Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The uric acid paradox, a confounder in cardiovascular research and experimental studies of cardioprotection
Time:2019-03-02
Number:13424
Author Affiliations

Conditioning Medicine, 2019. 2(1):40-49.
Abstract
Uric acid (UA) is the end product of purine metabolism in humans but not in experimental animals. Uric acid is an important antioxidant in the hydrophilic extracellular environment. However, within a lipophilic environment and intracellularly, it is a redox modulator that plays both pro- and anti-oxidant roles. Cell culture studies indicate that cellular uptake of UA takes place under certain conditions depending on the cell type. The physiological and pathophysiological roles of UA depend on the organ studied. The brain benefits from the anti-oxidant properties of UA. In the liver, an increase in UA production is a sign of metabolic stress. Experimental studies of ischemia reperfusion of the heart indicate a cardioprotective role, but it also has the ability to block preconditioning. In clinical and epidemiological studies, however, increasing blood UA concentration is significantly associated with cardiovascular disease, diabetes, and kidney disease. We conclude that this paradoxical role of UA is due to the crosstalk between organs and cells in the integrated organism and the unique role of UA in humans.
Keywords: uric acid, oxidation-reduction, species difference, ischemia reperfusion, cardiovascular disease
Abstract
Uric acid (UA) is the end product of purine metabolism in humans but not in experimental animals. Uric acid is an important antioxidant in the hydrophilic extracellular environment. However, within a lipophilic environment and intracellularly, it is a redox modulator that plays both pro- and anti-oxidant roles. Cell culture studies indicate that cellular uptake of UA takes place under certain conditions depending on the cell type. The physiological and pathophysiological roles of UA depend on the organ studied. The brain benefits from the anti-oxidant properties of UA. In the liver, an increase in UA production is a sign of metabolic stress. Experimental studies of ischemia reperfusion of the heart indicate a cardioprotective role, but it also has the ability to block preconditioning. In clinical and epidemiological studies, however, increasing blood UA concentration is significantly associated with cardiovascular disease, diabetes, and kidney disease. We conclude that this paradoxical role of UA is due to the crosstalk between organs and cells in the integrated organism and the unique role of UA in humans.
Keywords: uric acid, oxidation-reduction, species difference, ischemia reperfusion, cardiovascular disease
Extracellular uric acid and species variation
Humans, higher primates, and a few birds are the only species with uric acid (2,6,8 trioxypurine-C5H4N4O3; UA) as an endpoint of purine degradation pathways (Oda et al., 2002; Kratzer et al., 2014; Tan et al., 2016; Johnson et al., 2017). Other species, especially those used in experimental medicine (rats, mice, rabbits, and pigs), express functional urate oxidase (uricase) and metabolize UA into allantoin. One consequence of this is that the concentration of UA might be up to 10 times higher in human blood compared to other species, including those used for experimental research. Allantoin is named after the amnion cavity of the nonhuman fetus where allantoin is found in high concentrations. In the human fetus, UA is the dominant purine degradation product in the amniotic fluid. Allantoin is a highly soluble compound, freely filtered in glomerulus, and very effectively eliminated by the kidney. This is in contrast to UA. Nearly 90% of filtered UA is reabsorbed in the kidney. This is due to the UA membrane transporter URAT1 and other transporters present in epithelial cells of the nephron. The loss of expression of uric oxidase in humans is suggested to have occurred in the mid-Miocene age (between 16 and 11 million years ago) in early ancestral human life due to stepwise mutations leading to low urate oxidase activity (Kratzer et al., 2014). Most often it is assumed to be an advantageous natural selection. The benefit for such high plasma concentrations of UA in humans is open to speculation, but some scientists propose a link to changes in diet and habitat in those times. Other suggestions are an increased ability to maintain blood pressure control in a situation with reduced salt supply, and CNS purine receptors stimulation, which in turn would make humans more intellectually alert (Watanabe et al., 2002). Another hypothesis is that this was a thrifty gene change that is now associated with significant health problems due to modern diets that are rich in salt and sugar (Ndrepepa, 2018). A link to improved handling of cellular stress has also been proposed (Ristow and Schmeisser, 2011).
Wu et al. (1994) developed uricase knockout mice and showed that uricase deficiency caused pronounced hyperuricemia and UA nephropathy such that more than half of these mutant mice died before 4 weeks of age. Thus, a complete lack of uricase in mice evidently leads to death, although its absence in humans does not. Adding to this complexity is the differential expression of the URAT1 transporter in the nephron across species, and that URAT1 (gene SLC22A12) also seems to have developed during prehistoric human life (Tan et al., 2016). A study of individuals with loss of function mutations in the UA transporter URAT1 reported hypouricemia concomitant with significant endothelial dysfunction (Ruiz et al., 2018). A proportion of people have mutations in the proteins responsible for the excretion of UA by the kidneys. Variants within a number of genes involved in UA transport have been identified (Dehghan et al., 2008; Vitart et al., 2008; Kolz et al., 2009). Solute carrier family 2 member 9 (SLC2A9), also known as glucose transporter 9 (GLUT9), is known to transport UA, glucose, and fructose. It is an efficient urate transporter at the basolateral membrane in the tubulus responsible for urate reabsorption from tubulus fluid (Vitart et al., 2008). At pH 7.4 most of UA is present in blood as urate anion. Solubility of urate is low and sensitive to pH, temperature, and albumin concentration. With increasing concentrations there is risk of deposition of monosodium urate crystals in the extracellular environment, especially when pH is below 7.4 and/or temperature is reduced (Benn et al., 2018).
Serum concentrations of UA in men and women, and in species commonly used as experimental animals in cardiovascular research is found in Figure 1. There is a significant difference in concentrations between the sexes (Samimi et al., 2014). The reason for the discrepancy between genders is not fully understood, but lower renal clearance is seen in healthy men compared to healthy premenopausal women. The difference between the sexes is present also in non-human species (Sharma et al., 2015; Auberson et al., 2018), but data are more sparse due to male subject dominance in experimental studies.
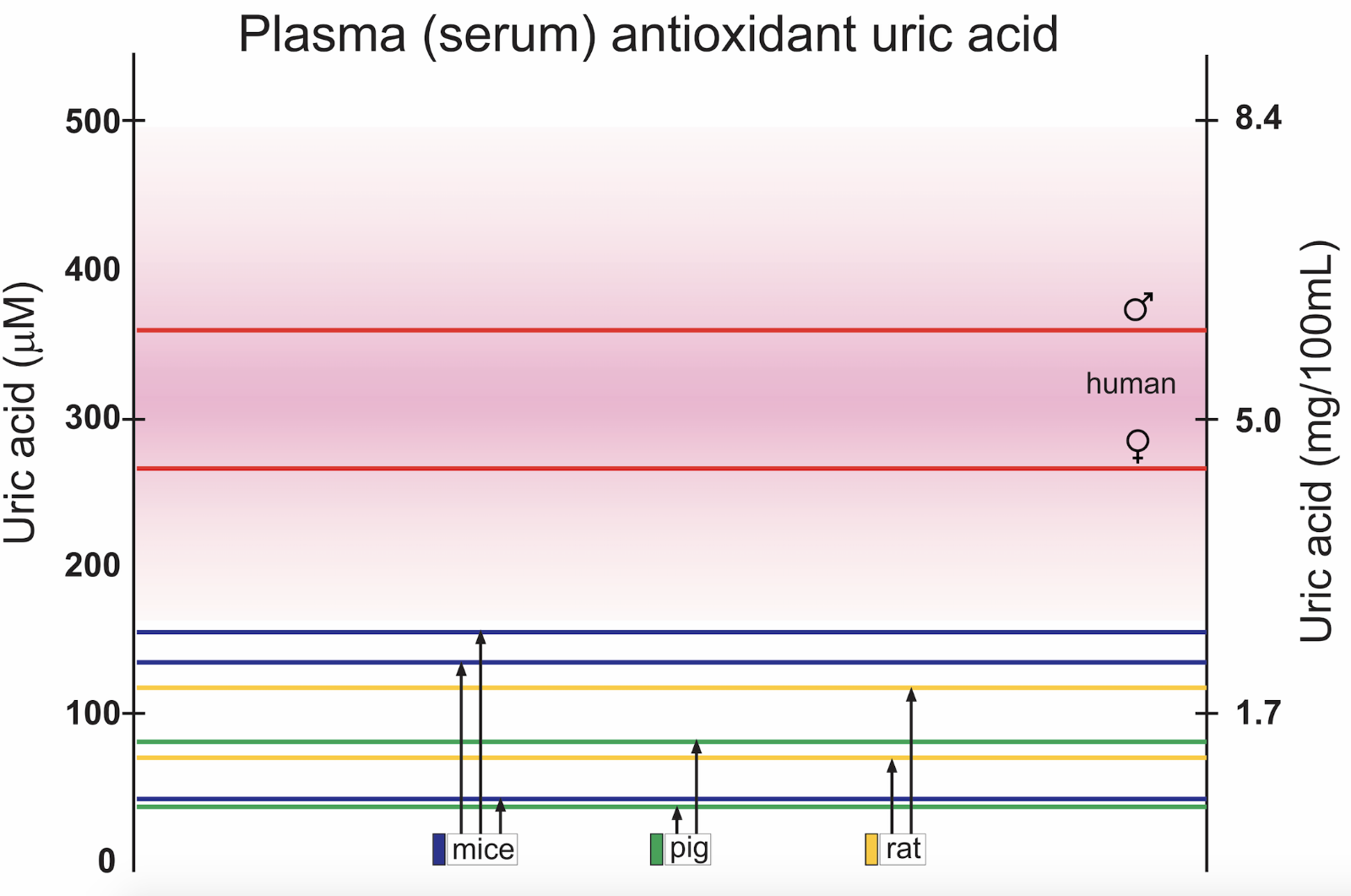
In a new window | Download PPT
Figure 1: Examples of reported mean control concentrations of plasma or serum UA in men and women (Norvik et al., 2017), and in studies using experimental animals, for example pigs (Stoltenberg et al., 1993; Szczurek et al., 2017), rats (Nakagawa et al., 2006; Yun et al., 2017), and mice (Iwama et al., 2012; DeBosch et al., 2014; Auberson et al., 2018). Male and female mice have significantly different serum values (Auberson et al., 2018). Pink shaded area represents upper and lower limits for normal reference values in humans.
Xanthine oxidoreductase
The enzyme xanthine oxidoreductase (XOR) is responsible for the conversion of hypoxanthine to xanthine and finally to UA. The enzyme exists as either a reductase or an oxidase but UA is produced independently of whether the enzyme is in the oxidase or reductase form. The presence, localization, and activity of XOR differ between species, organs, and situations (Kelley, 2015). In humans and most other species XOR is mainly located in the liver and intestines. Milk, including human milk, is also a rich source of the enzyme and contains UA. Human endothelial cells in culture also express the enzyme as demonstrated by Zweier et al. (1998). The enzyme can be released from where it is synthetized, enter the blood stream, and attach to vascular endothelial glycosaminoglycanes (Marti et al., 2004; Kelley, 2015). If and how much extrahepatic XOR contributes to UA levels in healthy individuals is not clear.
Xanthine oxidase (XO) is the product of proteolysis and sulfhydryl oxidation of xanthine dehydrogenase (XDH). The oxidase form of the enzyme, XO, produces the reactive oxygen species (ROS) superoxide and hydrogen peroxide (Kelley, 2015). Since superoxide rapidly dismutase spontaneously or due to the presence of superoxide dismutase (SOD), hydrogen peroxide is the dominant ROS product.
UA as an antioxidant
UA is a hydrophilic antioxidant that contributes markedly to antioxidant activity in plasma and the extracellular compartment in general. Up to two thirds of plasma antioxidant activity as measured by peroxyl scavenger capacity, is due to urate (Wayner et al., 1987), and the rest is ascribed to ascorbic acid (vitamin C), vitamin E, plasma proteins, and other compounds. Humans and several other primates are not able to synthesize vitamin C de novo but depend on dietary sources. Interestingly, the ability to synthesize vitamin C was also lost during evolution due to mutations in the gene coding for L-gulonolactone oxidase enzyme (Drouin et al., 2011). Experimental animals, with the exception of guinea pigs, synthesize vitamin C in the liver using D-glucose-1-phosphate as a precursor. Given the differences in UA concentration between species and individuals, the ability to combat oxidative stress might vary significantly. Increases in oxidative stress can lead to non-enzymatic oxidation of UA resulting in allantoin with H2O2 as a by-product. Plasma levels of allantoin have therefore been suggested as an indicator of oxidative stress in humans. In other species conversion to allantoin takes place enzymatically by uricase.
The antioxidant properties of UA in hydrophilic environments are experimentally well described. UA has the capacity to neutralize and scavenge several prooxidant molecules, such as hydroxyl radicals (.OH), hydrogen peroxide (H2O2), and peroxynitrite (ONOO_.). It can also suppress the Fenton reaction and chelate transition metals (Halliwell, 1999), which will indirectly prevent lipid peroxidation. However, urate radicals (UH−•) formed by its antioxidant action can induce peroxidation by abstraction of hydrogen. The antioxidant capacity of UA in lipophilic environments is, however, disputed (Patterson et al., 2003). Urate protects low-density lipoprotein (LDL) against oxidation, but has been proposed to switch to a prooxidant when the LDL is already partially oxidized (Patterson et al., 2003). A study using low urate concentrations (10.4 µM) found delayed peroxidation of phospholipid liposomes in the presence of cholesterol (Schnitzer et al., 2007). In the absence of cholesterol such effects are less visible and may explain, at least partially, the apparently conflicting conclusions reported previously (Patterson et al., 2003).
The potential link between ROS and aging has been studied in a wide variety of species (Ristow and Schmeisser, 2011; Liochev, 2013; Vina et al., 2013). The question of whether UA imparts beneficial antioxidant defense during healthy aging has been raised. In a human study examining reduction-oxidation (red-ox) biomarkers in aging, UA was identified as a key independent biomarker of aging, as were the plasma antioxidants cysteine and lycopene (Weber et al., 2017). In an experimental setting using insects, longevity was directly correlated with UA availability (Tasaki et al., 2017).
UA and peroxynitrite
The role of urate in ischemia reperfusion cannot be discussed without considering its relation to peroxynitrite (Pacher et al., 2005). Peroxynitrite (ONOO-) is produced when free radicals (superoxide) reacts with nitric oxide (NO). Nicotinamide adenine dinucleotide phosphate oxidase (NOX) and mitochondria are well known sources of superoxide. Therefore, conditions that activate NADPH oxidase (NOX) or increase mitochondrial ROS in conjunction with increases in NO synthase (NOS) activity will likely increase peroxynitrite. Peroxynitrite is an oxidant, and is relatively stable in contrast to other potent ROS related oxidants.
Peroxynitrite can mediate a variety of destructive interactions including oxidation, lipid peroxidation, and DNA strand breakage (Chen et al., 2014). Peroxynitrite depresses cardiac contractile performance and respiratory function (Xie et al., 1998). Peroxynitrite is responsible for nitration of cysteine and tyrosine residues on proteins. Since peroxynitrite is so easily formed from NO in this reaction, it is often difficult to determine whether an observed NOS effect is due to NO or due to production of peroxynitrite (Xie et al., 1998). Peroxinitrite reacts with sulfhydryl groups at the active sites of tyrosine phosphatases and is clearly involved in pathophysiological conditions. Peroxynitrite is detected in isolated buffer perfused hearts (Lee et al., 2003).
Whiteman and colleagues (2002) proposed that UA provides protection against peroxynitrite-dependent tyrosine nitration and compared the protection with that provided by glutathione and vitamin C. It has subsequently been shown that urate reacts with peroxynitrite and reduces its oxidant potential. Indeed, less nitrotyrosination occurs with peroxynitrite in the presence of urate (Xie et al., 1998).
Interestingly, the exact chemical reaction whereby urate inhibits peroxynitrite oxidation is not clearly known. One proposed mechanism is that it is only nitrotyrosination that is blocked, but not other targets of peroxynitrite. Another part of the story is the ability of peroxynitrite to oxidize urate, and the interference in biological solutions from molecules like vitamin C and CO2/HCO3- adds to the complexity.
By using a test system where the amino acid tyrosine was exposed to peroxynitrite and the resulting 3-nitrotyrosination measured by HPLC, a dose dependency of urate as an inhibitor of 3-nitrotyrosine formation was described (Whiteman et al., 2002). Concentrations corresponding to endogenous levels in human plasma reduced 3-nitrotyrosination, but with the addition of bicarbonate to UA reduction it was dampened. Concentrations of UA equal to what is endogenously present under animal experimental conditions, showed only minor effects. Thus, this might imply that nitrotyrosination is a minor problem in the human but likely to occur under experimental conditions in animals. Treatments aimed at reducing nitrotyrosination might fail to reliably show reduced effects in humans since higher levels of UA are present. Peroxynitrite can be detected by electrochemical sensors, by fluorescent probes, or by nitrotyrosine detection using Western blot technique. Teng et al. (2002) demonstrated that brain and heart homogenate responded differently to protein nitration.
Production of NO can also take place via the nitrate – nitrite – nitric oxide (NO3−-NO2—NO) pathway. Diet may play an important role as a nitrate source (Lundberg et al., 2008), with beetroot juice as an important example (Coggan et al., 2015; Hirai et al., 2017). Which role UA plays in these reactions is not fully clarified, but urate is naturally present in saliva in humans, and might interfere with the pathway. Nitration of pepsin has shown to be reduced by UA, indicating that less NO was produced from nitrite (Rocha et al., 2016). NO can also be produced with nitrite (NO2-) as a substrate under the influence of XOR. Hypoxic or anaerobic conditions are discussed as promotors of this pathway of NO production (Millar et al., 1998).
Cellular effects of UA
Several studies have examined the response when UA is added to the medium of in vitro cell cultures (Sanchez-Lozada et al., 2012). These studies document that UA is not an inert compound but interferes in various ways with cellular life. This becomes especially clear during prolonged incubation with UA at concentrations comparable to the upper plasma levels in humans i.e. above 5-600 µM (10mg/100ml) (Sautin and Johnson, 2008).
With the exception of kidney tubular epithelial cells, information regarding cellular uptake and release mechanisms is sparse (Benn et al., 2018). In addition to kidney, intestines and liver cells contain GLUT9, a confirmed UA transporter. GLUT9 expression has been detected in chondrocytes, hepatocytes, fibroblasts, endothelial cells, smooth muscle cells, and some cells in brain tissue (Price et al., 2006; Itahana et al., 2015; Tomioka et al., 2016; Begandt et al., 2017). Liu et al. (2017) using human umbilical vein endothelial cells (HUVEC) demonstrated uptake via GLUT9 using siRNA techniques. GLUT9 is involved both in uptake and in release of UA, which is assumed to be dependent on concentration gradients. GLUT9 is also a voltage sensitive transporter. In liver and kidney, GLUT9 is proposed to carry urate from the cytosol into the interstitium.
Due to the association of serum UA with hypertension (Sundstrom et al., 2005) and cardiovascular disease (Holme et al., 2009; So and Thorens 2010; Carluccio et al., 2018; Ndrepepa, 2018; Tseng et al., 2018), the response of endothelial cells in culture has often been studied. Sanchez-Lozada et al. (2012) documented lower basal concentrations of ATP in human aortic endothelial cells exposed to UA. Cells were exposed to UA at different concentrations (low normal, high normal, and hyperuremic), and significant changes were found especially with hyperuremic concentrations (714 µM, 12 mg/100ml). Adding 500 µM UA to the culture medium of HUVEC stimulated expression of inflammatory genes like monocyte chemoattractant protein-1 (MCP-1) (Figure 2). Another study demonstrated calcium related mitochondrial dysfunction and reduced NO/endothelial NOS function using HUVEC exposed to high levels of UA. Interestingly, in this study lower concentrations yielded the opposite result (Hong et al., 2012).
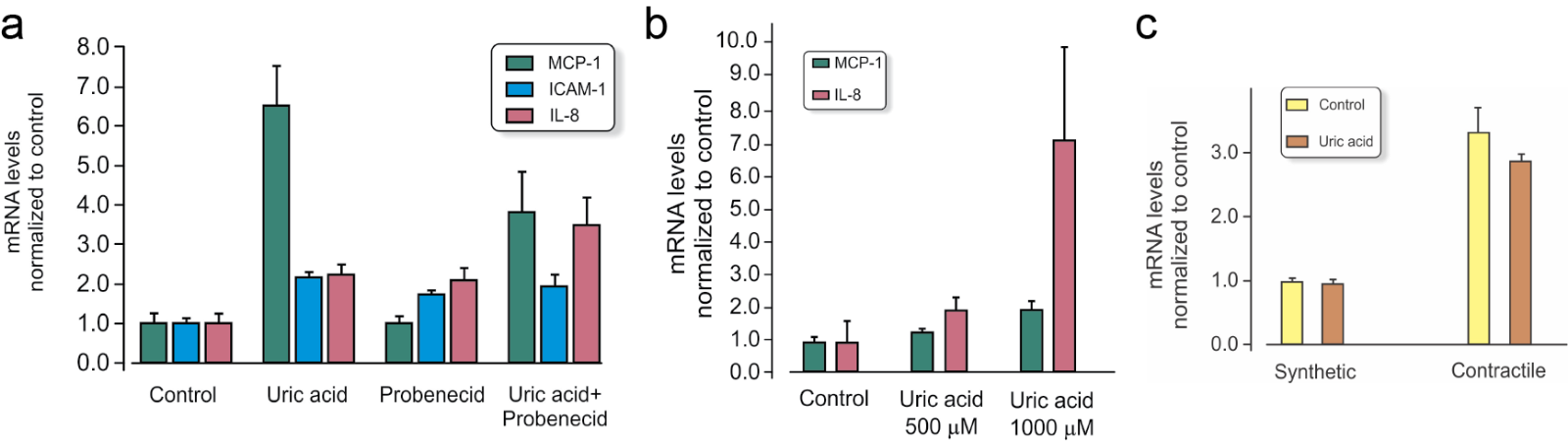
In a new window | Download PPT
Figure 2: Gene expression (mRNA) in cell cultures of vascular cells with and without 500 µM UA added to the cell culture medium for 4 hours (relative values compared to control and normalized to reference gene RPL13a). Values are mean ± SEM. Data presented are the authors unpublished observations. a) Monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), and interleukin-8 (IL-8) in HUVEC endothelial cells with and without the URAT1 inhibitor probenecid. b) MCP-1 and IL-8 in contractile phenotype of vascular smooth muscle in human coronary artery smooth muscle cells (HCASMC) and c) Glut9 gene Slc2a in synthetic and contractile phenotype HCASMC.
In primary cultures of smooth muscle cells obtained from male rat aorta, 200-300 µM UA interfered with angiotensin, gene expression and proliferation, and increased release of indicators of oxidative stress (Corry et al., 2008). The Figure 2b illustrates that the response to 500 µM UA is dependent on the subtype of smooth muscle cells.
Accumulation of monosodium urate crystals is a recognized damage associated molecular pattern able to activate nucleotide binding domain, leucine-rich containing family pyrin domain containing-3 (NLRP3) inflammasome and stimulate production of cytokines (Shi et al 2003; Kono et al 2010; Rock et al., 2013). In humans and the few other species without uricase, UA crystals are a significant problem. Some studies indicate that soluble UA also initiates inflammation (Braga et al., 2017)
UA is not acknowledged as an endogenous intracellular antioxidant. When added to cell cultures, UA enters the intracellular compartment. The intracellular presence of UA is not dependent on expression of XOR. Cells in the body of healthy individuals are exposed to some extracellular UA all the time, and it is thus difficult to make a definite conclusion about the role of UA in promoting disease. However, cell studies also demonstrate the complexity of red-ox modification in cellular life (Vina et al., 2013).
UA in ischemia reperfusion injury
Ischemia reperfusion, independent of the affected organ, implies an increased burden of ROS (Granger and Kvietys, 2015). Ischemia reperfusion affects various organs and cell types differently (Kalogeris et al., 2012). Endothelial cells (Laude et al., 2002), fibroblasts, smooth muscle cells in addition to parenchymal-cells like muscle cells (Wang et al., 2006), and neurons have been studied in experimental models mimicking ischemia reperfusion. Protective treatments of ischemia and ischemia reperfusion injury have also been studied experimentally for several decades to identify mechanisms behind the injury and to design better treatment. Among the different ROS sources described to date, mitochondria and enzymes xanthine oxidase, NOX, monoamine oxidase (MAO), and uncoupled NOS have gained a status as likely contributors to reperfusion-induced oxidative stress (Schulz et al., 2014). They have all been targets for therapeutic intervention against organ dysfunction, tissue damage, and cell death.
Treatment strategies against ischemia reperfusion injury are needed for many organs, including kidneys, brain, lung, liver, skeletal muscle, spinal cord, and the heart (Kalogeris et al., 2012). In this review, the difference between the heart, brain, and liver (Figure 3) in ischemia reperfusion and the role of UA is discussed further below.
In a new window | Download PPT
Figure 3: Three organs that differ in their response to UA and the role that UA play in these organs.
The brain: Exogenous administration of UA has repeatedly been shown to be neuroprotective in experimental models of CNS disease (Hooper et al., 2000), including brain ischemia, spinal cord injury, meningitis, and experimental allergic encephalitis. This is explained mechanistically by the limited antioxidant capacity of the brain. UA concentration in the cerebrospinal fluid is low. In human cerebrospinal fluid, it can be up to 10 times lower than the concentration in plasma, but there seems to be a positive correlation between concentrations in the two compartments. The blood brain barrier normally has limited permeability to UA, which explains the lower concentration in the brain compared to blood. Degenerative brain disease has been correlated to a higher level of nitrotyrosination in some studies (Hensley et al., 1998; Ischiropoulos and Beckman, 2003). UA has been assessed as an antioxidant against ischemia-reperfusion in clinical studies of acute ischemic stroke. In the case of fibrinolysis, some studies report better outcome with addition of UA (Llull et al., 2016). One example is the URICO-ICTUS study, which was a double-blind, placebo-controlled, phase 2b trial. In the study, the plasma UA level was increased by an average of 119 μmol/L above pretreatment level by intravenous (i.v.) infusions. Although not proven, it was assumed that this would also be reflected as an increase in interstitial UA in the brain. Interestingly, women who normally have lower levels of UA in plasma seemed to benefit more from the i.v. infusion compared to men. It was recently reported that UA enhances alteplase-mediated thrombolysis as an antioxidant (Kikuchi et al., 2018).
MAO-A and MAO-B are important in the brain and also present in other organs, including the heart. MAO metabolizes biogenic amines like the catecholamine dopamine with hydrogen peroxide as a by-product. The brain MAO system is tightly coupled to Parkinson’s disease (PD), which is characterised by reduced dopamine availability. Serum and brain tissue nigrostriatal UA levels are lower in PD compared to age-matched controls and the level is further decreased as the disease progresses (Wen et al., 2017). The use of the precursor inosine to increase plasma UA levels in PD has been proposed (Bhattacharyya et al., 2016), (Parkinson Study Group et al., 2014) to confirm a causal connection. In an experimental mice model of this disease produced by intraperitoneal injection of the lipophilic compound 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, or MPTP, a prodrug to the 1-methyl-4-phenylpyridium (MPP+) neurotoxin, extra UA supplement was neuroprotective (Huang et al., 2017). Neurotoxin leads to ROS related cell death in dopamine producing nigrostriatal neurons. Overexpression of uricase (urate oxidase) exaggerated injury in a similar mouse model. Whether these experimental findings have any relevance to human pathophysiology and to ischemia reperfusion or not is not clear. Considering the low level of plasma UA in experimental animals results in even lower levels in the CNS, direct translation to the human situation is very difficult.
The liver: Liver, kidney, pancreas, and lungs are subjected to ischemia-reperfusion in conjunction with transplantation. The gold standard of liver preservation solutions, the University of Wisconsin (UW) solution, contains allopurinol and the ROS scavenger glutathione. Of note, the UW solution is also used during lung, kidney, and pancreas transplantation (Parsons and Guarrera, 2014).
Liver hepatocytes are the main endogenous source of UA, especially in humans, where hepatocytes lack the ability to enzymatically degrade UA, resulting in a substantial intracellular load of this purine. Precursor purines from other organs released to the blood stream are metabolized in the liver and add to the metabolic burden of the liver, which is diet dependent. Both a diet rich in purines and table sugar with added fructose will increase liver UA. In contrast to the brain, the liver will release substantial amounts of UA when exposed to ischemia. Studies in humans as well as rats have demonstrated that serum concentrations of urate rise during reperfusion after ischemia (Layton et al., 1996).
The heart: During complete occlusion of a coronary artery, in the absence of collateral blood flow the affected area of the heart rapidly loses contractile force, and dangerous arrhythmias may develop. With complete ischemia lasting more than 25-30 minutes the first signs of irreversible cell death are present. Without reperfusion, cell death will progress in the ischemic area, and inflammation is followed by development of permanent scar tissue replacing necrotic cells after some time. Early reperfusion is thus the primary treatment goal. Initially, at reperfusion some additional injury takes place in the form of stunning or delayed recovery of contractile function, reperfusion arrhythmias, and there is risk of cell death by apoptosis, necroptosis, or uncontrolled autophagy (Kalogeris et al., 2012). Experimental studies have ascribed early reperfusion injury to increase in ROS coupled to reintroducing oxygen and calcium overload, but other mechanisms might also participate (Kalogeris et al., 2012). Inflammatory cells, polymorphonuclear cells (PMNs), macrophages, cytokines and disturbed microcirculation may also be involved in extending injury (Kalogeris et al., 2012).
Insulin given at reperfusion reduces injury after ischemia reperfusion both in buffer perfused hearts and in in situ blood perfused hearts in mice, rats, as well as pigs (Jonassen et al., 2000; Fuglesteg et al., 2008; Ji et al., 2010). Interestingly, Ji et al. (2010) demonstrated in an in situ rat heart that an intravascular injection of UA given at reperfusion conferred the same degree of infarct size limitation as insulin, and that the two treatments were not additive. The authors proposed that insulin or UA infused at reperfusion shared a cardioprotective mechanism, namely inhibition of peroxynitrite. Similarly, preischemic treatment with UA was also reported to be cardioprotective in rats, and the protection was not additive with a cardioprotective agent that activated phosphoinoside 3-kinase (PI3K) (Huang et al., 2013). Unfortunately, in these studies serum concentration of UA after exogenous addition was not measured and cannot be compared to concentrations in human blood (Khalpey et al., 2007) (Figure 1).
UA is always present in plasma and the extracellular space of the heart under in vivo conditions independent of the presence of XOR in the heart. It is, however, difficult to examine the role of UA without taking into account concomitant xanthine oxidase derived ROS. UA used experimentally as an antioxidant will reduce ischemia-reperfusion injury in ex vivo buffer-perfused rat hearts just as allopurinol in this species (Teng et al., 2002; Lee et al., 2003). Nevertheless, the cardioprotective effect of added UA has been taken as proof of the presence of ROS or peroxynitrite experimentally, further indicating ROS and peroxynitrite as contributing causal factors in ischemia reperfusion injury under experimental conditions. With elevated perfusion pressure, this effect is exaggerated. In 2011, Mozzafari et al. (2011) demonstrated that 100 µM UA reduced the infarct expanding effect of increased perfusion pressure in isolated rat heart. Corresponding findings using 500 µM UA added to the buffer of high-pressure perfused hearts confirmed the observation (Ytrehus, 2014). UA seems to reduce cell death without reducing the increased workload following high pressure perfusion. Mitochondria are a source of superoxide and hydrogen peroxide at reperfusion. Interestingly, these authors provide data pointing at mitochondria as an important source of the UA that inhibits ROS in high pressure buffer-perfused rat hearts. MAO, mainly the MAO-A subtype, is present in the heart and upregulated when exposed to increasing amounts of catecholamine (Kaludercic et al., 2011). The role of UA in dampening MAO derived ROS in the heart has not been investigated in detail. The higher UA level in humans compared to other species might indicate that ROS derived from mitochondria or MAO in ischemia-reperfusion of human hearts are less important compared to the situation in experimental animal heart models. Concentrations of UA used in experimental settings have varied widely, but it seems clear that concentrations corresponding to those normally present in human blood could have an infarct-sparing effect.
Conditioning of the heart: ROS has dual roles, regulatory in some situations and detrimental in other situations. Ischemic preconditioning is an example that nicely illustrates the dual roles of ROS. For decades, preconditioning has been an effective cardioprotective treatment against ischemia-reperfusion injury of the heart in experimental settings. Short lasting ischemia followed by reperfusion prior to a subsequent prolonged ischemic period followed by reperfusion reduced injury significantly compared to no pre-treatment. One to three cycles of five minutes of ischemia followed by five minutes of reperfusion are most often used to trigger the adaptation. The phenomenon was originally observed in 1986 to reduce infarct size in the hearts of anesthetized dogs, and was named ischemic preconditioning (Murry et al., 1986). It was later confirmed experimentally to be present in a wide variety of species and organs, and to protect against different types of injury (Hausenloy et al., 2016). Regulatory red-ox modulation during ischemic preconditioning appears to protect against further ROS related injury (Tullio et al., 2013). Increases in peroxides are detected during preconditioning (Starkopf et al., 1998), antioxidants block preconditioning, and oxidative agents can mimic the trigger of preconditioning (Tullio et al., 2013). By use of inhibitors and stimulating agents, the phenomenon has facilitated detailed description of intracellular signaling pathways that increase cell survival by delaying mitochondrial permeability transition (MPT) at reperfusion (Juhaszova et al., 2004; Hausenloy et al., 2016). UA also plays a dual role in ischemia reperfusion. In an isolated buffer-perfused heart preparation, ischemic preconditioning is blocked by adding 500 µM UA (Ytrehus, 2014). However, UA also blocked delayed protection by ischemic preconditioning against endothelial dysfunction in isolated rat hearts (Laude et al., 2002). In line with the role of ROS in the trigger phase of ischemic preconditioning, peroxynitrite preconditioning with and without concomitant UA infusion, was examined in dogs using arrhythmias as endpoint (Juhasz et al., 2011). In the in vivo study of the dog heart, additional UA conferred protection alone and blocked the peroxynitrite effect but did not block ischemic preconditioning. Another experimental protective conditioning strategy is postconditioning conferred by the application of short lasting cycles of ischemia reperfusion at reperfusion (Hausenloy et al., 2016). Rapid pacing-induced postconditioning has also been described to be cardioprotective (Babiker et al., 2017). This treatment increased formation of cardiac 3-nitrotyrosine measured in heart homogenate detected using enzyme-linked immunosorbent assay (Pipicz et al., 2015). In an isolated perfused rat heart, pacing-induced postconditioning was blocked by adding 500 µM UA to the perfusate (Babiker et al., 2017). Adding UA at reoxygenation of hypoxic isolated cardiomyocytes was protective but did not enhance the protection of hypoxic postconditioning (Wang et al., 2006).
UA and cardiovascular disease
In contrast to studies of some of the common brain diseases, epidemiological studies have repeatedly demonstrated that increasing serum level of UA is associated with increased presence of cardiovascular disease (Ndrepepa, 2018). Whether serum UA is a biomarker, a true risk factor, a disease modulating factor, or just a coincidental factor in the development of cardiovascular disease, is difficult to determine and is not fully settled (So and Thorens 2010; Carluccio et al., 2018). One reason for this ambiguity is the very frequent association and causal relationship between UA and established cardiovascular risk factors, such as metabolic syndrome, type 2 diabetes, and chronic kidney disease, and the roles played by reverse causality or simultaneity. An interesting study showed a U shaped relation between cardiovascular mortality and serum UA. Below 240 µM, cardiovascular mortality was increased in conjunction with malnutrition, and above 480 µM cardiovascular mortality gradually increased (Tseng et al., 2018).
The source of UA in the body is partly by intake through the diet and partly via endogenous purine degradation. The role of liver purine metabolism is significant. Diets high in sugar intake, fructose, and purine rich food, as well as caloric overload in general, are responsible for increases in serum UA if kidney function is not affected. Genetic factors (polymorphism, epigenetics) might add to variations in UA levels in the body (Awad et al., 2018; Benn et al., 2018). This is especially true when it comes to the kidney epithelial membrane transporters.
One way to examine the role of UA is with the use of XOR inhibitor, e.g. allopurinol (Pacher et al., 2006). The ability of allopurinol to reduce oxidative stress in the vasculature and improve heart function in clinical studies was extensively reviewed in 2009 (George and Struthers 2009) and in 2018 (Bredemeier et al., 2018). In studies using a limited number of heart failure patients, improved myocardial energy metabolism was observed (Hirsch et al., 2012; Ansari-Ramandi et al., 2017). The “Intravenous Allopurinol in Heart Failure; NCT00181155” study was able to demonstrate an effect on ATP flux through creatine kinase using (31)P magnetic resonance spectroscopy. However, this study did not examine the cellular mechanism, which potentially could be purine salvage, reduced UA, or reduced ROS (Hirsch et al., 2012). The EXACT-HF study failed, on the other hand, to detect any advantage of allopurinol in hyperuremic heart failure patients (UA > 9.5 mg/100ml or 564 µM) using mainly clinical endpoints (Givertz et al., 2015). In this study, the reduction in serum UA was on average 3.5 mg/100ml (206 µM) after 24 weeks of treatment. George and coworkers (2006) studied endothelial function in heart failure patients and circumvented the problem of ROS versus UA as causal factor by using the uricosuric agent probenecid. Their results indicated that lowering ROS was responsible for improving endothelial function since lowering UA alone had no effects.
Experimentally, the diabetic heart is relatively resistant to protection against ischemic injury by preconditioning. On the other hand, myocardial infarcts tend to be smaller in some of the models of diabetic cardiomyopathy. Hypertension, type 2 diabetes, and obesity are all major concerns in public health worldwide (So and Thorens 2010; Johnson et al., 2017; Borghi et al., 2018). Epidemiological studies repeatedly associate these cardiovascular risk factors with UA, but also with kidney disease, and non-alcoholic fatty liver disease (NAFLD). In a population in Northern Norway, ischemic stroke, diastolic dysfunction, metabolic syndrome, and atrial fibrillation all showed association with UA (Storhaug et al., 2013; Nyrnes et al., 2014; Norvik et al., 2016, 2017). Interestingly, with respect to coronary heart disease the data are less convincing. When risks for myocardial infarction were examined, no significant association was found with UA when adjusted for co-variates (Storhaug et al., 2013; Norvik et al., 2017). A study of subgroups with coronary heart disease found UA serum levels to be associated with the subtype suffering from oxygen supply imbalance but not with asymptomatic coronary calcification or acute coronary plaque rupture (Larsen et al., 2018).
Translational perspective
The UA paradox describes how UA is associated with pathophysiology in some situations, as a protective antioxidant, or inert in other situations. In ischemia reperfusion, endogenous UA is a potential confounder.
When translating results from animal experimental models to clinical studies the difference in purine metabolism and in UA concentrations between humans and other species might lead to the inability to confirm preclinical results in clinical studies. In studies where red-ox modulation is involved, the antioxidant effect of UA might change the response. Although very effective experimentally, pre- and postconditioning have been difficult to translate into clinical practice (Hausenloy et al., 2017). Several reasons for this discrepancy exist. Interference with conditioning mechanisms by higher levels of UA in humans could be an additional variable contributing to this lack of clinical translation. One way to circumvent the problem in the future would be to use UA as an additive to culture medium, buffers, and perfusion solutions for ex vivo and in vitro isolated heart studies and cell studies, prior to in vivo preclinical studies.
Another challenge is translation from chronic disease and clinical studies to preclinical research. To reveal the mechanisms behind the association between UA and cardiovascular disease, experimental models of hyperuricemia have been studied. Chronic elevation of UA in smaller animals is not well tolerated (Mene and Punzom, 2008). Levels far below those of healthy humans result in kidney dysfunction and hypertension.
UA enters and leaves the intracellular compartment depending on electrochemical gradients, expression of membrane transporters, and presence of competing substances. Knowledge of differences between species and cell types, other that kidney epithelial cells, is sparse. Mechanism of uptake and release, and the significance of these processes need to be investigated under in vivo conditions.
Conclusion
Despite longtime interest in UA, its role in the pathophysiology of cardiovascular disease, particularly coronary heart disease and ischemia reperfusion, remains unclear. UA is not an inert compound or metabolic rest-product, but takes part in regulation of ROS levels and red-ox reactions. The role of UA must be judged from the perspective of the most common comorbidities in ischemic heart disease (obesity, kidney disease, hypertension, and type 2 diabetes). Extracellular UA enters the intracellular compartment. Both mechanisms of uptake and release, and the significance of these processes under in vivo conditions need to be investigated. The large difference in purine metabolism between humans and species used in experimental research is difficult to circumvent, but most studies indicate that adding UA in amounts present in the blood of humans, reveal an antioxidant effect. Although such an effect is difficult to demonstrate in clinical studies, UA should be used more actively as standard additive to culture medium, buffers, and perfusion solutions for ex vivo and in vitro studies.
Acknowledgement
The authors have been supported by grants from Tromsø Research Foundation. We also acknowledge the contribution of the COST Action CA16225. The authors thank Knut Steinnes for help with illustrations.
References
Kirsti Ytrehus
1Cardiovascular Research Group, UiT The Arctic University of Norway.
Neoma Tove Boardman
1Cardiovascular Research Group, UiT The Arctic University of Norway.
Trine Lund
1Cardiovascular Research Group, UiT The Arctic University of Norway.
Svetlana Zykova
2Center for Quality Assurance and Development, University Hospital of North Norway.
3Department of Blood Bank and Medical Biochemistry, Innland Hospital Trust, Norway.
4Metabolic and Renal Research Group, UiT The Arctic University of Norway.
Trond Geir Jenssen
4Metabolic and Renal Research Group, UiT The Arctic University of Norway.
5University Hospital of Oslo, Department of Organ Transplantation and University of Oslo, Norway.
Corresponding author:
Kirsti Ytrehus
Email: Kirsti.ytrehus@uit.no
In a new window | Download PPT
Figure 1: Examples of reported mean control concentrations of plasma or serum UA in men and women (Norvik et al., 2017), and in studies using experimental animals, for example pigs (Stoltenberg et al., 1993; Szczurek et al., 2017), rats (Nakagawa et al., 2006; Yun et al., 2017), and mice (Iwama et al., 2012; DeBosch et al., 2014; Auberson et al., 2018). Male and female mice have significantly different serum values (Auberson et al., 2018). Pink shaded area represents upper and lower limits for normal reference values in humans.
In a new window | Download PPT
Figure 2: Gene expression (mRNA) in cell cultures of vascular cells with and without 500 µM UA added to the cell culture medium for 4 hours (relative values compared to control and normalized to reference gene RPL13a). Values are mean ± SEM. Data presented are the authors unpublished observations. a) Monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), and interleukin-8 (IL-8) in HUVEC endothelial cells with and without the URAT1 inhibitor probenecid. b) MCP-1 and IL-8 in contractile phenotype of vascular smooth muscle in human coronary artery smooth muscle cells (HCASMC) and c) Glut9 gene Slc2a in synthetic and contractile phenotype HCASMC.
In a new window | Download PPT
Figure 3: Three organs that differ in their response to UA and the role that UA play in these organs.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13424 | 34 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA