Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Loss of gonadal steroids as a negative conditioner for neurological disease
Time:2019-03-01
Number:11022
Author Affiliations

Conditioning Medicine, 2019. 2(1):50-62.
Abstract
The concept of hormesis in toxicology refers to a substance with a biphasic dose response in which a range of low doses is paradoxically beneficial to the organism, but higher doses are toxic. A similar concept exists in conditioning studies in which non-toxic exposure to a condition or factor affords protection from subsequent toxic exposure. For example, brief, mild ischemia can protect tissues from subsequent more severe or prolonged ischemia that would otherwise result in greater damage. Preconditioning is generally studied for positive benefits, whereas “negative conditioning” occurs when exposure to a factor or condition leads to detrimental effects greater than that caused by loss of the factor alone. Pleiotropic gonadal steroids display this negative conditioning effect in the vasculature, neurological disease, and cerebral ischemia. Indeed, gonadal steroids protect the central nervous system (CNS) during continuous physiological exposure, but this protection is loss with the decline in gonadal function such as occurs after menopause in women. Subsequent re-exposure to these hormones after a period of delay (i.e. 5-10 years in women) results not only in the loss of premenopausal benefit, but independent detrimental actions. The mechanisms underlying this negative conditioning are still not fully understood, but studies in animal models have begun to identify some of the key players leading to the detrimental effects of what would otherwise be considered beneficial hormones.
Abstract
The concept of hormesis in toxicology refers to a substance with a biphasic dose response in which a range of low doses is paradoxically beneficial to the organism, but higher doses are toxic. A similar concept exists in conditioning studies in which non-toxic exposure to a condition or factor affords protection from subsequent toxic exposure. For example, brief, mild ischemia can protect tissues from subsequent more severe or prolonged ischemia that would otherwise result in greater damage. Preconditioning is generally studied for positive benefits, whereas “negative conditioning” occurs when exposure to a factor or condition leads to detrimental effects greater than that caused by loss of the factor alone. Pleiotropic gonadal steroids display this negative conditioning effect in the vasculature, neurological disease, and cerebral ischemia. Indeed, gonadal steroids protect the central nervous system (CNS) during continuous physiological exposure, but this protection is loss with the decline in gonadal function such as occurs after menopause in women. Subsequent re-exposure to these hormones after a period of delay (i.e. 5-10 years in women) results not only in the loss of premenopausal benefit, but independent detrimental actions. The mechanisms underlying this negative conditioning are still not fully understood, but studies in animal models have begun to identify some of the key players leading to the detrimental effects of what would otherwise be considered beneficial hormones.
Loss of gonadal steroids and neurological disease
In addition to their roles in reproduction, gonadal steroids (estrogens and progestins in females, and the androgens testosterone and dihydrotestosterone in males) have nonreproductive effects. These nonreproductive roles include trophic and neuroprotective effects in the brain and cerebrovasculature. These actions are primarily observed through increased disease susceptibility and severity after natural or artificial loss of these steroids and reinstatement of protection with pharmacological replacement. Menopause is the age-associated absence of menstruation for a 12-month period or more (World Health Organization, 1996). Menopause, either natural or due to the surgical removal of the ovaries, is characterized by a significant drop in circulating concentrations of female gonadal hormones (Nappi et al., 1999; Laughlin et al., 2000). Reduced estradiol (E2) concentrations indicative of menopause are associated with cognitive decline, Alzheimer’s disease (Bove et al., 2014), and an increased risk of stroke (Bushnell, 2008). The increased risk of stroke is also correlated with the age at menopause, with early menopause further increasing risk (Yang et al., 2017; Soleimani et al., 2018). These effects are not purely age-dependent, as surgical menopause in younger women similarly increases neurological disease risk (Bove et al., 2014). Animal models of stroke support the protective nature of the ovaries, as ovariectomized (OVX) young rats and mice experience greater injury than intact and E2-treated animals (Gibson et al., 2006; Koellhoffer and McCullough, 2013). Similarly, OVX rats experience greater damage after traumatic brain injury (TBI) than intact or E2-treated rats (Bramlett and Dietrich, 2001). Cognitive deficits are apparent in some, but not all tasks in OVX rats (Su et al., 2012), an effect that might be age dependent (Savonenko and Markowska, 2003).
Unlike females undergoing menopause, the decline in androgens in men is not precipitous. However, several studies do reveal correlations between testosterone (T) levels and neurological disease. For instance, lower endogenous T levels are associated with worse performance in cognitive tasks in older men (Moffat et al., 2002; Wolf and Kirschbaum, 2002). A literature review by Zarotsky et al., (2014) concluded that stroke and other vascular diseases were associated with low T, but the cause was not assessed. Low T in older men is also associated with an increase in the risk of Alzheimer’s disease (Lv et al., 2016) and the incidence and severity of Parkinson’s disease in men (Okun et al., 2004). Studies in functionally gonadectomized men (as treatment for prostate cancer) also reveal detrimental effects from the loss of T that are not age-dependent. Men on long-term, but not short-term, androgen deprivation therapy have memory deficits compared to healthy controls (Beer et al., 2006; Chao et al., 2012). Furthermore, a recent meta-analysis concluded that androgen deprivation therapy increased the risk of stroke (Poorthuis et al., 2017). In animal studies, gonadectomized male rodents exhibited deficits in cognitive function tasks such as object recognition, the radial arm maze (Frye and Seliga, 2001; Kritzer et al., 2001; Galea et al., 2008), and the Barnes maze (Locklear and Kritzer, 2014). However, short-term castration (1 week) actually improved stroke outcomes in young male rats (Cheng et al., 2009). The role of androgens in cognition is further underscored by the administration of androgen receptor antagonist flutamide in animal studies of learning and memory. When flutamide was injected into the hippocampus of male rats, they showed significant deficits in the water maze and active avoidance tasks (Edinger and Frye, 2007).
Neuroprotective effects of gonadal steroids – Preclinical models
The neuroprotective role of gonadal steroids has been the subject of extensive study for several decades. These protective effects extend across numerous CNS injury models in rodents including cerebral ischemia (Engler-Chiurazzi et al., 2017), traumatic brain injury (Khaksari et al., 2018), spinal cord injury (Sengelaub and Xu, 2018), cognitive aging (Engler-Chiurazzi et al., 2017), Alzheimer’s disease (Lan et al., 2015), and multiple sclerosis (Kipp et al., 2012).
Activation of the classical estrogen receptor subtypes alpha (ERα) and beta (ERβ) and the membrane receptor G Protein-coupled Estrogen Receptor 1 (GPER 1) have all been shown to play some roles in the neuroprotective effects of estrogen (Dubal et al., 2001; Merchenthaler et al., 2003; Cheng et al., 2009; Zhao et al., 2016; Wu et al., 2018). The numerous mechanisms proposed for the protective actions of E2 include those associated with preconditioning. These include inhibition of oxidative stress (Lagranha et al., 2018), maintenance of mitochondrial function (Arnold and Beyer, 2009; Simpkins et al., 2010), immunomodulation (Petrone et al., 2014), increased expression of growth factors such as brain-derived neurotrophic factor (BDNF) (Singh et al., 1995; Bimonte-Nelson et al., 2004; Cheng et al., 2016), insulin-like growth factor-1 (Sohrabji, 2015), and nerve growth factors (Bimonte-Nelson et al., 2004), and actions on protective cell signaling pathways. Following global cerebral ischemia, E2-induced neuroprotection has been associated with increased activity of extracellular signal-regulated kinases (ERK 1/2) and Akt through GPER 1 activation, as observed in the CA1 region of the hippocampus and cortical neurons in both in vitro and in vivo studies (Choi et al., 2004; Zhang et al., 2006; Jover-Mengual et al., 2007; Tang et al., 2014). Akt signaling also appears to mediate the effect of estrogen on the key regulator of oxidative stress responses, Nrf2 (Zhu et al., 2015) and antiapoptotic proteins such as Bcl-2 (Honda et al., 2001; Yune et al., 2008).
Progesterone (P4), the other female principal sex hormone, and its metabolite allopregnanolone, have been shown to confer neuroprotection through both genomic and non-genomic mechanisms involving the progesterone receptor (PR)(Singh, 2001; Kaur et al., 2007). Animal stroke models using females have demonstrated the ability of P4 to reduce infarct size post stroke (Yousuf et al., 2016; Andrabi et al., 2017), and genomic-dependent increases in the expression of BDNF following P4 administration have been shown to play an integral role in P4-mediated neuroprotection (Singh et al., 1995; Gonzalez et al., 2004; Kaur et al., 2007). Like E2, P4 can protect neurons through activation of the phosphoinositide 3-kinase (PI3K)-Akt and mitogen-activated protein kinase (MAPK) pathways (Kaur et al., 2007). A non-classical progesterone receptor, progesterone receptor membrane component-1 (Pgrmc-1) has also been found to mediate the release of BDNF for neuroprotection (Kaur et al., 2007). When administered either before or after induced ischemic brain injury in animal studies, P4 holds high therapeutic potential against ischemic stroke even though its post-treatment effects are limited by a narrow therapeutic-time window (Jiang et al., 1996; Kumon et al., 2000; Morali et al., 2005; Wali et al., 2014). Preclinical studies also revealed the neuroprotective effects of P4 in TBI and spinal cord injury (SCI) (Brotfain et al., 2016), and amyloid beta toxicity in rats leading to improved cognitive function (Hu et al., 2016).
The testicular androgens T and dihydrotesterone (DHT) also show promise as neuroprotective agents in preclinical studies. T improves outcomes in stroke models (Pan et al., 2005; Fanaei et al., 2014), although there is evidence that this is an age-dependent effect (Cheng et al., 2009). T also improves cognition in Alzheimer’s disease models in both rats (Huo et al., 2016) and mice (Rosario et al., 2010). DHT is beneficial in rats experiencing SCI (Byers et al., 2012; Sengelaub et al., 2018) and in a model of chronic experimental autoimmune encephalomyelitis (Giatti et al., 2015). Other studies support beneficial effects of T on cognition (Frye and Seliga, 2001; Kritzer et al., 2001; Schneider-Rivas et al., 2007; Galea et al., 2008; Spritzer et al., 2011; Locklear and Kritzer, 2014; Pintana et al., 2015).
In addition to effects on neurons, all three gonadal steroids can promote stem growth and differentiation in the injured brain. E2 (Li et al., 2011) and T (Fanaei et al., 2014) both promote neurogenesis in the subventricular zone following experimental stroke, but the role of P4 remains unresolved (Lee et al., 2015; Jiang et al., 2016). E2 also enhances neurogenesis after SCI (Chen et al., 2010) and in mouse models of Alzheimer’s disease (Zheng et al., 2017). Interestingly, P4 enhances basal neurogenesis in the hippocampus, but suppresses neurogenesis after TBI (Barha et al., 2011). Gonadal steroids have also been shown to promote oligodendrocyte differentiation and remyelination in the brain (Hussain et al., 2013; Luo et al., 2016), spinal cord (Labombarda et al., 2009), and the peripheral nervous system (Chen et al., 2016). These beneficial effects on neural and oligodendrocyte precursors likely play a role in the chronic phases of injury and repair.
Neuroprotective effects of gonadal steroids – Clinical uncertainty
Despite the evidence for gonadal steroid mediated neuroprotection in preclinical models, results in human populations are much less clear. Epidemiological studies of several neurological diseases including stroke (Girijala et al., 2017), Alzheimer’s disease (Ferretti et al., 2018), and Parkinson’s disease (Georgiev et al., 2017), demonstrate gender differences that diminish following menopause, suggesting the benefits of female gonadal steroids. However, many recent interventional clinical studies have failed to recapitulate preclinical success. For example, a large Phase 3 clinical trial for P4 in TBI failed to show any benefit (Skolnick et al., 2014). However, some smaller studies have demonstrated benefits of gonadal steroids. In older men, T increased cognitive performance and men with lower T exhibited a decline in cognitive function (Janowsky et al., 2000). T showed very little benefit to men with Parkinson’s disease (Okun et al., 2006), although spatial benefits have been observed in men with Alzheimer’s disease (Cherrier et al., 2005). A study by the same group found minimal benefits in a larger cohort of men with mild cognitive impairment (Cherrier et al., 2015). Despite evidence that endogenous T levels <10% of normal value are associated with stroke risk in men, a large prospective cohort study did not find other relations between T or E2 and stroke (Holmegard et al., 2016).
The concept of a beneficial role of E2 in the brain was dramatically challenged by the results of the Women’s Health Initiative (WHI), the largest ever prospective study of postmenopausal hormone therapy (HT). The study included both interventional and observational arms, as well as treatment with oral conjugated equine estrogens (CEE) alone (in women without a uterus) or CEE + medroxyprogesterone acetate (MPA). The first results from over 16,000 postmenopausal women were published in 2002, approximately 5 years into the proposed 8.5-year study as it was stopped due to an increased rate of breast cancer (Rossouw et al., 2002). Numerous subsequent findings showed detrimental effects of HT on the brain. The ancillary WHI Memory Study (WHIMS) found that both CEE and CEE+MPA increased the risk of dementia, primarily Alzheimer’s disease (Shumaker et al., 2003; Shumaker et al., 2004). The WHIMS MRI study also revealed increased brain atrophy in those on HT (Resnick et al., 2009). Furthermore, both CEE and CEE+MPA increased the risk of stroke (Wassertheil-Smoller et al., 2014). When the observational and interventional arms of the WHI were combined, the risk of stroke was similarly increased with HT (Prentice et al., 2009). Results from the Nurse’s Health Study also found an increased risk of stroke with HT (Grodstein et al., 2008), and subarachnoid hemorrhage risk was slightly increased by HT in the WHI (Qureshi et al., 2016). The more concerning observation is that even 3 years after stopping HT, cognitive deficits persisted (Prentice et al., 2009).
Critiques of the WHI began almost immediately after the initial publications and identified numerous potential reasons for increases in stroke and dementia and other unexpected findings, but focused primarily on the average age of participants (63 years old), time since menopause (>10 years), hormone formulation (CEE instead of E2 and MPA instead of P4), the use of constant instead of cyclical MPA, and the route of administration (oral instead of transdermal) (Klaiber et al., 2005; Lacey, 2014). Singh et al. (2008) addressed these issues and their relations to preclinical studies, pointing out significant differences from the experimental designs demonstrating beneficial effects in the laboratory (Singh et al., 2008). Some of these issues, they argued, could have predicted the results of the WHI because preclinical models had already demonstrated differential neuroprotective effects in rodents that were dependent on age, steroid formulation, and route of administration (Singh et al., 2008). Importantly, the WHI study design did not optimize any of these parameters to enhance the likelihood of positive outcomes (Singh et al., 2008). Subsequent reanalysis and additional interventional studies sought to address these methodological issues and found either benefits or no detrimental effects on cardiovascular and cognitive risk (McCarrey and Resnick, 2015). The “critical window” hypothesis was examined in the WHI Memory Study–Young (WHIMS-Y) that reexamined women from WHIMS who had begun the trial between the ages of 50 and 55 with CEE only (Espeland et al., 2013). Unlike the WHIMS study, women in WHIMS-Y showed no increase in dementia over 14 years, but also no evidence of benefit (Espeland et al., 2013). The study did not address the role of MPA. The Kronos Early Estrogen Prevention Study (KEEPS) used CEE or transdermal E2 with oral P4 for 14 days each month in women within a year of menopause and the KEEPS-Cog arm examined numerous cognitive and affective outcomes over 4 years (Gleason et al., 2015). Like the WHIMS-Y study, no risk or benefit was observed for cognition (Gleason et al., 2015). Other smaller clinical studies of HT following surgical menopause showed mixed effects (McCarrey and Resnick, 2015).
In men, the role of T replacement in brain health is unclear, although detrimental effects such as those seen in the WHI have not been reported. Although some small studies show that T increases cognitive performance (Janowsky et al., 2000), other human studies did not show any relationship between maintaining normal T levels and cognitive function (Yonker et al., 2006; Martin et al., 2007). A small study of T intervention in Alzheimer’s disease did not find cognitive benefits (Lu et al., 2006). Similarly, a series of recent clinical trials, the “Testosterone Trials,” failed to find any cognitive benefit in a population of older hypogonadal men treated for 12 months with T gel whether they began the trial with impaired cognition or not (Resnick et al., 2017). These studies confirmed the results of the Testosterone Effects on Atherosclerosis in Aging Men (TEAAM) study which failed to show cognitive benefits with T treatment of 36 months in older hypogonadal and normal gonadal men (Huang et al., 2016). Although non-randomized studies show a lower risk of stroke, a recent meta-analysis of 8 randomized-controlled trials concluded that T therapy has no significant effect on stroke (Elliott et al., 2017). Specific studies examining long-term T deprivation and replacement in men have not been performed.
Window of Opportunity for Gonadal Steroids
Several studies using animal models support the concept of a “critical window of opportunity” for the benefits of gonadal hormone replacement in the brain. The most notable ones are those designed specifically to test this hypothesis following the WHI. In 2007, Suzuki et al., (2007) demonstrated that the neuroprotective effects of E2 on ischemic stroke could be abolished in mice by delaying HT 10 weeks or longer after OVX. The delayed HT also prevented the anti-inflammatory effect of E2 (Suzuki et al., 2007). A similar loss of neuroprotection after long-term E2 deprivation (LTED) was demonstrated by multiple groups in rats undergoing experimental focal cerebral ischemia (Selvamani and Sohrabji, 2010), global cerebral ischemia (Zhang et al., 2011; Wu et al., 2018), hippocampal amyloidogenesis (Zhang et al., 2013), and tests of cognition (Daniel et al., 2006). In aged mice with long-term low endogenous E2 levels, there is also a loss of E2 neuroprotection against focal cerebral ischemia (Cai et al., 2014), and the beneficial effects of E2 on hippocampal synaptic efficacy are lost after LTED (Bohacek and Daniel, 2009; Smith et al., 2010). Interestingly, acute centrally administered E2 still appears to maintain neuroprotective benefits under these conditions (Inagaki et al., 2012). No specific studies of P4 neuroprotection after long-term OVX have been published. However, 5-6 weeks, but not 1 week, of gonadal steroid deprivation through ovariectomy significantly reduces the ability of acute administration of E2 and P4 to induce sexual behavior in female rats, suggesting that the brain sensitivity to P4 is also diminished by the length of ovarian steroid deprivation (Clark et al., 1981).
Because men do not have a clear loss of T akin to menopausal estrogen loss in women, studies of long-term T deprivation and replacement in men are lacking. In preclinical studies, administration of T 1-week post-stroke can enhance functional recovery in young male rats (Pan et al., 2005). However, castration is protective to young male rats undergoing focal cerebral ischemia and T replacement reverses this benefit (Cheng et al., 2009). Studies using DHT support the concept that the detrimental effect of T is not due to conversion to E2 (Cheng et al., 2007; Uchida et al., 2009). Unlike young male rats, T replacement protects aged male rats with low T levels from stroke injury (Cheng et al., 2009). Another study demonstrated beneficial effects of T treatment on long-term memory in young, but not aged Wistar rats (Schneider-Rivas et al., 2007), and Pintana et al. (2015) showed that T replacement for 4 weeks improved water maze performance in rats 12 weeks after castration (Pintana et al., 2015). These few studies suggest that T replacement may be beneficial after long-term gonadectomy (GDX). Besides the loss of benefit after LTED, there is evidence that HT can be detrimental when it is delayed too long. Results of the WHI may be the most alarming clinical study to demonstrate these negative effects. Despite the many potential confounds associated with the WHI, older women not only “lost” the benefits of ovarian hormones, but also increased their risks of several diseases including stroke and dementia (see above). A few studies in rodents further support the negative effects of E2 after LTED. For example, OVX of reproductively senescent rats with low circulating gonadal hormones not only prevents the neuroprotective effects of E2 treatment on stroke injury, but exacerbates the injury (Selvamani and Sohrabji, 2010). Similarly, E2-induced inhibition of several cytokines and chemokines after stroke is reversed by LTED and the levels of monocyte chemoattractant protein-1 are actually increased by E2 treatment (Suzuki et al., 2007). Importantly, with respect to a postmenopausal population, gonadal hormones appear to exacerbate the deleterious effects of comorbid conditions both within and outside the CNS. For example, E2 treatment exacerbates ischemic injury in diabetic rats (Santizo et al., 2002), and high dose E2 can increase stroke injury in a permanent occlusion model even without LTED (Bingham et al., 2005). E2 and oral contraceptives both worsen hippocampal injury after global cerebral ischemia in nicotine exposed rats (Raval et al., 2011), and long-term E2 replacement exacerbates the inflammatory effects on cognitive tasks in female rats (Marriott et al., 2002). Outside the CNS, E2 exacerbates injury in the heart following myocardial infarction (de Almeida et al., 2018), and HT in older women and mice with low endogenous hormone levels enhanced hearing loss, an effect that was greater for E2 + P4 than for E2 alone (Price et al., 2009).
Dual beneficial and detrimental effects of gonadal steroids also appear to be dependent on timing. E2 treatment of OVX female mice prior to methamphetamine protects nigrostriatal dopamine neurons from dopamine depletion (Liu and Dluzen, 2006). In contrast, E2 treatment after an initial methamphetamine dose exacerbates dopamine cell loss (Liu and Dluzen, 2006). Similarly, in mouse primary cortical brain cultures, pretreatment with E2 protects neurons from NMDA-mediated toxicity, while E2 treatment starting 10 minutes after NMDA increased cell death (Spampinato et al., 2012). In vitro T has similar effects. Pretreatment of a dopaminergic cell line with T protects cells from oxidative stress, while T treatment after the oxidative stress increases cell death and enhances cellular oxidative and inflammatory responses (Holmes et al., 2016). These results further support the notion that the beneficial effects of gonadal steroids are dependent on their presence prior to homeostatic insults or injuries. Treatment following injury may not only be ineffective, but also has the potential to be detrimental. Some of the mechanisms underlying these shifts in action are reviewed by others, but likely involve numerous epigenetic changes and modulation of steroid receptor expression (Liu and Yang, 2013; Daniel et al., 2015).
Whether the detrimental effects of gonadal steroids result from preexisting disease, timing, formulation, or age, is still under investigation. The loss of receptor expression (and its persistence due to epigenetics) can explain the loss of the benefits of steroids, but is less able to explain detrimental actions. A possible explanation comes from cardiovascular studies in which the loss of gonadal hormones allows detrimental processes to progress. This progression may reveal detrimental effects of otherwise beneficial steroids. For example, a loss of gonadal hormones can lead to changes in circulating lipids that enhance atherosclerosis (Oliver-Williams et al., 2018). When combined with the thrombotic effect of estrogen, detrimental cerebrovascular events may be increased leading to cognitive decline, stroke risk, and enhanced inflammation. Thus, like aging, underlying conditions may alter the effects of the steroid.
Gonadal steroids and mechanisms of conditioning
Gonadal steroids beneficially influence several pathways associated with the effects of preconditioning. The loss of hormone action on these pathways could represent a loss of preconditioning. Ischemic preconditioning (IPC) for stroke injury has been recognized since the 1990’s. Brief sublethal ischemia induces tolerance of the brain and reduces neuronal death in response to a subsequent lethal ischemic insult in stroke models (Kitagawa et al., 1990; Kirino et al., 1991; Liu et al., 1992). Evidence also suggests that prior transient ischemic attacks in human patients acts to precondition the brain and results in better outcomes from subsequent ischemic stroke (Weih et al., 1999; Moncayo et al., 2000), consistent with animal studies. Besides IPC, other forms of preconditioning have been applied to many neurological diseases. For example, some animal studies have reported that preconditioning, with hyperbaric oxygen or N-methyl-D-aspartate protects against TBI (Hu et al., 2008; Costa et al., 2010; Hu et al., 2010). Other studies have shown that multiple preconditioning scenarios exert neuroprotective effects against amyloid beta toxicity in vitro (Mitchell et al., 2009; Zhang et al., 2018), a mouse model of Alzheimer’s disease (Tang et al., 2011), as well as in vitro and in vivo models of Parkinson’s disease (Cannon et al., 2005; El Ayadi and Zigmond, 2011). As reviewed by Stetler et al., (2014) a large number of interventions can act as preconditioning stimuli including ischemia, oxygen (hypoxic and hyperbaric conditions), temperature (hypothermia and hyperthermia), anesthetics/analgesics, ethanol, stimulants, neurotoxins, neuroinflammatory agents, systemic stress (physical exercise and caloric restriction), and subcellular stress (mitochondrial) (Stetler et al., 2014). Stetler et al., (2014) recognized two types of preconditioning, a rapid effect lasting a few hours and a delayed effect that begins as early as 24 hours that may last up to 7 days. The rapid effects involve modification of signaling or enzyme activity, whereas the delayed effects involve changes in gene transcription and de novo protein synthesis (Stetler et al., 2014).
Among the major pathways implicated in preconditioning are oxidative stress, enhancement of the antioxidant defense pathway, enhancement of anti-inflammatory pathways, and modification of signaling and transcriptional pathways. E2 has been widely reported to reduce oxidative stress by attenuating the production of reactive oxygen species (ROS) and increasing anti-oxidant activity (Stirone et al., 2005; Razmara et al., 2008; Guo et al., 2010; Kemper et al., 2013). OVX rats treated with P4 showed improved bioenergetic efficiency and balance, as well as attenuated oxidative stress in brain mitochondria (Irwin et al., 2008). Further, P4 promotes higher mitochondrial respiration and lower oxidative stress after stroke and TBI (Robertson and Saraswati, 2015; Gaignard et al., 2018). A recent clinical study has shown that diabetic men with lower T levels have increased mitochondrial ROS and reduced mitochondrial membrane potential and superoxide dismutase (SOD) expression (Rovira-Llopis et al., 2017).
It is well known that female sex hormones modulate inflammatory processes in the brain. E2 treatment is reported to attenuate the inflammatory response to ischemic stroke (Santizo et al., 2000), interleukin-1 beta (IL-1β) (Ospina et al., 2004), and lipopolysaccharide (LPS) (Sunday et al., 2006) in female OVX rats. The anti-inflammatory effect of E2 is mediated in part via nuclear factor-κB (NFκB) (Galea et al., 2002; Ospina et al., 2004), decreased levels of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) proteins, and attenuated production of prostaglandin E2 (PGE2) and nitric oxide (NO) (Ospina et al., 2004; Sunday et al., 2006; Sunday et al., 2007). Furthermore, this anti-inflammatory effect of E2 is reported to be age-related: E2 significantly suppressed LPS induced iNOS and COX-2 proteins, and production of NO and PGE2 only in young female OVX rats, not in middle-aged ones (Sunday et al., 2007). Additionally, Sohrabji (2005) also suggested that the effect on inflammation is dependent on the reproductive age of the individual receiving E2 replacement. E2 replacement is beneficial when given to young OVX females, while E2 replacement is deleterious to older reproductively senescent animals in a prolonged E2-deficient state (Sohrabji 2005). There are contradictory reports regarding the effects of P4 on inflammation in the brain. Sunday et al., (2006) found that P4 given to female OVX rats not only exacerbated the inflammatory response to LPS, but also diminished the anti-inflammatory effect of E2 when E2 and P4 were given as a combined treatment (Sunday et al., 2006). However, in another study, P4 was reported to reduce brain infarct volume and improve functional outcome in ischemic models, and these neuroprotective effects were associated with a suppression of the inflammatory response to ischemia (Gibson et al., 2005). The effects of androgens on brain inflammation differ under physiological and pathological conditions. In the absence of LPS stimulus, GDX with or without T replacement doesn’t affect cerebrovascular inflammation, i.e. COX-2 or iNOS, in male rats (Razmara et al., 2005). However, GDX rats with T replacement increased LPS-induced inflammatory markers in the brain, compared to GDX or intact male groups (Razmara et al., 2005). DHT treatment is pro-inflammatory via androgen receptor (AR)-dependent mechanisms under the physiological condition while anti-inflammatory by conversion of DHT to 3β-diol and activation of ERβ under pathological condition (Gonzales et al., 2009; Zuloaga and Gonzales, 2011; Zuloaga et al., 2012a; Zuloaga et al., 2012b).
E2 and P4 have been reported to activate the PI3K-Akt signaling pathway in explant cortical neurons, which is associated with neuroprotective action (Singh, 2001; Kaur et al., 2007). The expression of the cell survival/anti-apoptotic factor Bcl-2 decreases in the cerebrovascular in the middle-aged female OVX rats, while E2 replacement reverses this effect (Jesmin et al., 2003). Additionally, E2 exerted its neuroprotection by preventing the downregulation of Bcl-2 via ERβ in cerebral ischemia and glutamate induced injury (Dubal et al., 1999; Alkayed et al., 2001; Zhao et al., 2004). Moreover, E2 also inhibits the expression of the BAD gene, which is the antagonist of the Bcl-2 gene (Dubal et al., 1999; Alkayed et al., 2001; Zhao et al., 2004). DHT is reported to inhibit glial cell apoptosis by regulating Bcl-2 protein through activation of the PI3K-Akt signaling pathway (Yao et al., 2016). While preconditioning induced by isoflurane in males is reported to enhance Akt activation (Dudek et al., 1997; Brunet et al., 2001), it doesn’t alter Akt activation in females (Kitano et al., 2007). Research in other organs like the heart also supported the enhancement of Akt signaling by T treatment (Bai et al., 2005). It has also been suggested that isoflurane preconditioning induced neuroprotection in experimental stroke is male specific through an AR-dependent mechanism on Akt activation (Zhu et al., 2010). The gender differences in the response to isoflurane preconditioning in ischemic cortex may be mediated through the basal expression of neuronal inducible cell-death putative kinase (NIPK), a negative modulator of Akt activation (Kitano et al., 2007). Recently, NIPK was identified as an estrogen-responsive gene by microarray analysis (Terasaka et al., 2004; Ise et al., 2005), suggesting that the presence or absence of E2 could alter NIPK levels, leading to alterations in cortical Akt activation in the isoflurane preconditioned brain. Other in vitro studies have implicated a rapid protective preconditioning effect of E2 on hippocampal pyramidal neurons against oxygen-glucose deprivation 48 hours after a brief E2 exposure, mediated by calcium-calmodulin dependent protein kinase II (CAMKII) and mitogen-activated protein kinase (MAPK42/44) (Raval et al., 2006). Figure 1 depicts pathways shared by ischemic preconditioning and steroid hormones (particularly E2) in the brain. Although several other pathways and signaling mechanisms for both IPC and steroid actions in the brain have been examined, the overlap of these pathways in neuroprotection has not been fully elucidated.
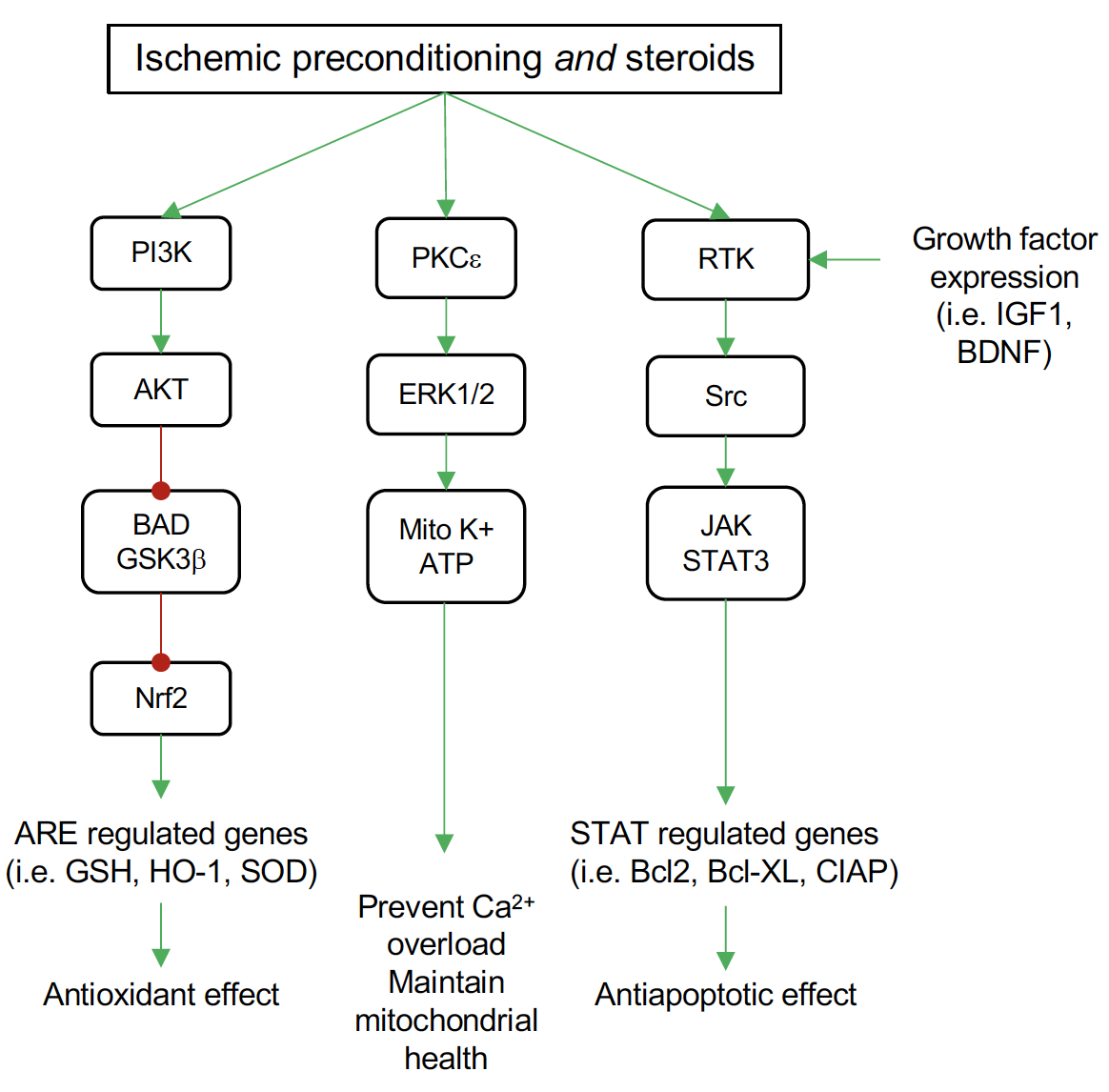
In a new window | Download PPT
Figure 1: Common pathways for ischemic preconditioning (IPC) and steroid neuroprotection by pretreatment in the brain. Both IPC and steroids activate the phosphatidylinositol 3-kinase (PI3K) signaling pathway to increase protein kinase B (Akt) activation. Among the targets for Akt is the proapoptotic mediator BAD and glycogen synthase kinase 3 beta (GSK3β), which Akt inhibits. This inhibition relieves the inhibitory effect of GSK3β on nuclear factor-like 2 (Nrf2), allowing Nrf2 to translocate to the nucleus where it binds antioxidant response elements (ARE) in the promoter of several antioxidant genes, including glutathione S-transferase (GSH), heme oxygenase-1 (HO-1), and superoxide dismutase (SOD). Both IPC and steroids activate protein kinase C epsilon (PKCξ) leading to phosphorylation of the mitogen activated protein kinases ERK1 and ERK2. These kinases enhance the activity of the mitochondrial ATP sensitive potassium channels (Mito K+ ATP). Activation of these channels maintains mitochondrial potential and prevents calcium overload and mitochondrial swelling. Signaling through growth factor receptors (receptor tyrosine kinases, RTK) occurs both in IPC and steroid treatment. Steroid actions are two-fold, both increasing transcription of growth factors, such as insulin-like growth factor (IGF-1) and brain-derived neurotropic factor (BDNF) that activate these receptors, and also activation of the SRC protooncogene directly. This leads to an increase in signaling through the janus kinases (JAKs)/signal transducer and activator of transcription 3 (STAT3) proteins. Among beneficial genes targeted by STAT3 are the antiapoptotic factors Bcl2 and BclXL and cellular inhibitor of apoptosis 1 protein (cIAP).
Hormetic and negative conditioning model for the detrimental effects of gonadal steroids
The loss of the protective effects of gonadal hormones we have described have been explained as a “critical window” for benefit. However, the physiological changes that accompany hormone depletion may also lead to toxic effects of the same hormones. This suggests a more complex relationship than a simple loss of action. These effects may be better described by the concept of hormesis, in which a same substance shows a biphasic response that is dose dependent (Figure 2A). However, this phenomenon can also be described as a therapeutic window for a beneficial response (Kendig et al., 2010). By adding the timing of the treatment to dose it could be applied to HT. Indeed, hormetic responses are often described as “U” or “J” shaped curves (Figure 2A). Such non-monotonic dose responses are well recognized in endocrine and other systems (Lagarde et al., 2015), and detrimental effects of high dose of estrogen have been reported on the heart, kidneys, and liver in young mice even with short term OVX (Meng et al., 2011).
A recent systematic review of cardiovascular and stroke risk concluded that higher doses and older age at initiation of HT significantly increases the risk of thromboembolism and stroke (Oliver-Williams et al., 2018). Cardiovascular disease increases with age, and clinical evidence suggests that higher E2 levels are associated with more vulnerable arterial plaques (Glisic et al., 2018). A new meta-analysis also revealed that the use of oral contraceptives increases the risk of subarachnoid hemorrhage and this risk increases with dose (Xu et al., 2018). In vitro and in vivo, Ma et al., (2013) demonstrated beneficial effects of E2 at physiological doses, but detrimental effects at supraphysiological doses in rats subjected to experimental stroke, again supporting a role for dose in the biphasic effect of E2 (Ma et al., 2013). Similar detrimental effects of high dose ethinyl E2 are observed for cognitive function in rats whereas low dose contraceptive levels have no effects but higher levels are detrimental (Mennenga et al., 2015). Low dose E2 also enhances long-term potentiation in the hippocampus of juvenile male rats, but high dose has the opposite effect (Tanaka and Sokabe, 2013). However, this dose effect may not hold for induced stroke in rats, at least not in the short term (Ingberg et al., 2016). Similar to E2, P4 neuroprotection against stroke is dose dependent, where the protective effects of low doses are reversed at supraphysiological concentrations (Yousuf et al., 2014) (Strom et al., 2009).
Strom et al., (2011) reviewed the hormetic nature of estrogens and progestins noting that despite evidence for inflammation in mediating the hormetic effects of female sex steroids in the brain and cardiovascular system, studies are far from conclusive (Strom et al., 2011). Nevertheless, hormetic effects of gonadal steroids have been demonstrated in a number of physiological systems (Strom et al., 2011). Calabrese et al., (2014) placed antioxidant pathways, including hemeoxygenase-1, in the factors underlying hormetic responses to sex hormones (Calabrese et al., 2014), and ROS are clearly sensitive to gonadal steroids. These hormetic concepts support the idea that long term hormone deprivation (LTHD) leads to potential shifts in the hormetic curve resulting in reduced benefit and enhanced adverse effects. If the optimal dose of HT lies near the peak of the hormetic benefit and within the physiological range (Figure 2A), only supraphysiological concentrations would likely cause harm. In contrast, LTHD could shift the hormetic curve up, not only reducing the range of benefits, but also decreasing the safe range of the treatment (Figure 2B). This shift may greatly reduce the range of concentrations at which benefits are observed or reduce the magnitude of the benefits, resulting in no observable effects. A third possibility is that LTHD shifts the hormetic curve left (Figure 2C). Under these conditions, what would be considered an otherwise optimal dose would now have no effect or be potentially detrimental. If LTHD both shifted the hormetic curve left and up, it is likely that only the detrimental effects would be observed at what would otherwise be considered therapeutic doses (Figure 2D).

In a new window | Download PPT
Figure 2: Shifts in the steroid hormone hormetic curve may lead to detrimental effects. A: “J” shaped hormetic curve for adverse effects. The physiological range of the hormone overlaps the beneficial hormetic dose range and results in a reduction in adverse effects that is maximized at an optimal dose falling at the peak of the curve. B: An upward shift in the hormetic curve reduces the hormetic range such that benefits are small and small deviations from the optimal dose lead to the enhancement of adverse effects. C: A leftward shift in the hormetic curve results in the optimal physiological dose falling in the range of enhanced adverse effects. D: A leftward and upward shift of the hormetic curve results in both a reduction of benefit and an enhancement of adverse effects even far below the optimal physiological dose.
Such shifts would be fully consistent with observations detailed above. First, there are dose dependent positive and negative effects of gonadal steroids in the brain. Second, supraphysiological doses can be detrimental in young individuals, but lower doses can be detrimental in older individuals or those with natural or surgical hypogonadism. Third, the negative effects are exacerbated by preexisting conditions that enhance ROS or inflammation. Although not widely examined, there are a few examples of very low dose benefits as would be expected in models C and D. Compared to standard HT, which increases thrombotic potential, ultra-low dose E2 has positive effects on hemostasis in postmenopausal women (Pirog et al., 2017).
The mechanisms underlying the shift in dose response with LTHD are not fully elucidated, and many studies correlate reduction in beneficial factors with a loss of hormonal benefit rather than a shift to detriment. However, no specific studies have examined the shift in dose response for neurological dysfunction. Importantly, studies showing reduced steroid hormone receptor expression i.e. (Zhang et al., 2011) do not conclude whether such changes are the result of permanent epigenetic changes that prevent a reinstatement of benefits, although estrogen and progesterone receptors clearly undergo epigenetic changes with age (Nugent et al., 2011; Westberry et al., 2011; Wilson et al., 2011). Whether such changes are permanent is also unknown, although at least one study showed that cognitive deficits that accompanied HT persisted at least three years after cessation of treatment (Prentice et al., 2009).
Conclusion
Menopause and other conditions that lead to hypogonadism in women and men are often treated with hormone therapy (HT), but both clinical and preclinical studies suggest that the benefits derived from these hormones is time and dose dependent. Importantly, under some conditions, such as preexisting cardiovascular disease, HT can have detrimental effects on the brain and other organ systems. Although conclusive evidence is lacking, the shift from beneficial to detrimental fits with a model of hormesis in which LTHD shifts the hormetic curve to minimize the benefits of gonadal steroids and reveal detrimental effects at otherwise beneficial doses.
References
Fen Sun
1Department of Physiology and Anatomy and the 3Institute for Healthy Aging, University of North Texas Health Science Center, 3500 Camp Bowie Boulevard, Fort Worth, Texas 76109.
Anthony Oppong-Gyebi
2Department of Pharmacology and Neuroscience, and the 3Institute for Healthy Aging, University of North Texas Health Science Center, 3500 Camp Bowie Boulevard, Fort Worth, Texas 76109.
Derek A. Schreihofer
2Department of Pharmacology and Neuroscience, and the 3Institute for Healthy Aging, University of North Texas Health Science Center, 3500 Camp Bowie Boulevard, Fort Worth, Texas 76109.
Corresponding author:
Derek A. Schreihofer
Email: Derek.Schreihofer@UNTHSC.edu
In a new window | Download PPT
Figure 1: Common pathways for ischemic preconditioning (IPC) and steroid neuroprotection by pretreatment in the brain. Both IPC and steroids activate the phosphatidylinositol 3-kinase (PI3K) signaling pathway to increase protein kinase B (Akt) activation. Among the targets for Akt is the proapoptotic mediator BAD and glycogen synthase kinase 3 beta (GSK3β), which Akt inhibits. This inhibition relieves the inhibitory effect of GSK3β on nuclear factor-like 2 (Nrf2), allowing Nrf2 to translocate to the nucleus where it binds antioxidant response elements (ARE) in the promoter of several antioxidant genes, including glutathione S-transferase (GSH), heme oxygenase-1 (HO-1), and superoxide dismutase (SOD). Both IPC and steroids activate protein kinase C epsilon (PKCξ) leading to phosphorylation of the mitogen activated protein kinases ERK1 and ERK2. These kinases enhance the activity of the mitochondrial ATP sensitive potassium channels (Mito K+ ATP). Activation of these channels maintains mitochondrial potential and prevents calcium overload and mitochondrial swelling. Signaling through growth factor receptors (receptor tyrosine kinases, RTK) occurs both in IPC and steroid treatment. Steroid actions are two-fold, both increasing transcription of growth factors, such as insulin-like growth factor (IGF-1) and brain-derived neurotropic factor (BDNF) that activate these receptors, and also activation of the SRC protooncogene directly. This leads to an increase in signaling through the janus kinases (JAKs)/signal transducer and activator of transcription 3 (STAT3) proteins. Among beneficial genes targeted by STAT3 are the antiapoptotic factors Bcl2 and BclXL and cellular inhibitor of apoptosis 1 protein (cIAP).
In a new window | Download PPT
Figure 2: Shifts in the steroid hormone hormetic curve may lead to detrimental effects. A: “J” shaped hormetic curve for adverse effects. The physiological range of the hormone overlaps the beneficial hormetic dose range and results in a reduction in adverse effects that is maximized at an optimal dose falling at the peak of the curve. B: An upward shift in the hormetic curve reduces the hormetic range such that benefits are small and small deviations from the optimal dose lead to the enhancement of adverse effects. C: A leftward shift in the hormetic curve results in the optimal physiological dose falling in the range of enhanced adverse effects. D: A leftward and upward shift of the hormetic curve results in both a reduction of benefit and an enhancement of adverse effects even far below the optimal physiological dose.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11022 | 14 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA