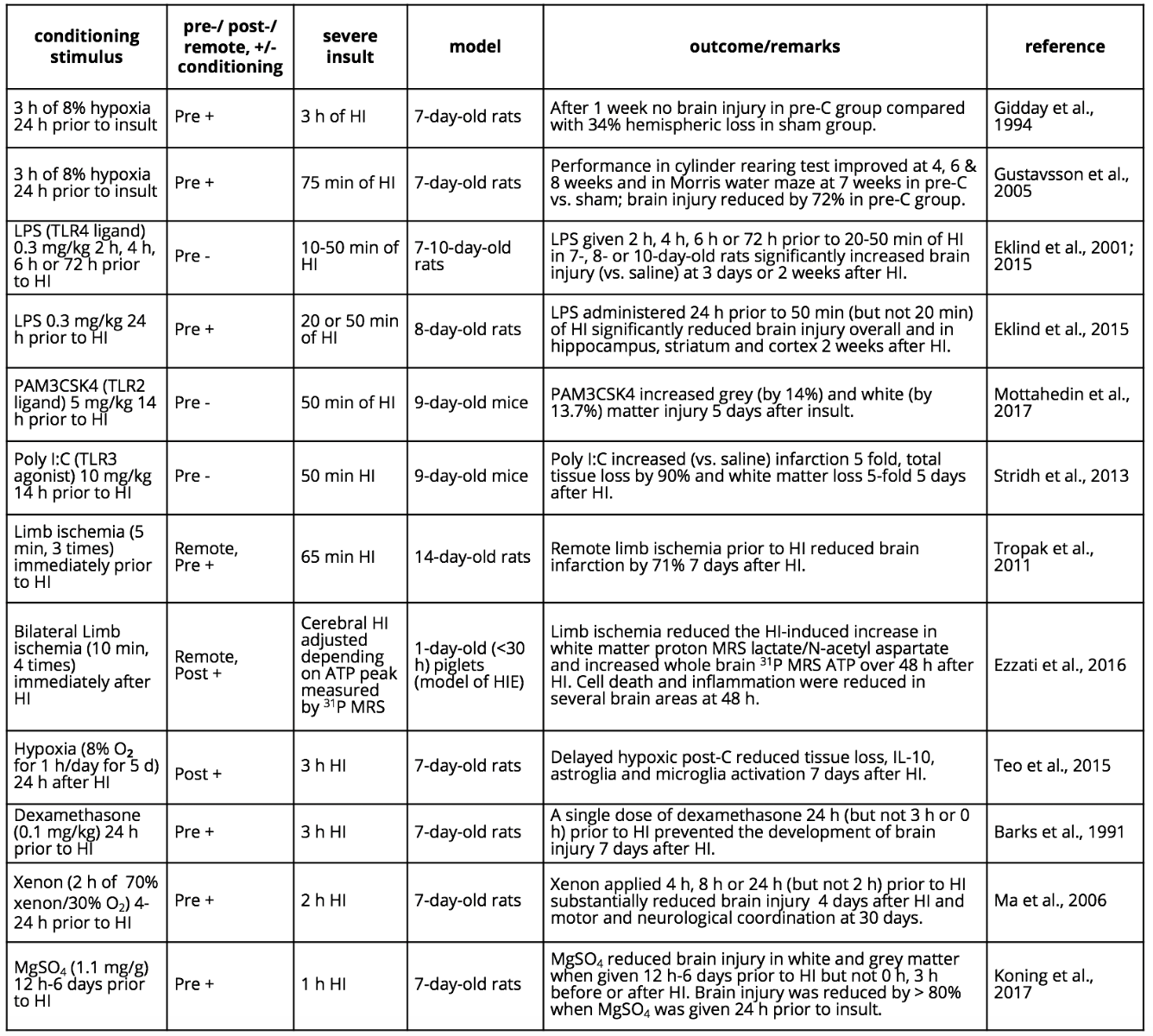
Abbreviations: Hypoxia-ischemia (HI); hypoxic-ischemic encephalopathy (HIE); lipopolysaccharide (LPS); magnetic resonance spectroscopy (MRS); magnesium sulphate (MgSO4); negative conditioning (-); polyinosinic: polycytidylic acid (poly I:C); positive conditioning (+).
Pre-C and post-C as modulators of the neurovascular unit in the injured immature brain
In the adult, CBF is not affected by pre-C itself, but the decrease in CBF during a subsequent episode of ischemia is attenuated (Nakamura et al., 2006). Similarly, we have found that hypoxic pre-C in the immature rat attenuates the drop in CBF during a severe HI insult induced 24 h after pre-C (Gustavsson et al., 2007). This effect was associated with upregulation of vascular genes (e.g. Angpt2, VEGF, Flt1, Kdr, Pdgfra) and an increase in vascular density. Recently, it was shown that the pre-C-induced preservation of vascular density after HI was accompanied by an increase in VEGFR2, and the pre-C effect was reversed by the angiotensin II receptor subtype 2 (AT2-R) inhibitor (PD123319), offering support for a vascular component in pre-C (Lopez-Aguilera et al., 2012). The pre-C effect of erythropoietin (EPO) could also at least partly be explained by vascular effects. Lipopolysaccharide (LPS)-induced pre-C diminished the extent of secondary perfusion deficits (rather than primary perfusion deficits) in adult stroke by induction of endothelial nitric oxide synthase (eNOS) (Dawson et al., 1999), and NOS inhibition by L-NAME abolished the pre-C effects, indicating a role for NOS in LPS-mediated positive pre-C. Similarly, in neonatal rats, Akt-mediated eNOS upregulation in neurons and vascular endothelial cells was required for LPS-induced positive pre-C against HI (Lin et al., 2010). However, in hypoxic pre-C, the CBF was not affected by L-nitroarginine (Gustavsson et al., 2007), implying NO has other targets besides the vasculature. There is no data yet in adult or immature brains with ischemia models demonstrating that pre-C neuroprotection predominantly depends on vascular adaptations (c.f. Gidday, 2006). However, in an excitotoxicity model, the protective effect of hypoxic pre-C was reversed by VEGFR2 antibodies, and VEGF reduced brain injury to a similar extent as hypoxic pre-C (Laudenbach et al., 2007). However, we can not rule out non-vascular actions mediated by VEGF-VEGFR2, considering VEGF also has neurotropic effects. Furthermore, the pre-C effect of MgSO4 was not associated with improvement in CBF either before or during a subsequent period of HI (Koning et al., 2017), suggesting that vascular modifications are not always required for induction of pre-C in the immature brain.
Disruption of blood-brain barrier (BBB) integrity is a major contributor to the pathophysiology of stroke. In adult stroke models, brief repetitive hypoxic episodes (Stowe et al., 2011) and exercise pre-C (Davis et al., 2007) were shown to limit BBB dysfunction, in part by downregulating endothelial adhesion molecules, limiting leukocyte infiltration, as well as by preserving basal lamina. The use of low thrombin concentrations (thrombin preconditioning; TPC) before middle cerebral artery occlusion (MCAO) attenuated brain edema, BBB disruption and brain hemorrhage (Stetler et al., 2009). Effects were mediated by mechanisms that include induction of heat shock proteins, particularly HSP27/HSP25, modulation of downstream cellular protective p44/42 MAPK/p90RSK/HSP25 pathways and protease-activated receptors (PAR) (Stetler et al., 2009). Pre-C also enhanced recognition and chaperoning of damaged or misfolded proteins, stabilizing actin filaments and limiting the stress-response cascade upstream of the mitochondrial cell death machinery (Stetler et al., 2009; Shi et al., 2017).
Literature has shown that BBB integrity greatly depends on the maturational stage of the brain at the time of stroke (Fernandez-Lopez et al., 2012; Kratzer et al., 2014), as well as the injury model used (Fernandez-Lopez et al., 2012; Ek et al., 2015; Mallard and Vexler, 2015). Individual components of the neurovascular unit, including endothelial cells, the extracellular matrix (ECM) and glial cells, undergo unsynchronized developmental changes and increases astrocytic and pericyte vessel coverage in postnatal brain, affecting both acute structural-functional BBB responses and delayed angiogenic processes.
Data on the role of the neurovascular unit in mediating pre-C in neonates, while scarce, suggest that pre-C protects the ECM and BBB integrity. One study showed that administration of plasminogen activator inhibitor-1 (PAI-1) before HI reduced brain injury in a dose-dependent manner (Yang et al., 2009). Protection was preceded by a marked reduction of tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator, affecting MMP-9 activation, brain edema and BBB permeability, and was associated with reduced axonal degeneration (Yang et al., 2009). Another study demonstrated that intra-cerebroventricular injection of a stable-mutant form of PAI-1 attenuates pro-inflammatory response and BBB disruption in, an LPS injury model followed by HI in P7 rats (Yang et al., 2013). Hyperthermic pre-C also prevented BBB disruption produced by HI in newborn rat (Ikeda et al., 1999).
Several studies have suggested that inflammation during gestation can prime the vasculature, producing long-lasting changes in BBB function but not necessarily short-term changes in BBB integrity. While LPS pre-C followed by HI induced significant changes in BBB integrity in P12 rats, there was little evidence of acute neutrophil infiltration into the brain or increased albumin leakage after a single injection of LPS (Brochu et al., 2011). Newborn rats given multiple intraperitoneal LPS injections at P0-P8 did not have significantly increased sucrose leakage through the BBB at either P9 or P20 (Stolp et al., 2011), but strikingly, LPS led to significantly higher brain sucrose concentration during adulthood (Stolp et al., 2005). Deficiency in omega-3 lipids during gestation negatively conditioned pups by making them more prone to HI postnatally by enhancing neuroinflammation, adversely affecting the vasculature and BBB integrity (Zhang et al., 2010; Zhang et al., 2015). Together, these results suggest that the brain ECM plays a modulatory role in BBB integrity and injury after neonatal HI.
Innate immune system modulation as positive and negative pre-C in the immature brain
Data have been accumulating on the role of innate immunity in injury to the immature brain and the possibility to attenuate injury by modifying immune responses. In utero infections, neonatal sepsis and chorioamnionitis, are linked to both preterm birth and neurological deficits later in life (Shatrov et al., 2010; Salas et al., 2013; Soraisham et al., 2013; Shankaran et al., 2014). Children born with bacteremia very preterm perform worse on neurocognitive tests in the first weeks of life (Bright et al., 2017) and demonstrate neurological dysfunction at school age (Kavas et al., 2017).
Induction of cerebral innate immunity via TLR has been thoroughly established in the perinatal brain injury field (Mallard et al., 2009). TLRs are innate immune receptors that sense “pathogen-associated molecular pattern molecules” (PAMP) expressed by viral and bacterial micro-organisms or “damage-associated molecular pattern molecules” (DAMP) that are released from injured tissue. Individual TLRs differ in their ligand specificity, signal transduction pathways they activate, and types of infection to which they respond. TLR4 mediates gram-negative bacterial infections (induced by LPS/endotoxin), TLR2 is mostly associated with gram-positive bacteria (mimicked by i.p. injection of the synthetic lipopeptide Pam(3)CSK(4); PAM), and TLR3 mediates viral infections (mimicked by i.p. injection of poly I:C). Several studies have shown that excessive activation of TLRs, either by bacterial stimuli or by injury, can have direct negative effects on the developing brain. For example, chronic exposure to LPS or PAM impairs both grey and white matter development in neonatal mice (Du et al., 2011), and TLR2 gene-deficient mice are protected from neonatal HI (Stridh et al., 2011). There is also growing evidence demonstrating that TLR-mediated mechanisms can modulate the vulnerability of the immature brain to other insults, resulting in either positive or negative pre-C. Negative pre-C effects on HI neonatal brain injury have been observed following activation of several TLR subtypes, including TLR2 (Falck et al., 2017; Mottahedin et al., 2017), TLR3 (Stridh et al., 2013) and TLR4 (Eklind et al., 2001; Eklind et al., 2005; Wang et al., 2007) (Table 1). Immune challenges during gestation have also demonstrated a broad range of negative conditioning effects in maturing rodents. Indeed, LPS administration during gestation increased brain cytokine levels and made pups more vulnerable to HI at P1 (Girard et al., 2008). The LPS-induced increased vulnerability to neonatal HI (negative pre-C) is dependent on the TLR adaptors MyD88 (Wang et al., 2009a) and TRIF (Stridh et al., 2013), while in adult stroke, positive pre-C after TLR4 signaling depends on TRIF but not on MyD88 signaling (Vartanian et al., 2011), emphasizing age-dependent differences in immune pre-C mechanisms.
The time interval between TLR activation and the main insult is another important factor that determines whether there are positive or negative immune pre-C effects on brain injury. For example, when administered 2-4 days before MCAO in adult rats, LPS enhanced protection, with maximal effect at 3 days, whereas no protection was observed when LPS was administered 1 day or 1 week before MCAO (Tasaki et al., 1997). The magnitude of TLR activation is also important in determining positive or negative outcome, with higher doses of LPS abolishing positive pre-C in MCAO-induced injury (Bordet et al., 2000). Similar to the adult, we have shown in neonates that, depending on the time interval between insults, the same dose of LPS can both increase and decrease vulnerability to HI in P7 rat pups. Administration of a low LPS dose 4 h, 6 h, or 3 days before HI exacerbated injury (Eklind et al., 2001) and produced short-term memory impairment (Ikeda et al., 2004), while a 24-h interval was protective (Eklind et al., 2005). Literature is also emerging on the lasting effects of brain reprogramming following gestational immune challenges affecting susceptibility to injury during adulthood. Remarkably, while intrauterine LPS administration sensitized (negative pre-C) to early postnatal HI, it served as a protective pre-C stimulus in adult HI (Wang et al., 2007).
The need for new protein synthesis for induction of pre-C has been shown in many settings including LPS-induced effects (Bordet et al., 2000). Protective pre-C LPS effects on MCAO injury were blocked by dexamethasone or indomethacin administration, supporting a role for inflammatory mechanisms (Bordet et al., 2000), whereas we reported that up-regulation of endogenous corticosterone protects the immature brain in LPS-induced pre-C 24 h prior to HI (Ikeda et al., 2006). Using microarray techniques, we compared the cerebral gene response following positive and negative LPS-induced pre-C. We showed that the increased vulnerability to HI (negative pre-C) seen at 6 h after LPS is associated with significant gene regulation, including genes associated with protein metabolism, immune and inflammatory response, chemotaxis, and cell death (Eklind et al., 2006). These changes in inflammatory genes were very prominent and distinct from the positive pre-C scenario (i.e., 24 h after LPS administration) (Eklind et al., 2006). Further, expression of transcript changes differed from those induced by HI only (Hedtjarn et al., 2004a; Hedtjarn et al., 2004b), indicating specificity in the pre-C response. Further evidence of involvement of immune responses in sensitizing effects on the immature brain was obtained in a study that utilized immunomodulator IDR-1018 in P9 mice subjected to LPS combined with HI (Bolouri et al., 2014). Importantly, IDR-1018 was protective when administered as pre-treatment or post-treatment (Bolouri et al., 2014); however, whether pre-C-specific responses were affected remains to be determined.
The source of the inflammatory response in the immature brain associated with TLR-induced pre-C has not been fully elucidated. While it is clear that microglia, the endogenous brain immune cells, respond to antenatal and postnatal systemic TLR activation, as well as non-infectious inflammation, the mechanisms that link peripheral signals to activated microglia remain obscure. We showed that microglia are the predominant source of TLR2 expression during normal postnatal brain development but that LPS stimulation and intracerebral IL-1β injection in P9-P10 mice produce stimuli-dependent induction of inflammatory mediators (Lalancette-Hebert et al., 2017). Administration of LPS at P5 leads to a transient increase in absolute number and cell density of Iba1-positive microglia, as well as a persistent alteration in hippocampal inflammatory status in microglia (Smith et al., 2014). Further, repeated injections of PAM at P3-P11 decreased the volume of cerebral grey and white matter in association with elevated levels of several pro-inflammatory cytokines and chemokines in the brain as well as increased microglial density (Du et al., 2011). In fetal sheep, LPS administered either to the fetus (Duncan et al., 2002; Mallard et al., 2003; Dean et al., 2011) or into the amniotic fluid (Nitsos et al., 2006) at a gestational age comparable to that of the preterm human infant led to CNS inflammation and increased numbers of microglia with activated morphologic phenotype. Comparative analysis of TLR2 and TLR4 stimulation has shown stimulus-dependent patterns of cytokine induction and leukocyte infiltration into immature brain (Mottahedin et al., 2017). The magnitude and particulars of induced infiltration of CD11+/CD45hi leukocytes were particularly prominent following PAM administration (Mottahedin et al., 2017). The role of increased microglia proliferation and leukocyte trafficking in preconditioning remains unclear, but, interestingly, it was recently shown that positive LPS pre-C induces a Ly6Chi monocyte response that protects the brain after MCAO (Garcia-Bonilla et al., 2018).
Intracellular mechanisms of hypoxic pre-C in the immature brain
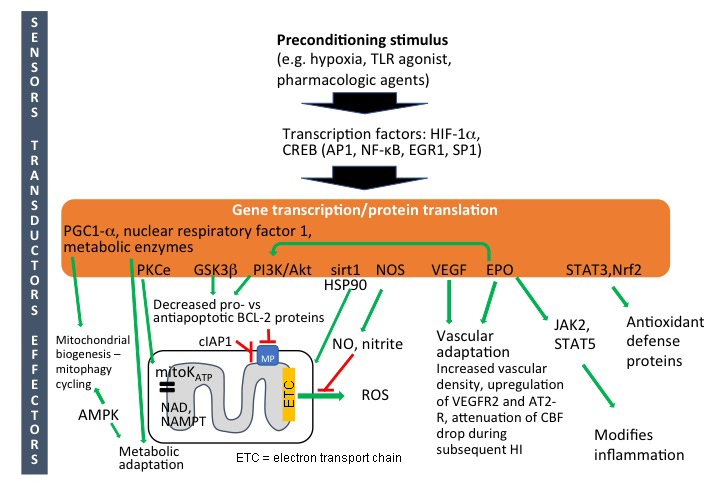
There is a vast amount of information on intracellular mechanisms of pre-C in response to various stress stimuli, and there seems to be a high degree of similarity across organs and species. It appears that some general endogenously protective cellular strategies have evolved in order to cope with dangerous exposures such as hypoxia/ischemia, substrate deficiency and infections. There isn't a single neuroprotective factor, but pre-C is the result of the combined action of multiple, partly overlapping cellular and molecular events that counteracts cell death, reduces the injurious effects of inflammation, adapts mitochondrial metabolism and enhances regenerative processes (Dirnagl et al., 2003; Gidday, 2006). In some reviews, the different components involved in pre-C have been divided into sensors, transducers, and effectors (Dirnagl and Meisel, 2008). However, in the immature brain, the sequence of events is still uncertain, but an approximate staging of the mechanisms is indicated in Figure 1.
In a new window | Download PPT
Figure 1: Intracellular mechanisms of preconditioning in neonatal brain. Abbreviations: Activator protein-1 (AP-1); AMP-activated protein kinase (AMPK); cellular inhibitor of apoptosis-1 (cIAP1); cyclic AMP response-element binding-protein (CREB); early growth response 1 (EGR1); erythropoietin (EPO); hypoxia-inducible factor-1α (HIF-1α); Janus kinase 2 (JAK2); mitochondrial permeabilization (MP); nicotinamide adenine dinucleotide (NAD); nicotinamide phosphoribosyltransferase (Nampt); nitric oxide synthase (NOS); nitric oxide (NO); nuclear factor kappa B (NFκB); peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α); phosphokinase B (PKB)/AKT; phosphoinositide 3 kinase (PI3K); phosphokinase C (PKC); reactive oxygen species (ROS); signal transducer and activator of transcription (STATs) specificity protein 1 (SP1); vascular endothelial growth factor (VEGF).
Gene activation and protein translation
It has been shown that pre-C induces a number of potentially neuroprotective genes in the brain (Jones and Bergeron, 2001; Bernaudin et al., 2002; Gustavsson et al., 2007), indicating that gene programming is important for development of pre-C also in the immature brain in addition to posttranslational and other adaptations (Gidday, 2006). Studies in the adult brain demonstrate that ischemic pre-C is inhibited by the protein translational inhibitor cycloheximide (Barone et al., 1998). Xenon-induced pre-C in the neonatal brain is also inhibited by cycloheximide (Ma et al., 2006), indicating that pre-C depends on gene transcription and de novo protein synthesis. However, no studies using transcription or translation inhibitors have been performed for hypoxic or other types of pre-C in the developing brain.
Hypoxic or ischemic pre-C induces a number of transcription factors including activating protein 1 (AP1), cyclic AMP response-element binding-protein (CREB), nuclear factor kappa B (NF-κB), early growth response 1 (EGR1), and the redox-regulated transcriptional activator specificity protein 1 (SP1) (Gidday, 2006). However, the best characterized transcription factor induced after hypoxic pre-C is probably hypoxia-inducible factor-1α (HIF-1α), which is strongly associated with preconditioning in the immature brain (Bergeron et al., 2000; Sharp and Bernaudin, 2004; Sheldon et al., 2009). Selective neuronal HIF-1α gene deletion blocked hypoxic pre-C in mice (Sheldon et al., 2009), indicating that pre-C depends on HIF-1α. However, the interpretation is complicated by the fact that HIF-1α gene deficiency confers heightened vulnerability also in response to the HI insult itself (Sheldon et al., 2009), and it is difficult to tell if HIF-1α is reducing vulnerability during the pre-C phase or during the HI insult or both. Ischemic pre-C in the neonatal brain was associated with a marked and sustained activation of CREB after 24 h (Lee et al., 2004). Intracerebroventricular infusions of antisense CREB oligodeoxynucleotides significantly reduced the pre-C neuroprotection and induction of CREB signaling with rolipram 24 h before HI protected newborn rats, supporting a role of CREB in neonatal pre-C (Lee et al., 2004).
Survival kinases
Several so-called survival kinases (e.g. protein kinase Cε (PKCε), phosphoinositide 3 kinase (PI3K)–AKT, and glycogen synthase kinase-3β (GSK3β)) are upregulated in the immature brain after hypoxic pre-C (Gustavsson et al., 2007) (Figure 1). PKCε phosphorylates and activates mitoKATP (see below) in the inner mitochondrial membrane (Raval et al., 2007; Bednarczyk, 2009), and inhibition of PKCε has been shown to reverse pre-C protection (Raval et al., 2007). PKCε is also critical for maintenance of mitochondrial nicotinamide adenine dinucleotide (NAD) or nicotinamide phosphoribosyltransferase (Nampt) pools (Morris-Blanco et al., 2014) known to be critical for immature brain integrity (Robertson et al., 2004; Fiskum et al., 2008; Galindo et al., 2017).
The PI3K/PKB/Akt/GSK3β pathway has been shown to reduce brain vulnerability in ischemia (Zhao et al., 2006b) and in the neonatal brain (Yin et al., 2007; Zhao et al., 2013), and has been implicated in pre-C of neuronal cells (Hillion et al., 2006) and the neonatal brain (Feng et al., 2010). Activation of this pathway leads to phosphorylation of Bcl-2 family proteins, decreases the pro- vs. anti-apoptotic balance, and prevents Bax-dependent mitochondrial permeabilization (Baines et al., 2003; Brywe et al., 2005; Maurer et al., 2006; Dirnagl and Meisel, 2008; Feng et al., 2010), which is implicated as an important cell death mechanism in the neonatal brain (Wang et al., 2009b) (Figure 1). Neuroprotection induced by hypoxic pre-C of the neonatal brain also induces changes in mRNA for Bcl-2 family proteins and other apoptosis-related proteins enforcing anti-cell death actions (Gustavsson et al., 2007). Recently, hypoxic pre-C was shown to also counteract caspase-Bax-dependent apoptosis induced by propofol (Lv et al., 2018). The cellular inhibitor of apoptosis-1 (cIAP1) is also important in pre-C of the immature brain, offering further support of the apoptotic pathway (Lin et al., 2013). Inhibition of PI3K/Akt using LY294002 attenuated pre-C neuroprotection and promoted the expression of NF-kappa-B, COX-2, and CD68. Proteomic microarray analysis revealed that pre-C inhibited expression of proinflammatory cytokines induced by HI (Yin et al., 2007).
The PI3K/AKT pathway is also stimulated by EPO which, in spite of being a HIF-1 target gene, was only moderately upregulated at the gene level (mainly in astrocytes) early after hypoxic pre-C in the immature brain compared to other organs (Bernaudin et al., 2002). The level of the EPO protein was not increased after hypoxic pre-C, at least not 0.5 h, 6 h, 12 h or 24 h after pre-C (Jones and Bergeron, 2001). However, EPO was induced by pharmacologic pre-C with deferoxamine (Mu et al., 2005) or MgSO4 (Koning et al., 2017), suggesting that EPO certainly could contribute to the development of tolerance, especially as it is well known to exert protective effects in the immature brain (Rangarajan and Juul, 2014). Furthermore, in the adult brain, EPO itself induces pre-C, and the protective effect of hypoxic pre-C is reversed by soluble EPO receptors (Prass et al., 2003) and EPO antisense (Liu et al., 2005), supporting its importance at least in some forms of pre-C.
Involvement of mitochondria: energy metabolism, mitoKATP and biogenesis/mitophagy
In the adult brain and heart, ischemic or hypoxic pre-C prevents mitochondrial swelling, protects membrane integrity, reduces ATP consumption and improves mitochondrial energy metabolism during a subsequent episode of cerebral ischemia (Zhang et al., 2003; Dirnagl and Meisel, 2008). Sirt1 and HSP90 have been proposed to be involved in mitochondrial protection via activation of PKCε (Thompson et al., 2015). In the neonatal brain, hypoxic pre-C also preserves mitochondrial function and attenuates the primary energy depletion during a subsequent period of HI (Brucklacher et al., 2002), and there is some evidence that AMP-activated protein kinase (AMPK) is involved (Rousset et al., 2015).
We found recently that a pre-C bolus of magnesium sulphate (MgSO4) applied 12 h to 6 days prior to neonatal HI reduces brain injury dramatically (Table 1). The protection was accompanied by downregulation of genes for metabolic enzymes, reduction of succinate during the subsequent HI, as well as preservation of high energy phosphates, mitochondrial respiration and attenuation of mitochondrial reactive oxygen species (ROS) production (Koning et al., 2017; Koning et al., 2018). It is important to emphasize that the serum levels of magnesium were normalized at the time of the HI insult and the treatment was ineffective if applied immediately prior, during or after the insult, suggesting that a pre-C mechanism is at play rather than a pre-treatment effect during HI (c.f. Riepe et al., 1997).
Opening of mitoKATP channels leads to influx of K+ into the mitochondrial matrix, increased oxygen consumption, and depolarization of the inner membrane (moderate uncoupling), reducing production of ROS mitochondrial Ca2+ accumulation, and Bax-dependent mitochondrial permeabilization (Bajgar et al., 2001; Liu et al., 2002; Bednarczyk, 2009). Brain mitochondria have higher concentrations of mitoKATP than do liver or heart mitochondria (Bajgar et al., 2001). Hypoxic pre-C in the neonatal brain induced upregulation of the protein level of the Kir6.2 isoform and enhanced current activities of mitoKATP channels (Sun et al., 2015). The mitoKATP opener diazoxide also reduced injury in the immature brain whereas inhibition of mitoKATP (using tolbutamide) blocked the protective effects of pre-C (Rajapakse et al., 2002; Wang et al., 2011; Sun et al., 2015).
Mitophagy of injured mitochondria and their replacement through biogenesis is an important process to maintain viability of postmitotic cells. A compelling notion is that cells with renewed mitochondria might be more resistant to severe stress. Ischemic pre-C of the heart depends on mitophagy through a parkin–SQSTM1-dependent mechanism (Huang et al., 2011), and mitophagy was recently also suggested to be involved in pre-C of neurons (Lizama et al., 2018). Under hypoxic conditions, HIF-1α (see above) induces BNIP3L-dependent and BNIP3-dependent mitophagy, probably through interaction with the outer mitochondrial membrane protein FUNDC1 (Ashrafi and Schwarz, 2013). Exposure of mice to hypoxic pre-C is also reported to increase mitochondrial DNA content, PGC-1α activity, and subsequent mitochondrial biogenesis (Gutsaeva et al., 2008). Hypoxic pre-C of mice induced expression of mRNA for Bnip3l and Bnip3, accompanied by an increase in the concentrations of the biogenesis markers PGC-1α and nuclear respiratory factors 1 and 2, suggesting that the mitophagy–biogenesis cycle might also be associated with pre-C of the immature brain (Gustavsson et al., 2008).
In summary, there is strong evidence that signaling cascades induced by pre-C converge on mitochondria in the brain of both adults (Liu et al., 2002) and neonates (Busija et al., 2005; Sun et al., 2015), as well as in other organs (Boengler et al., 2018). Interestingly, recent results in adult brain suggest that cross talk between mitochondria and endoplasmic reticulum with regard to Ca2+ regulation may be an additional critical mechanistic component in pre-C (Sisalli et al., 2015) that has to be addressed also in the immature brain in the future.
Nitric oxide (NO)-mediated conditioning
Nitric oxide (NO) synthase is upregulated after pre-C in the immature brain and generates NO and nitrite (Gidday et al., 1999; Gustavsson et al., 2007), and this inhibits production of ROS at complex 1 of the electron transport chain, improves mitochondrial respiration at complex II-IV and in turn reduces the threshold of mitochondrial permeabilization in heart and liver (Shiva et al., 2007). Indeed, non-specific inhibitors of NO synthase block hypoxic pre-C in the immature brain (Gidday et al., 1999). However, specific blockade of nNOS or iNOS had no effect whereas eNOS inhibition prevented hypoxic pre-C (Gidday et al., 1999), indicating that vascular factors are involved.
R-pre-C and R-post-C as conditioning paradigms in the immature brain
R-pre-C and R-post-C refer to a sublethal shear stress therapy by repetitive inflation-deflation of a blood pressure cuff on limbs before or after a major insult. R-pre-C and R-post-C have been demonstrated to be safe and are being actively studied in adult animal stroke models (Hess et al., 2013; Hess et al., 2015) and in small stroke clinical trials. A growing number of studies show that eNOS/NO/nitrite play an important role in improved perfusion in mice after stroke, and in protection conveyed by R-post-C (Hess et al., 2016) and effectiveness after embolic stroke in male and ovariectomized female mice (Hoda et al., 2014). Chronic R-post-C was recently demonstrated to induce vascular remodeling and protection in the adult (Khan et al., 2018). R-post-C was found effective alone and in combination with intravenous tPA in embolic stroke in adult mice (Hoda et al., 2012).
In neonates, while potential for protection by R-post-C has been demonstrated in a few studies, efficacy and the mechanisms of R-pre-C and R-post-C are not sufficiently understood. Available data have been discussed extensively in a recent review in this journal (Adstamongkonkul and Hess, 2017) and will not be discussed in detail in this review. As examples, in P10 rats subjected to HI, R-post-C induced immediately after the end of hypoxia by four 10-min ischemia/reperfusion cycles on both hind limbs reduced infarct volume at 48 h and improved functional outcomes at 4 weeks after HI, in part via involvement of the opioid receptor and PI3K/Akt signaling pathways (Zhou et al., 2011). In a piglet asphyxia model, immediate R-post-C reduced brain nitrotyrosine formation (Rocha-Ferreira et al., 2016) and protected oligodendrocytes in the corpus callosum and in the periventricular white matter but did not attenuate injury in grey matter (Ezzati et al., 2016).
In humans, effects of early and late R-pre-C were determined in infants who underwent surgery for congenital heart disease. In that pilot randomized controlled trial, R-pre-C proved to be feasible but did not produce significant differences in acute outcomes (Guerra et al., 2017). In another study conducted in neonates to 17-year-old children who underwent cardiac surgery, R-pre-C was not associated with improvements in clinical outcomes or physiological markers (McCrindle et al., 2014). Given the complexity of the studied effects and dependence of the effects on the type and severity of insult, age, the timing and exact protocol(s) of conditioning, additional animal studies and larger clinical trials are necessary to uncover the extent of the therapeutic potential for R-pre-C and R-post-C in the immature brain.
Noble gases and anesthetics as a pre-C strategy in injured immature brain
Anesthetic agents and noble gases have been tested for their efficacy in pre-C and post-C. The pros for such an approach are that anesthetic agents are commonly used in injured neonates and may provide additional benefits. The cons are that newborn brain is susceptible not only to widespread neuronal apoptosis resultant from ischemic, oxidative and excitotoxic injury (Ikonomidou et al., 1989; Ikonomidou et al., 1999; Hu et al., 2000) but also neuronal apoptosis induced by anesthetic agents (Olney et al., 2004; Schifilliti et al., 2010). In fact, models of neonatal toxicity were developed using prolonged exposure to the anesthetic agents isoflurane and sevoflurane (Shu et al., 2010).
Xenon (Xe), a noble gas that has been recognized as an anesthetic for more than 50 years, was found less neurotoxic to the developing brain compared to other anesthetics, effective as an anesthetic adjuvant in animals, and safe as a conditioning strategy in clinical trials in neonates with neurologic injury (Azzopardi et al., 2016; Alam et al., 2017). Xe has a small blood-gas partition coefficient, readily crosses the BBB and does not cause hemodynamic depression. Over the years, Xe was shown to exert neuroprotection (Preckel et al., 2000; Banks et al., 2010) and cardioprotection (Pagel, 2010; Schwiebert et al., 2010) in different adult animal models. Yet, Xe is not widely applied in clinical practice mainly because of its high cost and requirement for highly trained personnel.
Several mechanisms of Xe action have been identified. In neuronal-glial cell co-cultures subjected to oxygen-glucose deprivation, Xe pre-C produced a concentration-dependent reduction of lactate dehydrogenase release, an effect abolished by a protein synthesis inhibitor, and decreased cell death (Ma et al., 2006). Xe-induced pre-C reduced infarct size during subchronic injury and provided long-term neurological functional improvement in P7 rats subjected to HI (Ma et al., 2006). Xe pre-C and post-C was suggested to produce protection via induction of pro-survival proteins in models of neonatal HI (Ma et al., 2006), asphyxia (Luo et al., 2008) and prolonged (toxic) isoflurane exposure (Shu et al., 2010), such as Bcl-2 and BDNF. Xe enhanced CREB phosphorylation (Luo et al., 2008) and activity-dependent neuroprotective protein (ADNP) (Cattano et al., 2008) and decreased cytochrome-C release from mitochondria. Xe potently non-competitively inhibited NMDA receptors, with only minor effects on GABA receptors and non-NMDA receptors (Franks et al., 1998). At high dose, Xe inhibited the Ca2+-ATPase in rat brain synaptic plasma membranes (Franks et al., 1995) by inducing conformational changes in the lipid membrane (Lopez and Kosk-Kosicka, 1995). Xe administration alone showed neuroprotection after HI in neonatal rats (Dingley et al., 2006; Zhuang et al., 2012) and newborn pigs (Chakkarapani et al., 2010) as a post-C approach. Side-by-side use of Xe and hypoxic pre-C demonstrated positive pre-C by Xe but negative effects of hypoxic pre-C (i.e., injury exacerbation and lack of protective protein induction) (Shu et al., 2010). Additional protective mechanisms of Xe pre-C from ischemia/reperfusion injury, via mitochondrial K+-ATPase, were reported in immature rabbit heart (Li et al., 2013). Given the approval of TH for HIE in at-term human infants and inclusion of TH as a standard of care in many countries, efficacy of Xe post-treatment combined with immediate or delayed TH was examined. The presence or lack of beneficial effects of Xe administered after injury was shown to strongly depend on the severity of HI, Xe dosing, the timing of administration, and coordination between TH and Xe administration after injury. We limit the discussion of Xe effects to pre-C and do not focus on post-treatment in this review.
In cooled human newborns with neonatal encephalopathy, a small, single-arm, dose-escalation study showed the feasibility of using Xe in combination with TH (Dingley et al., 2014). No acute adverse respiratory, inflammatory or cardiovascular effects or adverse effects at 18 months' follow-up were observed (Dingley et al., 2014). Safety and feasibility of Xe was also confirmed in a recent TOBY-Xe trial. This open-label, randomized controlled trial compared effects of moderate whole-body cooling alone in moderately to severely injured 36-43-week-old infants to effects of moderate hypothermia combined with 30% Xe for 24 h (Azzopardi et al., 2016). At the same time, monitoring of multiple MRI/MRS markers showed lack of Xe-induced effects, suggesting that Xe is unlikely to enhance the neuroprotective effect of cooling after birth asphyxia (Azzopardi et al., 2016). While recent data in small and large newborn animal models and clinical trials have indicated safety and some neuroprotective potential of Xe, additional randomized clinical trials are necessary to corroborate these findings and confirm the feasibility of its routine use and its optimal timing, concentration, and duration for human neonatal HIE (Amer and Oorschot 2018).
Other noble gases, argon and helium, were also shown to be beneficial as pre-C approaches in animal studies. When administered at the same rate as Xe, argon and helium improved cell survival, brain structural integrity, and neurologic function at P40 in a model of neonatal asphyxia in rats (Zhuang et al., 2012). Argon reduced infarct volume following more severe HI injury (Zhuang et al., 2012). Protection was associated with increased expression of Bcl-2 and increased Bcl-xL and Bax expression as well (Zhuang et al., 2012). The safety of argon was established in newborn pigs under physiological conditions and following hypoxia (Alderliesten et al., 2014).
Testing of the commonly used anesthetic isoflurane as a post-C approach in HI revealed that brief exposure carries beneficial potential and that neuroprotection was concentration- and time-dependent (Xu et al., 2016). Short-term and long-term protection were partly mediated via the GluR2 AMPA receptor (Xu et al., 2016) and inhibition of the mitochondrial permeability transition pore following neonatal HI (Zhao et al., 2014). Sevoflurane post-C also improved long-term learning and memory following HI. As with hypoxic pre-C, protection was mediated via the PI3K/Akt-mPTP pathway (Lai et al., 2016). Dexmedetomidine post-C reduced brain injury after HI in neonatal rats as well (Ren et al., 2016). Taken together, the results show the central role of mitochondria in anesthetics-induced pre-C protection.
Effects of pre-C and post-C on injury after neonatal focal arterial stroke
In adult stroke models, while restoration of CBF is protective after a short ischemic episode, it can be damaging following sustained perfusion deficits because of hyperemia in previously occluded regions and subsequent secondary hypoperfusion known as “no-reflow phenomenon” (Aronowski et al., 1997). The latter phenomenon has been attributed to leukocytes, and to neutrophils in particular. Ischemic post-C induced by serial short interruptions of CBF during early reperfusion either interrupted hyperemia (Zhao et al., 2006a) or shortened its length (Wang et al., 2008) and reduced infarct volume, pointing to ischemic post-C as a neuroprotective strategy. Involvement of multiple mechanisms has been described, such as functional normalization of receptor and ion exchanger systems (Pignataro et al., 2011a; Pignataro et al., 2011b) and effects on inflammation (Joo et al., 2013). Effects of ischemic post-C on cell-cell interactions were also identified, such as neuronal VEGF regulation and microglial polarization (Esposito et al., 2018).
There is now ample evidence that the developmental stage of the brain at stroke onset plays a key role in injury (Ferriero, 2004; Yager and Ashwal, 2009; Semple et al., 2013; Fernandez-Lopez et al., 2014; Hagberg et al., 2015; Mallard and Vexler, 2015), including distinctions in the neuroinflammatory mechanisms, and the role of microglial cells and BBB responses between perinatal and adult stroke (Denker et al., 2007; Faustino et al., 2011; Fernandez-Lopez et al., 2012; Fernandez-Lopez et al., 2016). Compared to adult stroke models in which pre-C and post-C paradigms have been extensively studied, information on effectiveness and magnitude of conditioning-induced protection following neonatal arterial stroke is scant, largely due to too few laboratories performing neonatal arterial focal stroke models.
In contrast to the presence of hyperemia in adult stroke models, induction of focal arterial stroke in P7 rats by electrocoagulation of left MCAO combined with concomitant transient occlusion of both common carotid arteries (CCA) did not lead to hyperemia early reperfusion, as was evident from CBF measurement by 2D-color-coded ultrasound imaging and laser Doppler flowmetry (Leger et al., 2012). Post-C induced by a series of re-occluding single or both CCA for various short time periods (up to 5 min) did not reduce infarct size 72 h after MCAO (Leger et al., 2012). In the same model in P7 rats, pre-C with NO inhalation was beneficial, as it induced NO in the brain, improved CBF and protected neonatal brain against stroke (Charriaut-Marlangue et al., 2012). In contrast, based on increased infarct size, post-C with NO inhalation exacerbated injury (Charriaut-Marlangue et al., 2012). Taken together, these data are consistent with the notion that the pattern of CBF changes upon reperfusion is a critically important mediating mechanism of post-C protection. The constraint of these data is the contribution of both local and systemic perfusion deficits. In a transient 3-h suture MCAO occlusion in P7 rats, using perfusion-sensitive contrast-enhanced MRI, we observed partial to complete reperfusion of previously occluded regions at a single acute time point, 30 min after suture retraction, as well as perfusion of injured regions 24 h after reperfusion (Fernandez-Lopez et al., 2012). There was essentially no neutrophil infiltration 1-24 h after reperfusion and only a limited number of marginated neutrophils within previously occluded regions (Fernandez-Lopez et al., 2012). While post-C was not used in the latter study, the lack of neutrophil infiltration and only modest BBB leakage in neonatal tMCAO (Fernandez-Lopez et al., 2012; Fernandez-Lopez et al., 2016), as opposed to the presence of marked BBB disruption and apparent neutrophil margination that clog the vessels following tMCAO in adult brain, indirectly point to the importance of brain immaturity in the cerebrovascular responses that can affect efficacy of conditioning efforts in neonatal stroke.
Summary: progress and limitations in the understanding of beneficial effects of pre-C and post-C in the immature brain
In this review we focused on experimental models of pre-C and post-C that are most commonly applied, models that utilize brief hypoxic or ischemic exposures, immune conditioning or conditioning with pharmacological agents such as EPO, magnesium or anesthetic agents. We also discussed mechanistic insights at the intracellular levels, with emphasis on common and distinct model-specific mechanisms. Yet, the magnitude of conditioning-induced benefits and specifics in the immature brain are far from being well understood. One important lesson learned from these studies is that because of the immaturity of the brain, information can’t be easily transferred from the adult field. While the general conceptual framework is similar regardless of age—the same intracellular mechanisms can be involved in pre-C and post-C and the time interval between insults is important in determining the pathological outcome—the dynamic changes that occur in utero, in the early postnatal brain, and in immune system development create a delicate balance on the timing of sublethal exposure on the systemic, cell-to-cell and intracellular responses that can render the brain either more or less vulnerable to the severe insult. The field is evolving and other models of conditioning for the immature brain are being tested, namely hypobaric and hyperbaric hypoxia (Gamdzyk et al., 2016; Hentia et al., 2018), corticosteroids, resveratrol, and stem cells. Models where cells are conditioned prior to injection are also being developed (Sandvig et al., 2017). Overcoming some of the caveats should enhance our understanding. For example, the majority of rodent pre-C studies were performed in P7 rats, a rodent age that is no longer thought to correspond to brain development of at-term human infants, but rather to correspond to human brain at 32-36 gestation weeks (Semple et al., 2013). The HI injury manifests itself not in exactly the same ways in P7 and P10 rodents (Patel et al., 2015). Thus, there is a gap in knowledge of conditioning effects at-term. Another caveat is insufficient understanding of whether conditioning mechanisms can be expected to be similarly efficacious after arterial stroke as they are after HI. Literature on sex differences in pre-C and post-C effects in immature brain is essentially non-existent. Moving forward, while understanding of pre-C is important, post-C might be a more valuable strategy given the unknowns of the timing of insult in newborn humans and, as we discussed, that interventions that are effective as pre-C might not be effective or may even be damaging in the immature brain. Finally, identification of biomarkers of conditioning efficacy could make a difference in translating the findings from animal studies to humans.
Conflicts of interest statement
The authors have no conflicts of interest.
Acknowledgements
We gratefully acknowledge the support from ERA-net (EU;VR 529-2014-7551), Wellcome Trust (WT094823), Swedish Medical Research Council (VR 2015-02493, HH; VR-2017-01409, CM), Brain Foundation (HH, CM), Åhlen Foundation (HH, CM), ALF-GBG (426401, HH; 722491, CM), and the Leducq Foundation (DSRRP34404), Torsten Söderberg (M98/15, CM), RO1 NS44025 (ZSV), RO1 NS76726 (ZSV), R21NS098514 (ZSV), R01 HL139685 (ZSV, CM), R01 NS103483 (ZSV, CM), AHA17IRG33430004 (ZSV), CP Alliance PG0816 (ZSV).
References
Zinaida S. Vexler1
1Department of Neurology, University California San Francisco, San Francisco, California, USA.
Carina Mallard2
2Center of Perinatal Medicine and Health, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Henrik Hagberg3
3Center of Perinatal Medicine and Health, Institute of Clinical Sciences, Sahlgrenska Academy, University of Gothenburg, Sweden.
Corresponding author:
Zinaida S. Vexler
Email: zena.vexler@ucsf.edu
and
Henrik Hagberg
Email: henrik.hagberg@obgyn.gu.se
In a new window | Download PPT
Figure 1: Intracellular mechanisms of preconditioning in neonatal brain. Abbreviations: Activator protein-1 (AP-1); AMP-activated protein kinase (AMPK); cellular inhibitor of apoptosis-1 (cIAP1); cyclic AMP response-element binding-protein (CREB); early growth response 1 (EGR1); erythropoietin (EPO); hypoxia-inducible factor-1α (HIF-1α); Janus kinase 2 (JAK2); mitochondrial permeabilization (MP); nicotinamide adenine dinucleotide (NAD); nicotinamide phosphoribosyltransferase (Nampt); nitric oxide synthase (NOS); nitric oxide (NO); nuclear factor kappa B (NFκB); peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α); phosphokinase B (PKB)/AKT; phosphoinositide 3 kinase (PI3K); phosphokinase C (PKC); reactive oxygen species (ROS); signal transducer and activator of transcription (STATs) specificity protein 1 (SP1); vascular endothelial growth factor (VEGF).
Table 1. Examples of conditioning paradigms in the developing brain.
Abbreviations: Hypoxia-ischemia (HI); hypoxic-ischemic encephalopathy (HIE); lipopolysaccharide (LPS); magnetic resonance spectroscopy (MRS); magnesium sulphate (MgSO4); negative conditioning (-); polyinosinic: polycytidylic acid (poly I:C); positive conditioning (+).
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 12444 | 25 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA