Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Negative Conditioning of Mitochondrial Dysfunction in Age-related Neurodegenerative Diseases
Time:2019-03-02
Number:13997
Author Affiliations

Conditioning Medicine, 2018. 2(1):30-39.
Abstract
Mitochondrial dysfunction is regarded as one of the major causes of neuronal injury in age-associated neurodegenerative diseases and stroke. Mitochondrial dysfunction leads to increased reactive oxygen species production, causing mitochondrial DNA mutations, which then results in pathological conditions. Negative conditioning of mitochondrial dysfunction via pharmacological inhibition, phytochemicals, and dietary restriction serve as an avenue for therapeutic intervention to improve mitochondrial quality and function. Here, we focus primarily on mitochondrial biology, evidence for mitochondrial dysfunction in neurodegenerative conditions such as dementia and stroke, and the possibility of using negative conditioning to restore or preserve mitochondrial function in these diseases.
Keywords: aging, mitochondria, mitochondrial dysfunction, negative conditioning, dementia, stroke, phytochemicals, calorie restriction
Abstract
Mitochondrial dysfunction is regarded as one of the major causes of neuronal injury in age-associated neurodegenerative diseases and stroke. Mitochondrial dysfunction leads to increased reactive oxygen species production, causing mitochondrial DNA mutations, which then results in pathological conditions. Negative conditioning of mitochondrial dysfunction via pharmacological inhibition, phytochemicals, and dietary restriction serve as an avenue for therapeutic intervention to improve mitochondrial quality and function. Here, we focus primarily on mitochondrial biology, evidence for mitochondrial dysfunction in neurodegenerative conditions such as dementia and stroke, and the possibility of using negative conditioning to restore or preserve mitochondrial function in these diseases.
Keywords: aging, mitochondria, mitochondrial dysfunction, negative conditioning, dementia, stroke, phytochemicals, calorie restriction
Aging and neurodegenerative diseases
A phenomenal era of population aging has begun. In fact, the global aging population is expected to double by 2050 to an astounding 2 billion people (WHO, 2018). What seemed like a mere physiological phenomenon is becoming an increasing socioeconomic burden around the globe. Aging constitutes the accumulation of damage at the cellular and molecular level, which then translates into a decline in function of organs at a systems level (Payne & Chinnery, 2015). Examples of damages imposed at the molecular and cellular level include genomic instability, telomere shortening, epigenetic alterations, impaired protein homeostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, altered intracellular communication, and stem cell exhaustion (López-Otín et al., 2013). López-Otín and colleagues have thus described these nine damages as hallmarks of aging. Eventually, these factors contribute to an increased vulnerability to death. Cumulative effects of such damages over time is observed at the organ level where organs such as the heart, kidneys, and lungs are affected. The heart becomes more susceptible to hypertension and heart failure due to increased arterial stiffness, the kidneys are vulnerable to diffuse glomerulosclerosis and eventual impairment in filtering ability, and lung function becomes compromised with increased ventilation-perfusion mismatch (Khan et al., 2017). Like other organs, the function of the brain progressively declines with age. Cognitive decline, reduced cerebral perfusion and dopaminergic signalling are some effects of brain aging (Khan et al., 2017). Apart from its associated functional decline in a physiological state, aging also serves as a high-risk factor for neurodegenerative conditions like Alzheimer’s Disease (AD) and Parkinson’s Disease (PD), which are major neurodegenerative diseases (Mattson & Arumugam, 2018; Checkoway et al., 2011).
AD is the most common cause of dementia (Parachikova et al., 2007), with a new case occurring every 7 seconds in the world (Cornutiu, 2015). After 65 years of age, the rate of AD increases almost exponentially (Qiu et al., 2009). PD is the most common neurodegenerative movement disorder worldwide, and has a similar age-dependent increase (Driver et al., 2009). The incidence of neurodegenerative diseases will rapidly increase with an ever-growing aging population (Figure 1). Nonetheless, the pathophysiology of neurodegenerative diseases remains largely unknown. This further complicates the problem at hand and emphasizes the urgent need to research the underlying causes.
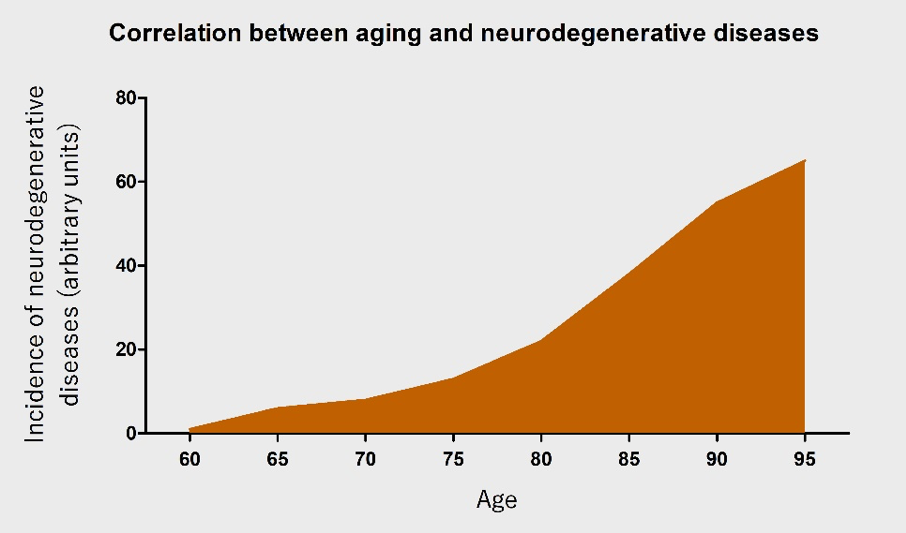
In a new window | Download PPT
Figure 1: An illustration of the correlation between age and the occurrence of neurodegenerative diseases. With increasing age, the incidence of neurodegenerative diseases is posited to increase almost exponentially.
In a bid to unravel how and why aging occurs, a plethora of different theories of aging have surfaced over the decades. A few well recognised theories include the telomere loss theory, somatic mutation theory, and the free radical theory. The telomere theory revolves around the idea that the loss of telomere causes replicative or cellular senescence of cells resulting in aging (Weinert & Timiras, 2003). Somatic mutation theory argues that DNA mutations accumulate as molecular damage leading to aging (Weinert & Timiras, 2003). The free radical theory, which was first proposed in 1957 is one of the most well-known and longstanding theories of aging (Payne & Chinnery, 2015). The free radical theory suggests that mitochondria play a crucial role in aging, as they are the main source of reactive oxygen species (ROS), leading to increased mitochondrial DNA (mtDNA) mutations (Payne & Chinnery, 2015). Such aging-associated mtDNA mutations thus perturb mitochondrial function resulting in pathological conditions (López-Otín et al., 2013). Mitochondria, the molecular batteries of the cell, play a crucial role in regulating the energy of the cells by producing adenosine triphosphate (ATP) through oxidative phosphorylation (Payne & Chinnery, 2015). Their prominent role in cell homeostasis in almost all tissues thus explains its postulated widespread effects on aging.
Mitochondrial function in a physiological state
The primary role of the mitochondria involves regulating the bioenergetics of the cell. Generation of ATP through oxidative phosphorylation in the mitochondria involves both the electron transport chain (ETC) and ATP synthase. Nutrient intake from the cells are converted to energy via key enzymes in the tricarboxylic acid (TCA) or Kreb’s cycle to produce reducing equivalents that feed the ETC (Figure 2). Electrons generated via redox reactions at ETC complexes are transferred from one member of the transport chain to another via redox reactions. These redox reactions are catalysed by nicotinamide adenine dinucleotide (NADH) dehydrogenase (Complex I), succinate dehydrogenase (Complex II), cytochrome bc1 (Complex III), and cytochrome c oxidase (Complex IV) (Johannsen & Ravussin, 2009). Complexes I, III, IV are involved when electrons flow from NADH to oxygen, while Complexes II, III and IV are involved when electrons flow from flavin adenine dinucleotide (FADH2) to oxygen. Energy released during this electron transfer is used to pump protons from the matrix and a proton gradient is thus established in the intermembrane space. The flow of protons back to the matrix through ATP is used for cellular activities. Oxidative phosphorylation in the mitochondria thus plays an important role in generating the ‘energy currency’ (ATP) of the body.
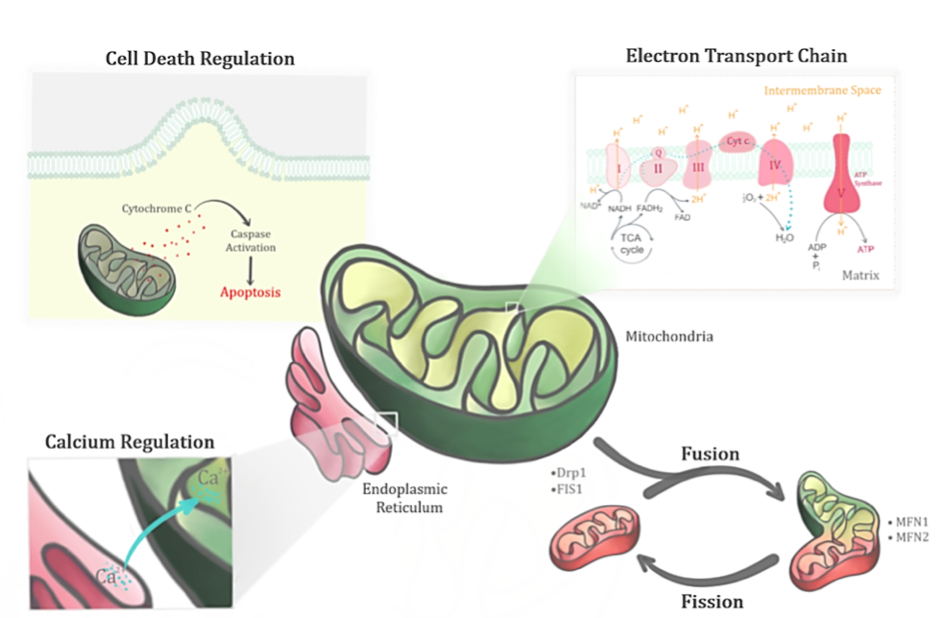
In a new window | Download PPT
Figure 2: Mitochondrion, a dynamic organelle, has several important functions in its physiological state. Apart from its primary role in regulating the bioenergetics of the cell through the electron transport chain, it also regulates the fusion-fission cycles, calcium levels and cell death.
The electrochemical proton gradient generated in the ETC provides a large driving force for the accumulation of calcium ions (Ca2+) as well (Rizzuto et al., 2012). Mitochondria is therefore involved in regulating calcium signaling to ensure that the balance between energy production and consumption of the cell is maintained. Calcium signals facilitate processes that are energy demanding such as movement, secretion, and electrical excitability (Osellame et al., 2012). Calcium is an essential cofactor for the activation of Kreb’s cycle enzymes where the intramitochondrial calcium concentration serves as regulators of bioenergetics (Osellame et al., 2012). Nonetheless, mitochondrial calcium overload leads to increased mitochondrial ROS production (Mallilankaraman et al., 2012), and thus dysregulation of mitochondrial matrix calcium levels leads to aberrant ROS production.
The bioenergetics of the mitochondria are seemingly dependent on mitochondrial morphology (Osellame et al., 2012). Mitochondria are heterogeneous in number and size within and among different tissues. Their shape is thus dependent on the dynamic fission and fusion events, which help to ensure homeostasis in the mitochondria. Mitochondrial fusion entails ‘combining’ of the intercellular contents of two mitochondrion through mitofusin proteins (MFN1/2). Mitochondrial fission, on the other hand, involves ‘division’ of a single mitochondrion into two mitochondria via proteins like dynamin-related protein 1 (DRP1) and mitochondrial fission 1 protein (Osellame et al., 2012). The former aids in motility and provides protection for mitochondrial function (Chen & Chan, 2010), while the latter is thought to facilitate clearance of damaged mitochondria via mitophagy and to increase the efficiency of distribution when ATP is in demand (Otera & Mihara, 2011).
Apart from energy metabolism, mitochondria also play a key role in cell death, which includes apoptosis and necrosis. Apoptosis is a form of programmed cell death that involves caspase activation through a cascade of proteolytic events. Mitochondria play a role in both the intrinsic and extrinsic pathways of apoptosis. Upon activation, the cascade of signaling events eventually leads to cell death by activating numerous substrates of apoptosis. Oxidative stress and calcium overload primes the outer mitochondrial membrane to become permeable, resulting in the release of pro-apoptotic factors such as cytochrome c triggering apoptosis (Vakifahmetoglu-Norberg et al., 2017). Impairment of homeostatic calcium regulation results in the release of pro-apoptotic factors. Overload of calcium either leads to necrosis or opens a mega channel, the mitochondrial permeability transition pore (mPTP), which disrupts mitochondrial function (Vakifahmetoglu-Norberg et al., 2017). The mPTP is defined as an abrupt loss of mitochondrial membrane potential due to Ca2+ overload leading to an increase in membrane permeability to molecules larger than 1.5 kDa (Hunter & Haworth, 1979). The formation of mPTP is also catalysed by increased oxidative stress in addition to increased matrix Ca2+ levels, which uncouples oxidative phosphorylation by dissipating the membrane potential (Crompton et al., 1999).
The Ca2+ overload-induced opening of the mPTP, coupled with aging, results in the release of cytochrome c (Petrosillo et al., 2006) and other proteins from mitochondria to the cytosol. This eventually triggers the apoptotic cascade, ultimately leading to programmed cell death or necrosis. The role of mitochondria in regulating cell death is thus evident.
As the mitochondria play crucial roles in breaking down nutrients, providing energy for the cells, and driving cell death pathways, deviance from such physiological states of the mitochondria thus increases the vulnerability to pathogenic conditions. Given the functions of the mitochondria and the involvement of numerous components, mitochondrial dysfunction takes on many forms. Changes in mitochondrial morphology, mutations or dysfunction in the fusion/fission proteins of mitochondria, impairment of mitochondrial complexes, reduction in coupling efficiency, and decline in mitochondrial numbers are some examples. These mitochondrial dysfunctions essentially lead to functional changes such as metabolic disorders and neurodegeneration.
Mitochondrial dysfunction in neurodegenerative conditions
Mitochondrial dysfunctions are implicated in numerous pathological conditions. However, their role in neurodegenerative diseases is more prominent perhaps due to the high energy demands of the brain. While there are many other contributing factors, mitochondrial dysfunction seems to be an overlapping factor in all if not most neurodegenerative conditions. In fact, it seems to play a central role in these pathological conditions (Figure 3). The heterogeneity in how the various types of neurodegenerative disease manifest could perhaps stem from the different types of mitochondrial dysfunction that exist. The role of mPTP has been observed to trigger apoptotic and necrotic pathways of cell death in several studies (Di et al., 2005; Grimm et al., 2007; Ott et al., 2007). However, the exact mechanism of mPTP opening and the core components of mPTP involved in neurodegenerative conditions remain unclear. However, several factors seem to contribute to the formation of mPTP, such as impaired Ca2+ homeostasis and increased oxidative stress conditions (Dirks et al., 2002). For instance, mitochondrial matrix-localised cyclophilin D (Cyp-D) and inner mitochondrial membrane-localised adenine nucleotide translocase (ANT) were proposed to be essential for mPTP formation (Halestrap et al., 2002, Zoratti et al., 2005, Gutierrez-Aguilar et al., 2015). In addition, other factors including high phosphate and peroxidation of cardiolipin conditions were observed to modulate the mPTP (Crompton et al., 1999, Leung et al., 2008). Some controversies do exist where, for example, initial studies proposed the voltage-dependent anion channel (VDAC) as a component of the mPTP (Szabó et al 1993), but subsequent gene knock-out studies ruled out its role in mPTP formation (Baines et al., 2007).
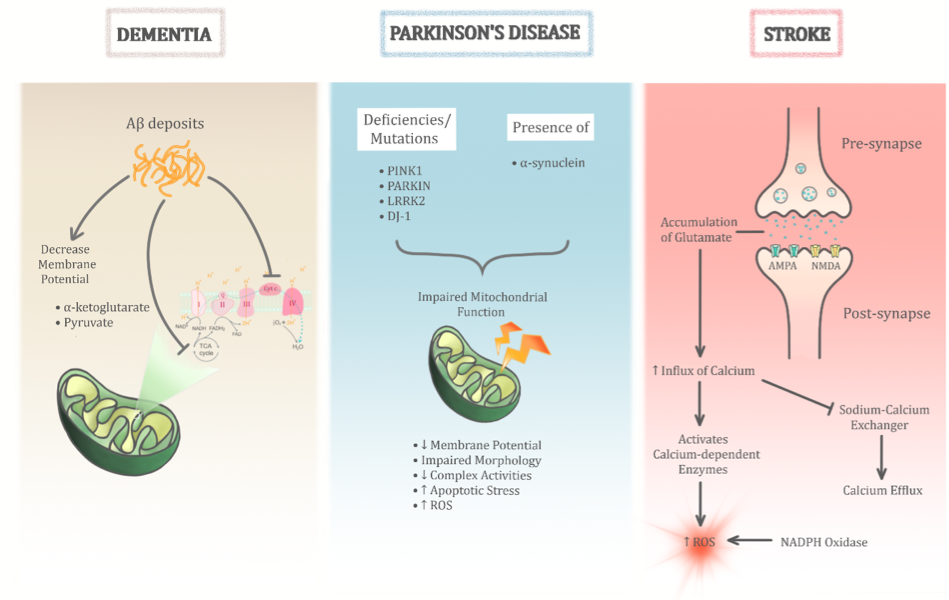
In a new window | Download PPT
Figure 3: The role of mitochondrial dysfunction in age-related neurodegenerative diseases is significant. Its heterogenous role is evident through three main age-related neurodegenerative diseases as illustrated above (dementia, Parkinson’s disease, and stroke). Despite sharing common pathological insults to the mitochondria, the permutations of these different pathways help differentiate between the different diseases.
mPTP mediated cell death plays a major role in the pathophysiology of age related diseases. Increased mPTP formation has been observed in a variety of aged cells such as lymphocytes, neurons, hepatocytes, and cardiomyocytes (Goodell et al., 1998, Mather et al., 2000, Jahangir et al., 2001, Mather et al., 2002). In addition, significant expression of Cyp-D expression (Du et al., 2008), reduced threshold to Ca2+ overload, and mitochondrial oxidative stress in affected brain were observed (Gandhi et al., 2009). Ca2+ mediated mPTP formation in aging brain (Mather et al., 2000) and heart tissues (Petrosillo et al., 2010) suggests its potential role in neurodegenerative diseases associated with aging.
Dementia
Dementia is the world’s most prevalent neurodegenerative disease, affecting over 50 million people worldwide as of 2018 (World Alzheimer Report, 2018). This number is expected to triple by 2050. The cost to treat dementia is expected to double to 2 trillion U.S. dollars by 2030. Dementia is an umbrella term that describes a group of symptoms associated with a decline in cognitive function and eventual memory loss (Prince et al., 2013). Clinically, the common causes of dementia are AD, vascular dementia (VaD), lewy body disease, and frontotemporal dementia (Prince et al., 2013), in order of their prevalence. Around 70% of dementia cases arise from AD followed by about 20% due to VaD (Korolev, 2014), which underscores the reason researchers focus on these two domains to find a cure for dementia.
AD is characterised by an accumulation of amyloid-β (Aβ) plaques, formation of neurofibrillary tangles (NFT), and clinically by cognitive decline (Patrick, et al., 2017). At the cellular level, there is a loss of cortical neurons and pyramidal cells. Despite the Aβ and tau hypotheses, which are deemed to be the hallmarks of AD, there exists an oxidative stress hypothesis as well. It states that the neuronal dysfunction in AD is attributed to free radical production from dysfunctional ETC (Mohandas et al., 2009). This hypothesis stemmed from the observed structural abnormalities in mitochondria (Hirai et al., 2001), early impairment in glucose usage (Blass et al., 2002), and defects in mitochondrial ETC (Changon et al., 1995). More recently, mitochondrial dysfunction has been shown to play a key role in the early stage of AD pathogenesis (Su et al., 2008). The exact roles of these mitochondrial dysfunctions remain diverse, with the types of mitochondrial abnormalities existing in different forms in AD. For instance, Aβ has been proposed to dampen mitochondrial respiratory function possibly by inhibiting Complex IV activity as well as by enhancing ROS production (Kish et al., 1992). More specifically, an increased intracellular Aβ load has been shown to lead to declining mitochondrial membrane potentials and ATP levels (Hauptmann et al., 2009). With these observations being made under the presence of intracellular and not that of extracellular Aβ deposits, it suggests a role for mitochondrial dysfunction in the early stages of AD. Interestingly, in addition to the role of oxidative phosphorylation at complex IV, defects in citric acid cycle enzymes such as isocitrate dehydrogenase (ICDH), pyruvate dehydrogenase (PDH), and α-ketoglutarate dehydrogenase (alpha-KGDH) have also been implicated in AD patients (Sorbi et al., 1983). These enzymes, which are involved in the TCA cycle in the matrix of the mitochondria, are key contributors of oxidative phosphorylation, and thus their deficiency results in pathogenic conditions. Changes in mitochondrial morphology and distribution have also been noted in AD fibroblasts. These have been associated with decreased levels of dynamin-like protein 1, which are regulators of mitochondrial fission and distribution (Moreira et al., 2010). Furthermore, in AD, increased mtDNA and proteins were observed in vacuoles related to lipofuscin (Hirai et al., 2001). These mitochondria-derived lysosomes and lipofuscin were found to be present in different sizes and density (Zhu et al., 2004). Lipofuscin, a lysosome involved in autophagy is thought to be a means for clearance of damaged mitochondria (Hirai et al., 2001), hence its associated increase.
Aβ also has the ability to elevate levels of intracellular Ca2+ and free radicals, indirectly contributing to mPTP formation. Aβ was also shown to trigger mitochondrial swelling, loss of transmembrane potential, and finally release of cytochrome c, indicating its direct role in regulating mPTP formation (Moreira et al., 2001). The translocation of Cyp-D affects the ability of mitochondria to buffer Ca2+ when treated with Aβ. Aβ is known to trigger mPTP formation along with oxidative stress in mitochondria (Shevtzova et al., 2001) and exhibited mitochondrial swelling in the presence of mPTP inducers (Du et al., 2008). These findings indicate that Aβ plays a vital role in mPTP formation. While it is clear that the mPTP plays a crucial role in age-associated neurodegenerative diseases like AD, several studies examined mPTP components as potential therapeutic targets for AD. Neuronal protection against oxidative stress and Aβ induced damages were effectively achieved by genetic diminution and inhibition of components associated with mPTP formation (Du et al., 2008). Indentifying and understanding the complexity of the cascading events that regulate mPTP formation in aging associated disorders, could potentially lead to targeted drug therapeutics.
While the prevalence of dementia due to VaD is less than that of AD, it is still of significance, as it is often associated with a higher mortality rate than AD (Fitzpatrick et al., 2005). Moreover, VaD shares most of the myriad of mitochondrial dysfunctions mentioned above with AD. In fact, oxidative stress serves as a common pathology between AD and VaD, given the increase in evidence of its role in VaD found in atherosclerotic pathologies (Bennett et al., 2009). VaD patients were found to have systemic upregulation of oxidative stress (Bennett et al., 2009). In addition, another study identified a decline in PDH protein levels and cytochrome c oxidase, which was associated with increased oxidative stress and decreased cellular respiration, respectively in VaD (Du et al., 2013). Essentially, both AD and VaD, which are common causes of dementia, incur defects in mitochondrial bioenergetics contributing to its dysfunctional state.
Parkinson’s Disease
PD is the most common progressive neurodegenerative movement disorder that ultimately leads to decline of motor coordination. Currently, PD affects more than 10 million people worldwide (Abdullah et al., 2014). It is characterised by the loss of dopaminergic neurons (DA) in the substantia nigra pars compacta (SNpc) of the midbrain. The loss of such neurons results in a deficiency of dopamine, a major neurotransmitter involved in the regulation of voluntary movement. This loss results in the primary motor symptoms of PD, which includes resting tremor, bradykinesia, muscle rigidity, and postural instability (Dickson, 2012).
With the mechanisms of PD yet to be elucidated, the pathogenesis remains multifactorial, ranging from genetic to environmental factors. Different hypotheses were proposed to explain the possible mechanisms behind PD. Once such hypothesis is the medical hypothesis where patients with PD experience rapid eye movement sleep with motor behaviours that correlate with the accumulation of Lewy bodies in their brain (Arnulf et al., 2000). Braaks’ hypothesis, on the other hand, postulates that sporadic PD begins in the gut due to the presence of an unknown pathogen, which then spreads to the brain (Rietdijk et al., 2017). Thereafter, the dual-hit hypothesis of PD surfaced where PD was posited to be initiated in two sets of neurons, in the nasal cavity and in the gut (Rietdijk et al., 2017). Several other hypotheses exists, such as the Prion hypothesis, protein propagation hypothesis, and spiral inflammation hypothesis. These vastly distinct hypotheses arise from the ambiguity of the possible pathogenesis of PD.
Out of the many widely discussed hypotheses regarding the etiology of PD, mitochondrial dysfunction is an important one which needs to be further explored. The mitochondrial cascade hypothesis places mitochondrial dysfunction as a central player in PD. The observation that 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine (MPTP), an inhibitor of complex I in the ETC of mitochondria, induces PD-like symptoms (Langston et al., 1983) supports the mitochondrial cascade hypothesis. This was the first piece of evidence that inducing damage to mitochondria caused in vivo phenotypes resembling PD. Since then, extensive research has been carried out supporting the pivotal role of mitochondrial dysfunction in PD. For instance, mitochondrial complex activities were found to be significantly reduced in the SNpc region of PD patients resulting in increased oxidative stress (Winklhofer & Haass, 2010). Due to DNA repair mechanisms being less efficient in mtDNA, the mitochondria are more susceptible to mutations. The more common forms of mutations in mitochondria observed in PD patients include those in PTEN-induced putative kinase (PINK1), Parkin, DJ-1, Leucine-rich repeat kinase (LRRK2) and α-synuclein.
PINK1 contains an N-terminal mitochondrial targeting sequence that confers neuroprotective activity from cellular stress induced death. Hence, mutations in PINK1 impairs mitochondrial membrane potential, mitochondrial morphology, complex activities, and eventually increases apoptotic stress (Winklhofer & Haass, 2010). Parkin is a cytosolic E3 ubiquitin ligase associated to the other mitochondrial membrane that facilitates proteasomal degradation via ubiquitination. Therefore, deficiency of Parkin results in adverse effects to the mitochondria (Park et al., 2018), which includes effects on mitochondrial transcription, replication, and morphology in terms of cristae structure, and swelling of mitochondria. These negative effects lead to increased sensitivity to oxidative stress and ultimately to loss of neurons (Banerjee et al., 2009). DJ-1 is known for its protective function in the mitochondria and its deficiency subjects the mitochondria to oxidative stress-induced damage (Winklhofer & Haass, 2010). Although DJ-1 is known to function as a redox sensor, the exact mechanism by which it mediates antioxidant activities are unclear, and its mutations are rather rare (Banerjee et al., 2009). LRRK2 is known to bind to the outer mitochondrial membrane and its mutations are the most common causes of familial PD (Winklhofer & Haass, 2010). However, LRRK2’s role in mitochondrial integrity, and its physiological and pathological mechanisms remain unknown. α-Synuclein is a major constituent of Lewy bodies, which is known to be involved in sporadic PD through mitochondrial dysfunction and protein aggregation (Banerjee et al., 2009). α-Synuclein is present in abundance in the nervous system and its presence and binding to the mitochondria implies deviance from the physiological state. Cytosolic acidification drives binding of α-synuclein to mitochondria, which has been found to downregulate complex I activity, consequently increasing oxidative stress (Winklhofer & Haass, 2010). While the mechanisms on how exactly mitochondrial dysfunction occurs in PD is unclear, the evidence regarding the diverse roles of mitochondrial dysfunction in PD pathology affirms its significant involvement (Franco-Iborra et al., 2015).
Stroke
Stroke disrupts blood flow in the cerebral artery of the brain (Zheng et al., 2003), and is the leading cause of serious, long-term disability worldwide. Patients with stroke experience symptoms like slurred speech, vision impairment, facial numbness, and hemiparesis (Yew & Cheng, 2009). Stroke ranks as the second most common cause of death and third in causing disability worldwide (Bennett et al., 2014). Individuals become more vulnerable to stroke as they age, and the incidence doubles every ten years after the age of 55, emphasising the gravity of the issue (Chong & Sacco, 2005). The two major types of stroke are ischemic stroke and hemorrhagic stroke. Ischemic stroke involves clots, either cerebral thrombosis or cerebral embolism, obstructing the blood vessels to the brain, thereby reducing the blood supply. Hemorrhagic stroke, on the other hand, occurs when the weakened blood vessels rupture (American Stroke Association, 2018). The major risk factors and the possible triggers for stroke have been postulated based on large scale studies. Some of the modifiable risk factors include hypertension, diabetes, smoking, and hypercholesterolemia. These risk factors are believed to affect the structure and function of blood vessels, alter the vasculature, and alter the regulation of cerebral blood flow, thus facilitating the occurrence of stroke (Moskowitz et al., 2010).
Mechanistically, cell death pathways and inflammatory responses play a role in mitochondrial dysfunction, and the associated oxidative stress contributes to the pathogenesis of stroke. Essentially, stroke is observed to prime the mitochondria by activating ROS-generating enzymatic systems (Moskowitz et al., 2010). Glutamate neurotoxicity is one such means of contributing to the ischemic death of neurons. The accumulation of glutamate in the extracellular region due to decreased ATP levels or impaired ion pumps prolongs the stimulation of ionotropic receptors. This increases the influx of calcium, sodium, and water into neurons. The resulting calcium overload activates calcium dependent enzymes that contributes to the production of ROS (Woodruff et al., 2011). Such excitotoxicity uncouples oxidative phosphorylation, increases ROS production, further reduces ATP, and lays out the possible path to stroke via mitochondrial dysfunction, resulting in cell death (Choi & Rothman, 1990). Influx of calcium occurs due to dysregulation of the Na+/Ca2+ exchanger that controls calcium efflux, as well as other channels and pumps, such as acid-sensing ion channels and transient receptor potential channels (Moskowitz et al., 2010). Furthermore, ROS is not only generated by mitochondria but there is also contribution from the plasma membrane associated NADPH oxidase (Moskowitz et al., 2010). Hence, such oxidative stress, especially that of vascular ROS, is induced by risk factors for stroke, serving as a potential mechanistic explanation underlying the pathogenesis of stroke.
Fixing the batteries of the cell: improving mitochondrial quality and function
In light of the wide-ranging effects of aging and the associated neurodegenerative diseases on mitochondrial dysfunction, negative conditioning thus surfaces as a solution to tackle the problem (Figure 4). Despite the fact that the mechanism of action of neurodegenerative conditions on mitochondrial dysfunction remains to be elucidated, the possible mechanism(s) and the potential key molecules involved have been narrowed, and could lead to new avenues for therapeutic intervention to improve mitochondrial quality and function.
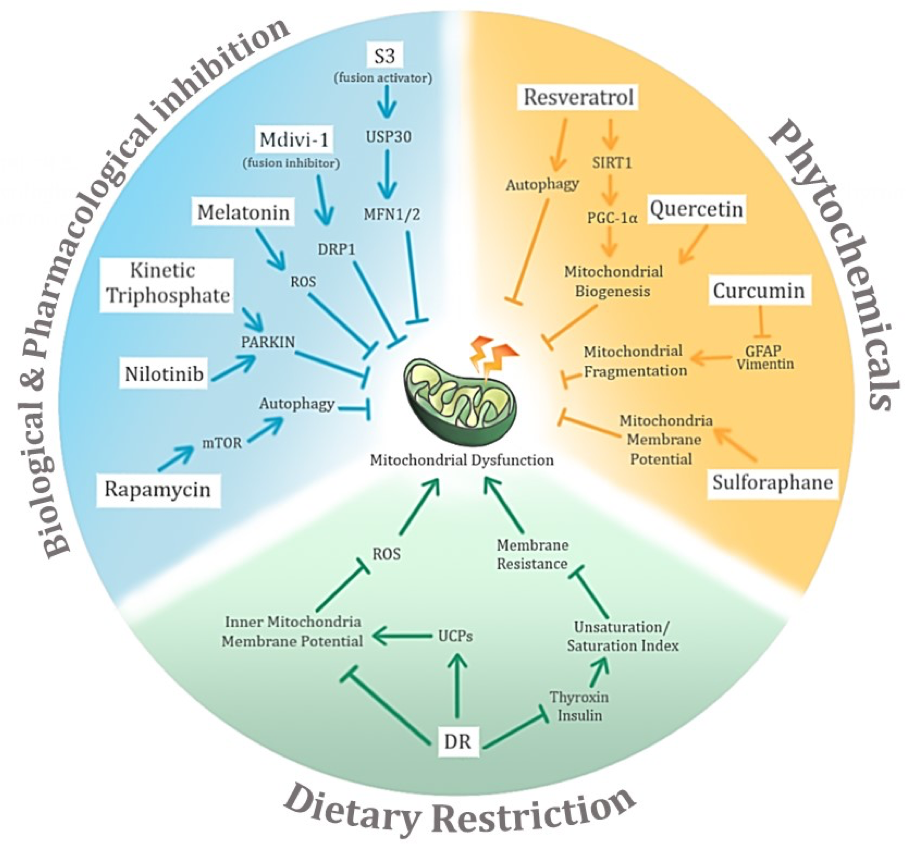
In a new window | Download PPT
Figure 4: Potential avenues of negative conditioning of mitochondrial dysfunction in age-related neurodegenerative diseases and their associated potential mechanisms of action. The three main proposed modes of negative conditioning are biological and pharmacological inhibition, use of phytochemicals, and dietary restriction.
(1) Biochemical and Pharmacological interventions
Molecular evidence of mitochondrial dysfunctions opens up possibilities for targeting specific molecules or complexes for biochemical or pharmacological therapeutic interventions. Given the neuroprotective function conferred by Parkin and PINK1, their deficiencies could be targeted to restore mitochondrial function in PD patients. For instance, nilotinib, a c-Abl tyrosine kinase inhibitor that is able to cross the blood brain barrier, can be used to increase Parkin levels (Karuppagounder et al., 2014) and Parkin recruitment could also be increased by upregulating mutant PINK1 activity via kinetin triphosphate, an ATP analogue (Hertz et al., 2013). Rapamycin is well-known to specifically inhibit the mammalian target of rapamycin (mTOR), which is a master regulator of growth and metabolism (Li et al., 2014). The therapeutic potential of rapamycin against neurodegenerative disorders lies in its ability to reduce injury by activating autophagy (Li et al., 2014). Experimental evidence has shown that rapamycin reduced mitochondrial dysfunction after cerebral ischemia and this reduction was linked to significantly upregulated mitophagy (Li et al., 2014). Melatonin, a hormone produced by the pineal gland, which regulates wakefulness, is postulated to protect mitochondria by acting as an antioxidant, ensuring the homeostatic state of mitochondrial functions and its membrane potential (Tan et al., 2016). Melatonin seems to regulate the morphology of the mitochondria as well, downregulating fission and upregulating fusion activity. Moreover, given that some of the main mitochondrial fusion and fission proteins are known, it is possible to target them directly. As discussed previously, upregulating fusion activity protects against stress-induced cell death. Hence, biochemical and pharmacological interventions have been designed to inhibit fission and upregulate fusion activity. For example fission inhibitors like Mdivi-1 have been developed to inhibit DRP1 (Cassidy-Stone, 2008). Small peptides like P110 are used to inhibit binding of DRP1 to its outer membrane receptor blocking the Drp1/Fis1 interaction (Qi et al., 2013). The activity of the fusion protein MFN1/2 has been experimentally improved by 15-oxospiramilactone (S3), which acts by inhibiting the deubiquitinase USP30 (Yue et al., 2014).
(2) Phytochemicals
Recently, researchers have looked at phytochemicals, natural compounds of vegetal origin, as a potential means of therapy. This approach is perceived to be closer to ‘natural’ treatment since the compounds are consumed in the diet, occur at physiological concentrations, or are known as traditional medicine (Gibellini et al., 2015). Notably, resveratrol, curcumin, quercetin, and sulforaphane are phytochemicals with the ability to contribute to negative conditioning of mitochondrial dysfunction. They do so by altering mitochondrial function and processes.
Resveratrol is naturally produced by plants when subjected to stress or lesion, but is also present in fruits (Gibellini et al., 2015). It is known to contribute to longevity and to improve mitochondrial function as well. Resveratrol has been shown to activate autophagy, which is otherwise impaired under oxidative stress, thus conferring protection against mitochondrial dysfunction (Zhang et al., 2017). In another study, resveratrol is postulated to induce peroxisome proliferator–activated receptor-γ coactivator-1α (PGC-1α), the central inducer of mitochondrial biogenesis, through activation of Sirtuin 1 (Lagouge et al., 2006). The protective role of resveratrol in preventing mitochondrial dysfunction is thus evident from these experiments. Curcumin is obtained from Curcuma longa rhizome. Curcumin has significant medicinal potential as it has been shown to inhibit cancer growth through inhibition of cell proliferation and tumorigenesis (Gibellini et al., 2015). A study by Daverey and Agrawal shows that curcumin is able to inhibit mitochondrial fragmentation under stress conditions, reverse the oxidative-stress induced damage to the mitochondria, and protect against apoptosis by inhibiting the action of glial fibrillary acidic protein (GFAP) and vimentin (Daverey & Agrawal, 2016). Moreover, other studies affirm the role of curcumin in restoring mitochondrial function and its ability to prevent dysfunction from occurring (Eckert et al., 2013; Soto-Urquieta et al., 2014). Quercetin is one of the major dietary flavonoids found in vegetables, fruits, and seeds (Gibellini et al., 2015). It has been reported to exert neuroprotective effects by improving complex I activity under pathogenic conditions. Experimentally, quercetin has been shown to stimulate mitochondrial biogenesis and to upregulate mitochondrial bioenergetics, thus conferring protection to neurons (Ay et al., 2017). Sulforaphane is an isothiocyanate released from glucosynolates of cruciferous vegetables by myrosinase (Grabacka et al., 2014). Research shows that sulforaphane impairs mitochondrial membrane potential, inhibits cell proliferation, and causes apoptosis in cancer cells, but contrarily restores mitochondrial function in normal cells after stress conditions. More specifically, the mitochondrial membrane potential is restored by sulforaphane in normal cells subjected to trauma or stress (Grabacka et al., 2014).
(3) Dietary restriction
Dietary energy restriction (DR) by daily calorie reduction (CR) or intermittent fasting (IF) has been shown to extend lifespan and health span in various animal models (Manzanero et al., 2011; Mercken et al., 2012). In addition, both CR and IF protect against age-related cardiovascular diseases and neurodegenerative diseases (Mercken et al., 2012; Wahl., 2016; Fann et al., 2017). Under CR, ROS generation has been observed to decrease especially at the liver and heart mitochondrial complex I in several studies (reviewed by Merry, 2002 and Manzanero et al., 2011). Such finding sheds light on how decreasing ROS can reduce disease occurrence. In an attempt to elucidate the molecular mechanism involved, numerous hypotheses have been put forth to explain how CR reduces ROS. One such hypothesis is that lowering the inner mitochondrial membrane potential along with the associated proton leak, may lead to a reduction in ROS generation (Merry, 2002; Sreekumar et al., 2002; Hagopian et al., 2005; López-Lluch et al., 2006). Due to reduced plasma concentration of hormones like thyroxin (T4) and insulin by CR, loss of double bonds in the membrane phospholipids is induced, resulting in a decline in the unsaturation/saturation index in several animal models tested (Merry, 2002; Fontana et al., 2010). Such reduction increases membrane resistance to peroxidation injury thus lowering oxidative damage. Another potential explanation involves the increased activity of uncoupling proteins (UCPs) induced by CR. However, uncoupling the proton motive force via uncoupling proteins (UCP) such as UCP1 has also been shown to reduce ROS production (Oelkrug et al., 2010).
The time period of CR also seems to have a heterogeneous effect on mitochondrial dysfunction. While CR is shown to induce mitochondria biogenesis, and hence bioenergetic efficiency (López-Lluch et al., 2006), prolonged CR improved mitochondrial function independent of mitochondrial biogenesis (Lanza et al., 2012). Hence, mounting evidence highlight the crucial role of reducing ROS production to extend healthy lifespan, and the central role of CR in doing so despite the varied proposed mechanisms.
The caveat of aging
Aging, commonly accepted as a natural phenomenon, causes changes at all levels from molecules to morphology (Peters, 2005). The decline in function due to these changes eventually leads to organ failure and subsequent death. The process of aging involves a myriad of pathways and mechanisms that deviates from physiological function. It is therefore important to acknowledge the possible influence of aging when potential negative conditioning strategies as above are devised. Theoretically, negative conditioning measures should yield beneficial effects as it addresses the problem directly. However, often a negative baseline or a sub-optimal response is returned, and this could possibly be due to the influence of aging. For instance, resveratrol is known to have beneficial anti-aging effects where it reduces oxidative stress and has anti-inflammatory properties (Li et al., 2017). However, while evidence exists, in isolation or in different models, that resveratrol can be a potential therapeutic intervention for age-related diseases, it is often not the case. The complexity of aging and its wide-ranging effects compromises the effect of pre-conditioning stimuli. It is therefore equally important to address the negative effects of age when examining potential treatment options.
Conclusion
With increasing healthcare and research innovations over the years, neurodegenerative diseases have become more prevalent as the lifespan has extended. Essentially, aging and associated neurodegenerative diseases are a result of numerous pleiotropic changes that lead to downstream effects, such as that on mitochondrial function. While numerous unresolved questions persist about the mechanistic link between neurodegenerative diseases and mitochondrial dysfunction, the fact that mitochondrial dysfunction plays a crucial role in the pathogenesis is clear. Mitochondrial dysfunction is a wide-ranging phenomenon that is triggered by a cohort of molecules, often incurring damage via multiple pathways. Despite decades of research on neurodegenerative diseases, treatment options remain purely symptomatic due to the unknown etiology. Given the common role played by mitochondrial dysfunction in neurodegenerative conditions, it provides a potential avenue for effective therapeutic intervention, and hopefully a platform for early intervention, providing a solution to this “age-old” problem.
References
Sharmelee Selvaraji
1Department of Physiology, Yong Loo Lin School Medicine, National University of Singapore, Singapore.
2NUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, Singapore.
Luting Poh
1Department of Physiology, Yong Loo Lin School Medicine, National University of Singapore, Singapore.
Venkateswaran Natarajan
1Department of Physiology, Yong Loo Lin School Medicine, National University of Singapore, Singapore.
Karthik Mallilankaraman
1Department of Physiology, Yong Loo Lin School Medicine, National University of Singapore, Singapore.
Thiruma V. Arumugam
1Department of Physiology, Yong Loo Lin School Medicine, National University of Singapore, Singapore.
3School of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea.
4Neurobiology/Ageing Programme, Life Sciences Institute, National University of Singapore, Singapore.
Corresponding author:
Thiruma V. Arumugam
Email: phstva@nus.edu.sg
In a new window | Download PPT
Figure 1: An illustration of the correlation between age and the occurrence of neurodegenerative diseases. With increasing age, the incidence of neurodegenerative diseases is posited to increase almost exponentially.
In a new window | Download PPT
Figure 2: Mitochondrion, a dynamic organelle, has several important functions in its physiological state. Apart from its primary role in regulating the bioenergetics of the cell through the electron transport chain, it also regulates the fusion-fission cycles, calcium levels and cell death.
In a new window | Download PPT
Figure 3: The role of mitochondrial dysfunction in age-related neurodegenerative diseases is significant. Its heterogenous role is evident through three main age-related neurodegenerative diseases as illustrated above (dementia, Parkinson’s disease, and stroke). Despite sharing common pathological insults to the mitochondria, the permutations of these different pathways help differentiate between the different diseases.
In a new window | Download PPT
Figure 4: Potential avenues of negative conditioning of mitochondrial dysfunction in age-related neurodegenerative diseases and their associated potential mechanisms of action. The three main proposed modes of negative conditioning are biological and pharmacological inhibition, use of phytochemicals, and dietary restriction.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13997 | 54 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA