Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Cardioprotection with Ischemic Conditioning: The Diabetes Dilemma
Time:2019-03-02
Number:9555
Author Affiliations

Conditioning Medicine, 2019. 2(1):10-17.
Abstract
Ischemic conditioning paradigms used to reduce infarct size are largely based on data obtained from preclinical models that are devoid of the risk factors and comorbidities typically seen in patients with coronary artery disease. In this review, we focus on diabetes mellitus, an established risk factor for cardiovascular disease, and summarize our current understanding of the impact of type-2 and type-1 diabetes on conditioning-induced cardioprotection.
Keywords: Ischemic preconditioning, postconditioning, remote conditioning, diabetes, heart, infarct size
Abstract
Ischemic conditioning paradigms used to reduce infarct size are largely based on data obtained from preclinical models that are devoid of the risk factors and comorbidities typically seen in patients with coronary artery disease. In this review, we focus on diabetes mellitus, an established risk factor for cardiovascular disease, and summarize our current understanding of the impact of type-2 and type-1 diabetes on conditioning-induced cardioprotection.
Keywords: Ischemic preconditioning, postconditioning, remote conditioning, diabetes, heart, infarct size
Introduction
A wealth of preclinical evidence has established that ischemic conditioning – encompassing the phenomena of preconditioning, postconditioning, and remote conditioning – is profoundly cardioprotective, evoking a significant infarct-sparing effect in models ranging from cardiomyocytes in culture to isolated buffer-perfused hearts to in vivo models of myocardial ischemia-reperfusion (I-R) (Hausenloy et al., 2016; Przyklenk, 2013). These data have provided the impetus for the launch of Phase II and Phase III trials that seek to translate the concept of conditioning-induced cardioprotection to patients with cardiovascular disease (Heusch, 2013; Heusch et al., 2016; Ovize et al., 2013). However, in contrast to the consensus among preclinical studies that ischemic conditioning reduces infarct size, the outcomes of clinical trials completed to date have been variable (ranging from positive to neutral to deleterious). Thus progress toward clinical translation has aptly been described as “somewhere between frustrating and disappointing” (Schevchuck et al., 2013).
Various explanations have been raised to explain the apparent incongruity between the efficacy of ischemic conditioning in preclinical and clinical studies (Garratt et al., 2016; Heusch et al., 2016; Heusch et al., 2017; Przyklenk et al., 2017). However, one issue that we believe merits scrutiny and discussion is the clinical relevance of the preclinical models that have been utilized. In this regard, it is noteworthy that the overwhelming majority of preclinical studies have used healthy juvenile or adult animal cohorts that are devoid of the constellation of clinically relevant risk factors and comorbidities seen in patients with coronary artery disease (Ferdinandy et al., 2014; Heusch, 2017; McCafferty et al., 2014; Miki et al., 2012; Przyklenk, 2011; Przyklenk, 2013; Przyklenk, 2015). The importance of this issue extends beyond simple choices in study design: i.e., there is a growing body of literature suggesting that these risk factors and comorbidities (including, most notably, diabetes, aging, hypertension, and hypercholesterolemia) are accompanied by dysregulation of multiple components of the signal transduction pathways that play requisite mechanistic roles in reducing infarct size with ischemic conditioning (Ferdinandy et al., 2014; Pipicz et al., 2018; Przyklenk, 2011; Przyklenk, 2013; Przyklenk, 2015; Saeid et al., 2018; Varga et al., 2015).
In the current review, we focus specifically on diabetes mellitus, an established risk factor for cardiovascular disease that affects ~8.5% of adults worldwide and is associated with a significant ~3-fold higher risk of acute myocardial infarction (WHO, 2018; Wider et al., 2014). Our goal is to provide a state-of-the-art summary of our present understanding of the impact of type-2 and type-1 diabetes on conditioning-induced cardioprotection.
Efficacy of ischemic conditioning in animal models of diabetes: a preclinical consensus?
Studies on ischemic conditioning in the context of diabetes have used various established animal models. Type-1 diabetes has been induced in rodents, rabbits, and in large animal (canine) models by single or repeated injection of the cytotoxic glucose analogues streptozotocin and alloxan. Although the mechanisms of toxicity are distinct, diabetogenic doses of streptozotocin and alloxan cause necrosis of insulin-producing beta cells, resulting in sustained hyperglycemia and insulinopenia within 48 hours (Lenzen, 2008). For protocols that have focused on ischemic conditioning in type-2 diabetes, rodent models in which key genes have been mutated (in particular, the leptin gene or its receptor) or wild-type mice fed high-fat and high-glucose diets have largely been used. In these models, hyperglycemia is caused by insulin resistance and is associated with additional metabolic derangements (including symptoms of metabolic syndrome such as hyperlipidemia), which may affect the response of heart ischemic conditioning or ischemia-reperfusion (I-R) injury per se (Fuentes-Antras et al., 2015).
Effect of experimental diabetes on sensitivity to myocardial I-R
Infarct size and clinical outcome are decidedly worse in the type-1 and type-2 diabetic population (Alegria et al., 2007; Chowdhry et al., 2007; Frustaci et al., 2000; Go et al., 2014; Haffner et al., 1998; Krempf et al., 2010; Marso et al., 2007; Mukamal et al., 2001; Murcia et al., 2004; Zia et al., 2014). In apparent contrast, the cardiac consequences of diabetes and hyperglycemia are less clear in animal models: diabetes has been reported to exacerbate the sensitivity to I-R, and increase myocardial infarct size, to render the heart resistant to I-R injury, or to have no effect (Liu et al., 1993; Miki et al., 2012; Paulson, 1997; Wider et al., 2018). This variation in outcomes may be explained by differences in experimental conditions including: i) the duration and severity of diabetes; ii) the glycemic index; iii) the presence or absence of dyslipidemia and obesity; as well as iv) the severity and duration of ischemia (Paulson, 1997; Whittington et al., 2012).
The big picture: ischemic conditioning in preclinical models of diabetes
Despite the aforementioned variations in experimental design among models and the accompanying differences in the consequences of I-R in control cohorts, there is a growing preclinical consensus that the efficacy of ischemic conditioning (including preconditioning, postconditioning, and remote conditioning) is diminished in the setting of diabetes. Of the 32 studies published to date that have measured infarct size (the acknowledged gold standard for the assessment of cardioprotection (Botker et al., 2018; Lindsey et al., 2018), 27 (84%) reported that the infarct-sparing effect of ischemic conditioning was partially or completely attenuated in models of type-1 and type-2 diabetes (Badalzadeh et al., 2012; Bouhidel et al., 2008; Drenger et al., 2011; Fan et al., 2012; Galagudza et al., 2007; Hausenloy et al., 2013; Hjortbak et al., 2018; Ji et al., 2013; Katakam et al., 2007; Kersten et al., 2000; Kiss et al., 2014; Kristiansen et al., 2004; Lacerda et al., 2012; Liu et al., 2013; Liu et al., 1993; Liu et al., 2018; Nieszner et al., 2002; Oosterlinck et al., 2013; Potier et al., 2013; Przyklenk et al., 2011; Shi-ting, 2010; Tsang et al., 2005; Vinokur et al., 2013; Wagner et al., 2008; Wang et al., 2018; Whittington et al., 2013; Wider et al., 2018; Xue et al., 2016; Yadav et al., 2010; Zhou et al., 2017; Zhu et al., 2012; Zhu et al., 2011); summarized in Tables 1-3. Moreover, among these, only one preclinical study concluded that type-2 diabetes did not alter the efficacy of ischemic conditioning in reducing infarct size (Hjortbak et al., 2018).
Taken together, these preclinical data provide three additional and overarching insights into ischemic conditioning in diabetic models. First, the concept of an attenuation in the efficacy of ischemic conditioning was a consistent finding, irrespective of the species and model that was used (Tables 1-3), thereby suggesting that cardiac sensitivity to metabolic dysregulation is a common and consistent theme (Miki et al., 2012; Wider et al., 2014). Second, diabetes may not abrogate the infarct-sparing effect of ischemic conditioning but, rather, may increase the threshold required to achieve protection: i.e., a stronger stimulus (increased number of ischemic conditioning cycles) may be required to reach the protective threshold. For example, there are three reports that one cycle of ischemic preconditioning had no protective effect, whereas amplification to three cycles of brief I-R reduced infarct size in Goto-Kakizaki rats, a non-obese model of diabetes (Hausenloy et al., 2013; Tsang et al., 2005; Whittington et al., 2013). Finally, and perhaps not surprisingly, there is an apparent temporal or aging component to the diabetes-associated loss in efficacy of ischemic conditioning. This concept is illustrated by evidence that ischemic preconditioning effectively reduced infarct size in 3- to 8-month old Goto-Kakizaki rats but had no benefit after 12 months of age (Whittington et al., 2013). Furthermore, in the small number of studies that reported a persistent benefit of ischemic conditioning in the setting of diabetes, all utilized animals in the very early stage of type-1 diabetes; i.e., diabetic cohorts had been injected with streptozotocin 5-10 days before the ischemic event (Lacerda et al., 2012; Zhu et al., 2011). These results provide further support for the concept that the effect of diabetes on infarct size reduction with ischemic conditioning is dependent on the duration of the disease.
A deeper dive: effect of diabetes on preconditioning-, postconditioning-, and remote preconditioning-induced infarct size reduction
Ischemic preconditioning is the archetype among the conditioning paradigms and thus, is the benchmark and gold standard of conditioning-induced cardioprotection. Of the 15 studies that assessed the efficacy of ischemic preconditioning in diabetic models, 13 (87%) concluded that the infarct-sparing effect of ischemic preconditioning is attenuated or eliminated: (Galagudza et al., 2007; Hausenloy et al., 2013; Hjortbak et al., 2018; Ji et al., 2013; Katakam et al., 2007; Kersten et al., 2000; Kristiansen et al., 2004; Liu et al., 1993; Liu et al., 2018; Nieszner et al., 2002; Shi-ting, 2010; Tsang et al., 2005; Vinokur et al., 2013; Whittington et al., 2013; Yadav et al., 2010; Zhu et al., 2011); see Table 1.
Table 1: Summary of Published Preclinical Studies: Ischemic Preconditioning
|
Author |
Year |
Species |
|
Protective? |
Mechanistic Insights? |
|
|
|
|
|
|
|
|
Type-1 diabetes * |
|
Duration |
|
|
|
|
Liu |
1993 |
Rat: Wistar |
11-12 months |
YES |
|
|
Kersten |
2000 |
Dog |
3 weeks |
NO |
|
|
|
|
|
|
|
|
|
Nieszner |
2002 |
Rabbit: New Zealand White |
4-5 weeks |
NO |
Impaired mito-K ATP channel opening |
|
Galagudza |
2007 |
Rat: Wistar |
6 weeks |
Attenuated |
|
|
Shi-Ting |
2010 |
Rat: Sprague Dawley |
4 and 8 weeks |
Attenuated |
Efficacy attenuated with increased duration of diabetes |
|
Yadav |
2010 |
Rat: Wistar |
6 weeks |
Attenuated |
Resistance to GSK-3 inhibition. |
|
Vinokur |
2013 |
Rat: Sprague Dawley |
4 weeks |
NO |
Ferritin loss after preconditioning |
|
Ji |
2013 |
Rat: Sprague Dawley |
|
NO |
Impaired Akt phosphorylation and GLUT4 translocation |
|
Liu |
2018 |
Rat: Sprague Dawley |
8 weeks |
NO |
Impaired Akt phosphorylation; overactive autophagy |
|
|
|
|
|
|
|
|
Type-2 diabetes |
|
Age |
|
|
|
|
Kristiansen |
2004 |
Rat: Zucker Fatty |
16 weeks |
NO |
|
|
Kristiansen |
2004 |
Rat: Goto-Kakizaki |
16 weeks |
NO |
|
|
Tsang |
2005 |
Rat: Goto-Kakizaki |
|
Attenuated |
Efficacy attenuated; impaired Akt phosphorylation Amplified preconditioning stimulus required to achieve protection. |
|
Katakam |
2007 |
Rat: Zucker Obese |
10-12 weeks |
NO |
Zucker Obese rats are normoglycemic. |
|
Hausenloy |
2013 |
Rat: Goto-Kakizaki |
|
Attenuated |
Efficacy attenuated; amplified preconditioning stimulus required to achieve protection. Co-administration of glimepimide restored protection, possibly by activation of Akt |
|
Whittington |
2013 |
Rat: Goto-Kakizaki |
3,8,12,18 months |
Attenuated |
Impaired Akt phosphorylation; amplified stimulus was protective in 3 and 8 month old rats; complete loss in efficacy at > 12 months |
|
Hjortbak |
2018 |
Rat: Zucker Fatty |
6, 12, 24 weeks |
YES |
|
* induced by injection of streptozotocin or alloxan
Investigations into the molecular mechanisms that may contribute to the diabetes-associated loss in efficacy of ischemic preconditioning have, in most studies, focused on possible defects in elements of the so-called reperfusion injury salvage kinase (RISK) and survival activating factor enhancement (SAFE) pathways – i.e., the two pathways, in addition to nitric oxide/protein kinase G (NO/PKG) signaling, that are considered to play pivotal roles in conditioning-induced cardioprotection (Hausenloy et al., 2016; Heusch, 2015; Przyklenk, 2013). In this regard, impaired phosphorylation of Akt was identified in both diabetic Goto-Kakizaki rats and streptozotocin-induced diabetes in Sprague Dawley rats (Ji et al., 2013; Tsang et al., 2005), whereas glycogen synthase kinase-3β (GSK-3β), the inhibition of which is involved as a distal component in the RISK pathway, is reportedly activated in diabetic myocardium (Eldar-Finkelman et al., 1999; Gross et al., 2004). Interestingly, while ischemic preconditioning was not protective in streptozotocin-induced diabetic rats, direct pharmacologic inhibition of GSK-3β reduced infarct size, supporting the concept that the defect in cardioprotective signaling is upstream of GSK-3β (Yadav et al., 2010). Finally, the loss in efficacy of ischemic preconditioning in the setting of diabetes has also been attributed to defects in mitochondrial-associated mechanisms of protection (including impaired activation of mitochondrial KATP channels and aberrant hexokinase translocation (Gurel et al., 2013; Hassouna et al., 2006; Katakam et al., 2007), as well as a diabetes-associated upregulation of autophagy (Liu et al., 2018).
Similarly, the majority of preclinical studies that have used ischemic postconditioning as the protective stimulus found that cardioprotection was lost or attenuated in diabetic models (12 of 13 studies; 92%: see Table 2). As with preconditioning, dysregulation of one or more components of the RISK, SAFE, and NO/PKG pathways in diabetic models has been implicated to contribute to the compromised cardioprotection, with defects in a diverse array of candidates, including Akt, GSK-3β, extracellular signal-regulated kinase (ERK), p70s6 kinase, and 5' adenosine monophosphate-activated protein kinase (AMPK) (Bouhidel et al., 2008; Fan et al., 2012; Liu et al., 2013; Przyklenk et al., 2011; Wagner et al., 2008), as well as signal transducer and activator of transcription 3 (STAT3) and NO synthase (Badalzadeh et al., 2012; Drenger et al., 2011; Fan et al., 2012) having been identified. An additional, novel culprit may be phosphatase and tensin homolog (PTEN), a negative regulator of phosphoinositide 3-kinase/Akt signaling. There is evidence that, in diabetic myocardium, PTEN is resistant to deactivation by ischemic postconditioning (thereby exerting a molecular brake on the upregulation of cardioprotective signaling) – a defect that purportedly can be mitigated (and cardioprotection restored) by pharmacologic inhibition of PTEN (Xue et al., 2016). Lastly, aberrations in autophagy have also been implicated. However, contrary to the aforementioned upregulation in autophagy in hearts from rats with streptozotocin-induced diabetes (Liu et al., 2018), others have concluded, using the same model, that: i) autophagy is repressed in the setting of diabetes, and ii) the infarct-sparing effect of postconditioning is re-established in response to genetic and pharmacologic upregulation of autophagy (Zhou et al., 2017). The reasons for this discrepancy are unclear, but may reflect the complexities of this still poorly understood phenomenon, particularly under pathophysiologic conditions of stress and cardioprotection (Dong et al., 2010; Przyklenk et al., 2012).
Table 2: Summary of Published Preclinical Studies: Postconditioning
|
Author |
Year |
Species |
|
Protective? |
Mechanistic Insight? |
|
|
|
|
|
|
|
|
Type-1 diabetes * |
|
Duration |
|
|
|
|
Dregner |
2011 |
Rat: Sprague Dawley |
4-5 weeks |
NO |
Inhibited p-STAT3 nuclear translocation |
|
Przyklenk |
2011 |
Mouse: C57 |
2 weeks |
NO |
Impaired ERK phosphorylation |
|
Lacerda |
2012 |
Mouse |
5, 10 days |
YES |
|
|
Fan |
2012 |
Rat: Wistar ** |
4 weeks |
NO |
Impaired eNOS, Akt phosphorylation |
|
Badalzadeh |
2012 |
Rat: Wistar |
8 weeks |
NO |
Impaired NO synthesis |
|
Liu |
2013 |
Rat: Sprague Dawley |
12 weeks |
NO |
Impaired Akt phosphorylation, increased PTEN |
|
Potier |
2013 |
Mouse: C57 |
4-5 weeks |
NO |
|
|
Xue |
2016 |
Rat: Sprague Dawley |
8 weeks |
NO |
Impaired Akt, STAT3 and GSK-3β phosphorylation; impaired inhibition of PTEN |
|
Zhou |
2017 |
Rat: Sprague Dawley |
8 weeks |
NO |
Defect in AMPK/mTOR-mediated activation of autophagy |
|
|
|
|
|
|
|
|
Type-2 diabetes |
|
Age |
|
|
|
|
Wagner |
2008 |
Rat: WOKW |
28 weeks |
NO |
Impaired ERK, GSK-3-β phosphorylation |
|
Bouhidel |
2008 |
Mouse: ob/ob |
8-10 weeks |
NO |
Impaired Akt, ERK, p70S6 kinase, AMPK phosphorylation |
|
Przyklenk |
2011 |
Mouse: db/db |
12-14 weeks |
NO |
Impaired ERK phosphorylation |
|
Zhu |
2012 |
Mouse: db/db |
10-12 weeks |
NO |
Differential expression of F1-ATPaseγ, Echs1 and HSP20 |
|
Oosterlinck |
2013 |
Mouse: ob/ob |
24 weeks |
Attenuated |
|
* induced by injection of streptozotocin; ** injection of streptozotocin + high fat diet
Remarkably, there are at present only four published studies that have investigated infarct size reduction with remote preconditioning in preclinical models of diabetes. Among these, two have reported persistent and significant cardioprotection, while the remaining two studies found a loss in efficacy (Table 3).
Table 3: Summary of Published Preclinical Studies: Remote Preconditioning
|
Author |
Year |
Species |
|
Protective? |
Mechanistic insight? |
|
|
|
|
|
|
|
|
Type-1 diabetes * |
|
Duration |
|
|
|
|
Zhu |
2011 |
Rat: Wistar |
1 week |
YES |
Maintained superoxide dismutase activation, xanthine oxidase deactivation |
|
Kiss |
2014 |
Rat: Sprague Dawley |
4-5 weeks |
NO |
Impaired eNOS phosphorylation, arginase activity, ROCK activity |
|
Wang |
2018 |
Rat: Sprague Dawley |
8 weeks |
YES |
Maintained PKC-ε deactivation; Akt, STAT3 activation |
|
|
|
|
|
|
|
|
Type-2 diabetes |
|
Age |
|
|
|
|
Wider |
2018 |
Rat: Zucker Fatty |
10-12 weeks |
NO |
Impaired humoral communication of protective signal |
* induced by injection of streptozotocin
Our group provided the first and only study to date conducted in the setting of type-2 diabetes (Wider et al., 2018), Using 10-12 week old Zucker Fatty rats (an early stage time point characterized by modest elevations in non-fasting blood glucose), our results revealed that remote preconditioning, initiated by the standard stimulus of four 5-minute episodes of bilateral hindlimb ischemia, failed to reduce infarct size (Figure 1). This loss in protection did not correlate with plasma glucose concentration, thereby suggesting that the defect was not caused by hyperglycemia per se (Wider et al., 2018). Rather, focusing on the hallmark of remote conditioning (that is, the communication of the cardioprotective signal from the site of the remote conditioning stimulus to the at-risk myocardium (Hausenloy et al., 2016; Pickard et al., 2015; Przyklenk, 2013), we found evidence that the production or transfer of humoral blood-borne protective factor(s) in response to the preconditioning stimulus was impaired in Zucker Fatty rats when compared with matched normoglycemic Zucker Lean cohorts. Specifically, serum harvested from Zucker Lean rats following hindlimb ischemia and applied to cultured HL-1 cardiomyocytes rendered the cells resistant to a subsequent episode of hypoxia-reoxygenation, whereas serum from Zucker Fatty rats either had no cytoprotective effect or, for a specific sub-fraction of serum, exacerbated HL-1 cell death (Wider et al., 2018). Despite an exploratory proteomic analysis (Wider et al., 2018), the identity of the protective humoral factor(s) released in normoglycemic rats in response to the remote preconditioning stimulus and the identity of the toxic circulating factor(s) released in the diabetic Zucker Fatty rats remain unknown. Interestingly, one previous study used a similar experimental strategy but, in this case, collected plasma from diabetic human subjects following a remote conditioning stimulus and assessed its cardioprotective efficacy in isolated buffer-perfused rabbit hearts subjected to global I-R. Diabetes was associated with a defect in the humoral transfer of a protective trigger, but this defect was limited to a subset of diabetic patients displaying peripheral neuropathy (Jensen et al., 2012) – a factor that, in all likelihood, did not contribute to our observations made using Zucker Fatty rats treated before the development of neuropathy in our model (Oltman et al., 2005).
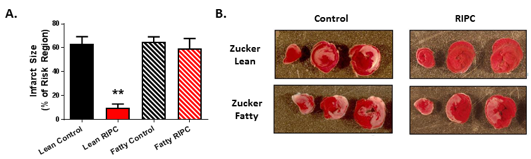
In a new window | Download PPT
Figure 1: (A) Infarct size, expressed as a % of the myocardium at risk (mean ± SEM), for Zucker Lean and Zucker Fatty rats randomized to receive remote ischemic preconditioning (RIPC) or a time-matched control period. **p<0.01 versus the Zucker Lean control group. (B) Images of heart slices obtained from one control and one RIPC-treated rat from the Zucker Lean and Zucker Fatty cohorts. Heart slices were incubated in triphenyltetrazolium chloride; using this method, viable myocardium stains red while areas of necrosis remain unstained and thus appear pale. Reprinted with permission from Wider et al., 2018.
Among the remaining three studies, all of which used the rat model of streptozotocin-induced type-1 diabetes, outcomes have been mixed. One study reported that the reduction in infarct size with remote preconditioning was diminished 4-5 weeks after induction of diabetes, citing a defect in the generation of cardioprotective NO (Kiss et al., 2014). Conversely, in rats in which remote ischemic conditioning was commenced 8 weeks after streptozotocin injection, significant protective was still observed; however, an atypical stimulus – hindlimb ischemia applied repeatedly (daily for 3 days) was used (Wang et al., 2018). These results are consistent with the concept that an amplified (in this case, repeated) conditioning stimuli may be capable of achieving a protective threshold in diabetic myocardium, either by augmenting conventional cardioprotective signaling or via mechanisms that differ from acute kinase phosphorylation. Although the paradigm of repeated remote preconditioning is a recent development and, thus, its mechanisms are not well-understood (Thijssen et al., 2016), evidence suggests that repeated stimulus influences vascular function, myocardial gene expression, circulating factors, and effectors that are distinct from the acute standard stimulus (Epps et al., 2016; Luca et al., 2013; Wang et al., 2018; Yamaguchi et al., 2015). The fourth study also reported persistent cardioprotection using an amplified preconditioning stimulus (Zhu et al., 2011), however, as animals in this latter protocol were subjected to the conditioning stimulus only 1 week after induction of diabetes, the efficacy of conditioning-induced cardioprotection may not yet have been compromised.
Clues into clinical relevance?
Taken together, and despite the substantial heterogeneity in experimental design and mechanistic endpoints among the small number of published reports, the majority of preclinical studies have concluded that the infarct-sparing effect of ischemic conditioning is attenuated or lost in genetic and drug-induced models of diabetes (Tables 1-3; Figure 1). Nonetheless, it must be acknowledged that these data were obtained in models that do not fully mimic the scope and often years-long duration of the disease in patients. Thus, the obvious question is: are the aforementioned observations of a diabetes-associated defect in conditioning-induced cardioprotection clinically relevant?
A handful of Phase II trials took the proactive step, presumably based in part on these emerging preclinical concerns, and prospectively excluded diabetic patients from enrollment (Heusch et al., 2012; Kottenberg et al., 2012; Thielmann et al., 2010; Venugopal et al., 2009) – a practice that has also been applied to studies of ischemic conditioning in other organs (Venugopal et al., 2010). In terms of more direct evidence, there are clinical data, albeit limited, that appear to corroborate the preclinical outcomes. For example, prospective subset analyses of larger Phase II studies revealed that preinfarct angina (considered a proxy for ischemic preconditioning) failed to limit infarct size (assessed by cardiac enzyme release) in the cohort with type-2 diabetes, while there was a trend toward exacerbation of infarct size with postconditioning in diabetic subjects (Ishihara et al., 2001; Yetgin et al., 2014). Similarly, remote preconditioning was reportedly ineffective in attenuating cardiac enzyme release in diabetic patients undergoing surgical Rcoronary revascularization (Kottenberg et al., 2014), whereas in the setting of elective percutaneous coronary intervention (PCI), the incidence of peri-procedural myocardial infarction was either unchanged (rather than decreased: (Xu et al., 2014) or exacerbated (Carrasco-Chinchilla et al., 2013) in patients with diabetes. Finally, as an interesting corollary in apparent support of preclinical reports that cardioprotection can be re-established by direct pharmacologic activation of key signaling elements, intracoronary administration of adenosine during PCI has been shown to act as an effective ‘conditioning-mimetic’ in diabetic patients (Shehata, 2014). However, and of potential importance: despite the aforementioned outcomes, a meta-analysis assessing the aggregate data from five trials concluded that there was no diabetes-associated loss in efficacy of remote preconditioning in patients undergoing elective PCI (D'Ascenzo et al., 2014).
Limitations, conclusions, and future directions
It could be argued that studies conducted using preclinical models of type-2 and type-1 diabetes (and, in fact, all preclinical models) are too simplistic, and will not be helpful in advancing the clinical translation of ischemic conditioning. The preclinical models clearly do not duplicate the complexities of patients with cardiovascular disease – elements of complexity that include both the presence, in some cohorts, of multiple comorbidities, as well as the potential confounding effects of the pharmacologic therapies administered as a standard of care for the clinical management of these diseases. Indeed, this latter concept is supported by evidence that anti-platelet drugs, statins, nitrates, and opiates may, in themselves, evoke significant cardioprotection and mimic or re-initiate the infarct-sparing effect of ischemic conditioning (Ferdinandy et al., 2014; Heusch, 2013; Przyklenk, 2011; Przyklenk, 2015). Moreover, there is evidence that, in surgical studies, the choice of anesthetic regimen (and, in particular, the use of propofol) can profoundly affect outcomes and conclusions regarding cardioprotection (Behmenburg et al., 2018; Garratt et al., 2016; Heusch et al., 2016; Heusch et al., 2017), most notably in diabetic populations (Ansley et al., 2016). Nonetheless, despite these limitations, the majority of studies conducted in our simplistic preclinical models of diabetes have found a loss in efficacy of conditioning-induced cardioprotection – an observation that has largely been corroborated in clinical studies and, we believe, warrants continued prospective investigation.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
Joseph M. Wider
1Departments of Emergency Medicine and Molecular & Integrative Physiology, University of Michigan Medical School, Ann Arbor, MI, USA.
Karin Przyklenk
2Cardiovascular Research Institute and Departments of Emergency Medicine and Physiology, Wayne State University School of Medicine, Detroit, MI, USA.
Corresponding author:
Karin Przyklenk
Email: karinp@wayne.edu
In a new window | Download PPT
Figure 1: (A) Infarct size, expressed as a % of the myocardium at risk (mean ± SEM), for Zucker Lean and Zucker Fatty rats randomized to receive remote ischemic preconditioning (RIPC) or a time-matched control period. **p<0.01 versus the Zucker Lean control group. (B) Images of heart slices obtained from one control and one RIPC-treated rat from the Zucker Lean and Zucker Fatty cohorts. Heart slices were incubated in triphenyltetrazolium chloride; using this method, viable myocardium stains red while areas of necrosis remain unstained and thus appear pale. Reprinted with permission from Wider et al., 2018.
Table 1: Summary of Published Preclinical Studies: Ischemic Preconditioning
|
Author |
Year |
Species |
|
Protective? |
Mechanistic Insights? |
|
|
|
|
|
|
|
|
Type-1 diabetes * |
|
Duration |
|
|
|
|
Liu |
1993 |
Rat: Wistar |
11-12 months |
YES |
|
|
Kersten |
2000 |
Dog |
3 weeks |
NO |
|
|
|
|
|
|
|
|
|
Nieszner |
2002 |
Rabbit: New Zealand White |
4-5 weeks |
NO |
Impaired mito-K ATP channel opening |
|
Galagudza |
2007 |
Rat: Wistar |
6 weeks |
Attenuated |
|
|
Shi-Ting |
2010 |
Rat: Sprague Dawley |
4 and 8 weeks |
Attenuated |
Efficacy attenuated with increased duration of diabetes |
|
Yadav |
2010 |
Rat: Wistar |
6 weeks |
Attenuated |
Resistance to GSK-3 inhibition. |
|
Vinokur |
2013 |
Rat: Sprague Dawley |
4 weeks |
NO |
Ferritin loss after preconditioning |
|
Ji |
2013 |
Rat: Sprague Dawley |
|
NO |
Impaired Akt phosphorylation and GLUT4 translocation |
|
Liu |
2018 |
Rat: Sprague Dawley |
8 weeks |
NO |
Impaired Akt phosphorylation; overactive autophagy |
|
|
|
|
|
|
|
|
Type-2 diabetes |
|
Age |
|
|
|
|
Kristiansen |
2004 |
Rat: Zucker Fatty |
16 weeks |
NO |
|
|
Kristiansen |
2004 |
Rat: Goto-Kakizaki |
16 weeks |
NO |
|
|
Tsang |
2005 |
Rat: Goto-Kakizaki |
|
Attenuated |
Efficacy attenuated; impaired Akt phosphorylation Amplified preconditioning stimulus required to achieve protection. |
|
Katakam |
2007 |
Rat: Zucker Obese |
10-12 weeks |
NO |
Zucker Obese rats are normoglycemic. |
|
Hausenloy |
2013 |
Rat: Goto-Kakizaki |
|
Attenuated |
Efficacy attenuated; amplified preconditioning stimulus required to achieve protection. Co-administration of glimepimide restored protection, possibly by activation of Akt |
|
Whittington |
2013 |
Rat: Goto-Kakizaki |
3,8,12,18 months |
Attenuated |
Impaired Akt phosphorylation; amplified stimulus was protective in 3 and 8 month old rats; complete loss in efficacy at > 12 months |
|
Hjortbak |
2018 |
Rat: Zucker Fatty |
6, 12, 24 weeks |
YES |
|
* induced by injection of streptozotocin or alloxan
Table 2: Summary of Published Preclinical Studies: Postconditioning
|
Author |
Year |
Species |
|
Protective? |
Mechanistic Insight? |
|
|
|
|
|
|
|
|
Type-1 diabetes * |
|
Duration |
|
|
|
|
Dregner |
2011 |
Rat: Sprague Dawley |
4-5 weeks |
NO |
Inhibited p-STAT3 nuclear translocation |
|
Przyklenk |
2011 |
Mouse: C57 |
2 weeks |
NO |
Impaired ERK phosphorylation |
|
Lacerda |
2012 |
Mouse |
5, 10 days |
YES |
|
|
Fan |
2012 |
Rat: Wistar ** |
4 weeks |
NO |
Impaired eNOS, Akt phosphorylation |
|
Badalzadeh |
2012 |
Rat: Wistar |
8 weeks |
NO |
Impaired NO synthesis |
|
Liu |
2013 |
Rat: Sprague Dawley |
12 weeks |
NO |
Impaired Akt phosphorylation, increased PTEN |
|
Potier |
2013 |
Mouse: C57 |
4-5 weeks |
NO |
|
|
Xue |
2016 |
Rat: Sprague Dawley |
8 weeks |
NO |
Impaired Akt, STAT3 and GSK-3β phosphorylation; impaired inhibition of PTEN |
|
Zhou |
2017 |
Rat: Sprague Dawley |
8 weeks |
NO |
Defect in AMPK/mTOR-mediated activation of autophagy |
|
|
|
|
|
|
|
|
Type-2 diabetes |
|
Age |
|
|
|
|
Wagner |
2008 |
Rat: WOKW |
28 weeks |
NO |
Impaired ERK, GSK-3-β phosphorylation |
|
Bouhidel |
2008 |
Mouse: ob/ob |
8-10 weeks |
NO |
Impaired Akt, ERK, p70S6 kinase, AMPK phosphorylation |
|
Przyklenk |
2011 |
Mouse: db/db |
12-14 weeks |
NO |
Impaired ERK phosphorylation |
|
Zhu |
2012 |
Mouse: db/db |
10-12 weeks |
NO |
Differential expression of F1-ATPaseγ, Echs1 and HSP20 |
|
Oosterlinck |
2013 |
Mouse: ob/ob |
24 weeks |
Attenuated |
|
* induced by injection of streptozotocin; ** injection of streptozotocin + high fat diet
Table 3: Summary of Published Preclinical Studies: Remote Preconditioning
|
Author |
Year |
Species |
|
Protective? |
Mechanistic insight? |
|
|
|
|
|
|
|
|
Type-1 diabetes * |
|
Duration |
|
|
|
|
Zhu |
2011 |
Rat: Wistar |
1 week |
YES |
Maintained superoxide dismutase activation, xanthine oxidase deactivation |
|
Kiss |
2014 |
Rat: Sprague Dawley |
4-5 weeks |
NO |
Impaired eNOS phosphorylation, arginase activity, ROCK activity |
|
Wang |
2018 |
Rat: Sprague Dawley |
8 weeks |
YES |
Maintained PKC-ε deactivation; Akt, STAT3 activation |
|
|
|
|
|
|
|
|
Type-2 diabetes |
|
Age |
|
|
|
|
Wider |
2018 |
Rat: Zucker Fatty |
10-12 weeks |
NO |
Impaired humoral communication of protective signal |
* induced by injection of streptozotocin
Abbreviations for Tables 1-3: AMPK =5' adenosine monophosphate-activated protein kinase; Echs1 = enoyl coenzyme A hydratate, short chain 1; eNOS
= endothelial nitric oxide synthase; ERK = extracellular signal-regulated kinase; GLUT4 = glucose transporter type 4; GSK-3β = glycogen synthase kinase-3β; HSP20 = heat shock protein 20; mito KATP = mitochondrial ATP-sensitive potassium channel; mTOR = mammalian target of rapamycin; NO = nitric oxide; PKC-ε = protein kinase C-ε; PTEN = signal transducer and activator of transcription 3; ROCK = rho-associated coiled containing protein kinase; STAT3 = signal transducer and activator of transcription 3
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 9555 | 20 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA