Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Multifactorial neuroprotection: Does the brain have an answer?
Time:2019-05-03
Number:14069
Author Affiliations

Conditioning Medicine, 2019. 2(2):75-89.
Abstract
Massive research efforts to develop effective neuroprotective therapy against stroke until now produced unsatisfactory results. It has been suggested that monotherapeutic approaches may not be sufficient. Investigations over the last three decades convincingly demonstrated the existence of powerful endogenous protective mechanisms. One of the innate protective mechanisms includes several brain structures, which when activated render the brain tolerant to various damaging stimuli. The best studied today is the cerebellar fastigial nucleus, neurons of which when activated, initiate coordinated multifactorial response providing long lasting neuroprotection. Numerous protective mechanisms induced by fastigial nucleus stimulation and other conditioning maneuvers are shared. In this review we summarize current knowledge of the neurogenic neuroprotection system related to the cerebellar fastigial nucleus and its commonalities with other forms of conditioning. Unveiling the systemic neuroprotective mechanisms will allow development of therapeutic approaches targeted toward activation/amplification of innate protective multifactorial mechanisms.
Keywords: cerebellar fastigial nucleus; neuroprotection; stroke; preconditioning
Abstract
Massive research efforts to develop effective neuroprotective therapy against stroke until now produced unsatisfactory results. It has been suggested that monotherapeutic approaches may not be sufficient. Investigations over the last three decades convincingly demonstrated the existence of powerful endogenous protective mechanisms. One of the innate protective mechanisms includes several brain structures, which when activated render the brain tolerant to various damaging stimuli. The best studied today is the cerebellar fastigial nucleus, neurons of which when activated, initiate coordinated multifactorial response providing long lasting neuroprotection. Numerous protective mechanisms induced by fastigial nucleus stimulation and other conditioning maneuvers are shared. In this review we summarize current knowledge of the neurogenic neuroprotection system related to the cerebellar fastigial nucleus and its commonalities with other forms of conditioning. Unveiling the systemic neuroprotective mechanisms will allow development of therapeutic approaches targeted toward activation/amplification of innate protective multifactorial mechanisms.
Keywords: cerebellar fastigial nucleus; neuroprotection; stroke; preconditioning
Introduction
In 2013 6.5 million people globally died of stroke and almost 25.7 million stroke survivors suffered different degrees of chronic disability (Feigin et al., 2015). In spite of massive efforts (according to PubMed just in the last 5 years, 18,956 studies on “ischemic or hemorrhagic stroke” have been published), the progress in treatment of this severe condition remains limited (Dirnagl and Endres, 2014). There is a compelling need to develop new therapeutic options to improve treatment and recovery after stroke. As stressed by Dirnagl and Endes (2014), monotherapeutic approaches seem to be non-productive while fact-based multitargeted approaches may be more fruitful. During evolution, a variety of complex mechanisms have developed to help organism to survive hostile, potentially damaging conditions. Knowledge of these mechanisms may provide us with new therapeutic tools and approaches.
Preconditioning (PC) is a naturally occurring survival mechanism. Generally, the phenomenon of PC can be defined as increased tolerance of cells, organs, and organisms to the damaging effects of strong impacts of various nature following pre-exposure to sublethal doses of insulting agents (Dirnagl et al., 2003; Dirnagl et al., 2009; Iadecola and Anrather, 2011a). Recently, significant amounts of research have been devoted to understanding the mechanisms underlying PC due to its potentially wide applications: from neurological diseases and myocardial infarction to organ transplantation. Investigators unveiled various mechanisms underlying PC (see (Dirnagl et al., 2009; Gidday, 2015; Meller and Simon, 2015; Cheng et al., 2017; Jasova et al., 2017; Lee et al., 2017; Lepiesza et al., 2017; Veighey et al., 2017; Yang et al., 2017). Pre-, peri- or postconditioning have become “catch-all” terms designating increased tolerance or decreased damage by the severe insult via pre-, peri- or post-ictal application of weaker or non-harmful action. While numerous mechanisms are involved in the conditioning phenomenon, there are numerous commonalities between its various forms. We still do not fully understand the interplay between various forms of conditioning. In this review focused mostly on neuroprotection, we provide synopsis of the major “subtypes” of PC from the cellular to organismal levels. However, special attention is devoted to so called, “neurogenic neuroprotection”, which we suggest may embody multifactorial organismal protective mechanisms, often triggered in the anticipation of adverse insult, coordinated by the nervous system and sharing numerous common features with other forms of conditioning.
Innate self-defense
Cellular self-protection. Neurons, as well as other cells, are able to mount limited defense against anoxic or other adverse conditions depending on the type and cell origin (mammals, reptiles, amphibians etc.) (Hochachka et al., 1996; Gidday, 2006; Perez-Pinzon, 2007) (Fig.1). The immediate defensive cellular response is denoted as acute PC (Perez-Pinzon, 2007), indicating fast-developing often short-lasting increases in cell tolerance to the subsequent potentially lethal stimuli (Dirnagl et al., 2009; Iadecola and Anrather, 2011a; Koch et al., 2012). An acute defensive cellular response is referred to as an immediate PC as compared to the so-called delayed PC, an effect which can last for days and weeks often requiring changes in gene expression (Dirnagl et al., 2009; Iadecola and Anrather, 2011a). The phenomenon of PC can be observed in vitro in near pure neuronal cultures indicating the existence of protective mechanisms at the single cell level, i.e. when neurons (or other cells) directly affected by sublethal insult acquire the ability to withstand the subsequent lethal insult (Meloni et al., 2002).
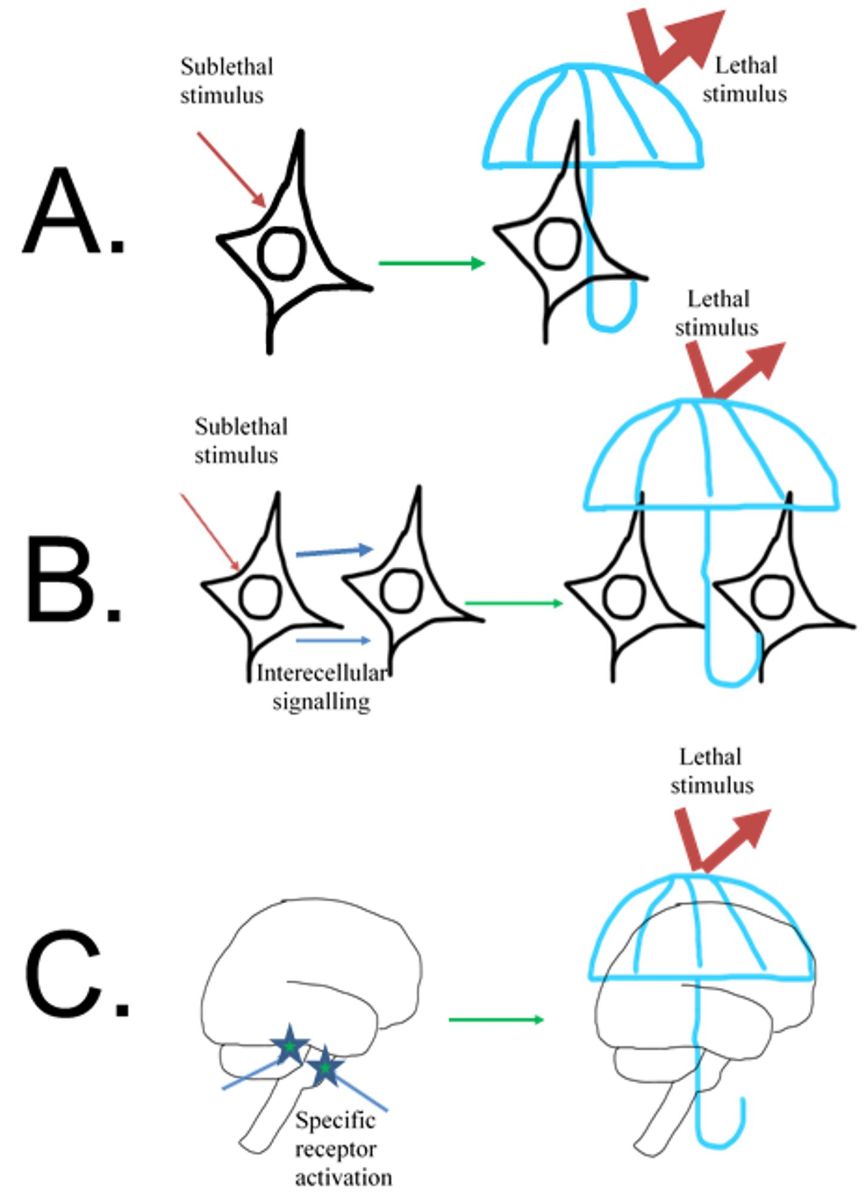
In a new window | Download PPT
Figure 1: Different levels of innate self-defense. A. Innate self-defense can be triggered in a single cell by a strong but not lethal signal. In response to cellular changes triggered by the sublethal stimulus, the cell temporarily mobilizes protective mechanisms and becomes tolerant to the stimulus of lethal strength. B. The cell can be sensitive to the potentially dangerous stimulus and not only mount its own defense but also trigger changes in a neighbor cell making both cells tolerant to the subsequent lethal strength stimulus. C. In brain specific cells, receptors can sense changes in the environment before their own or their neighbor’s metabolism becomes affected and trigger a coordinated multicomponent response, which would make the brain or the whole organism protected against lethal stimulus.
PC neuroprotection is not modality-specific and can be evoked by various potentially lethal insults (see Dirnagl et al., 2003). For example, in vitro exposure to combined oxygen-glucose deprivation (OGD) (Bruer et al., 1997; Khaspekov et al., 1998; Xu et al., 2002a), hypothermia (Yuan et al., 2004), hyperthermia (Kelty et al., 2002), excitotoxic insult (NMDA, kainate) (Pringle et al., 1999; Tremblay et al., 2000), 3-nitropropionic acid (Weih et al., 1999; Nakagawa et al., 2003), and other factors (Meloni et al., 2002) render neurons tolerant to the subsequent noxious stimuli of the same or different nature, a phenomenon known as cross-tolerance (Gidday, 2006). Acute cellular defense mechanisms are multifactorial (Bickler and Donohoe, 2002; Kirino, 2002; Dirnagl et al., 2009; Kitagawa, 2012) and involve a number of various mechanisms such as modification of mitochondrial KATP channels (Heurteaux et al., 1995; Cohen et al., 2000; McLaughlin et al., 2003), G-protein coupled E-prostanoid receptors (McCullough et al., 2004), GABA(A) receptors (Grabb et al., 2002), adenosine receptors (Heurteaux et al., 1995; Perez-Pinzon et al., 1996), caspases inactivation (McLaughlin et al., 2003), reactive oxygen species scavenging (McLaughlin et al., 2003), and protein synthesis (Gage and Stanton, 1996; Ravati et al., 2001) etc.
Inter-cellular protective mechanisms. The ability to initiate cellular protective mechanisms is not restricted to insult directed to the cell. Other cells affected by the insult can initiate or facilitate cellular protective cascades in their neighbors, e.g., neurons. In multicellular systems with different co-existing specialized cell types, mutually protective mechanisms seem to be involved. Thus lipopolysaccharide (LPS), which does not affect neurons directly (Bronstein et al., 1995), when administered in vivo induces ischemic tolerance (Bordet et al., 2000; Zimmermann et al., 2001). This indicates that in multicellular systems noxious insult does not necessarily have to act directly on neuronal cells to render them tolerant to subsequent damaging insult. Microglia activated by LPS promote neuronal survival (Zhou and Spittau, 2018) probably by converting microglia toward a prosurvival M2 phenotype (Ajmone-Cat et al., 2013). Neurons co-cultured with astrocytes, which underwent conditioning OGD, acquired tolerance to OGD (Narayanan and Perez-Pinzon, 2017). This effect seems to be mediated through glia activation and subsequent release of cytokines (Boche et al., 2003). Various mechanisms appear to be involved in the intercellular neuroprotection, such as pre-synaptic suppression of neuronal glutamate release (Tauskela et al., 2012), adenosine receptors (Yun et al., 2014), chemokines (Shin et al., 2014), thrombin's endogenous inhibitor, protease nexin-1 (PN-1)(Mirante et al., 2013), HIF-1α (Jones et al., 2013), and others (see Obrenovitch, 2008)). An important role seems to belong to mitochondria, which are considered to be a vital hub of conditioning (Pamenter, 2014; Prendes et al., 2014; Thompson et al., 2015; Silachev et al., 2016).
Neurons are highly susceptible to hypoxic/ischemic damage, while glial cells are not only more tolerant to adverse conditions, but are capable of providing neuronal support by regulating the neural environment (Xing and Lo, 2017). Neurons, in turn, are capable of regulating the activity of micro- and astroglia, preventing their inflammatory response to LPS (Bjorklund et al., 2010). The bidirectional protective interaction between neighboring cells has recently been elegantly formulated as the concept of “help me” signaling (Xing and Lo, 2017; Esposito et al., 2018), which suggests generation of “help me” signals by the “victim” cell to induce neighboring cells to activate protective mechanisms. Various molecules convey “help me” signals: danger associated molecular patterns (DAMPS), such as ATP (An et al., 2014), chemokines (Conductier et al., 2010), and others as reviewed by (Xing and Lo, 2017).
Direct and indirect PC at the organismal level. In vivo in 1990 Kitagawa and colleagues (1990) discovered that pre-exposure of the brain to short-term ischemia, which alone does not induce cell damage, significantly attenuates the subsequent effect of extended damage-inducing ischemia. Subsequent studies demonstrated that exposure of the brain to the non-lethal potentially damaging insults of various forms initiate a cascade of events rendering the brain tolerant against later application of damaging insult (see reviews by Kirino, 2002; Dirnagl et al., 2003; Perez-Pinzon, 2007; Obrenovitch, 2008; Dirnagl et al., 2009; Iadecola and Anrather, 2011a; Kitagawa, 2012; Koch et al., 2012; Dirnagl and Endres, 2014). This phenomenon became widely known as PC. Similar factors, which activate intrinsic cellular protection in neurons when applied to cells directly, are also effective when applied to the whole organism. In vivo pre-exposure to sub-lethal global (Kitagawa et al., 1990), or focal (Glazier et al., 1994; Toyoda et al., 1997) ischemia, hypoxia (Vannucci et al., 1998; Bernaudin et al., 2002), 3-nitropropionic acid (Sugino et al., 1999; Weih et al., 1999), hyperthermia (Xu et al., 2002b), or hypothermia (Nishio et al., 2000; Yunoki et al., 2002) induced PC rendering the brain tolerant to subsequent potentially lethal insult application.
In multicellular organisms, PC extends not only to neurons/cells immediately affected by hurtful stimulus. Multiple stab wounds to the brain increase animal survival after brain ischemia (Takahata and Shimoji, 1986). Two-hour reversible unilateral occlusion of the middle cerebral artery (MCA) renders the contralateral hippocampal neurons tolerant to subsequent global ischemia (Belayev et al., 1996). Cortical spreading depression not involving the hippocampus induces ischemic tolerance in hippocampal neurons (Kawahara et al., 1997). These observations suggest that in the whole organism neuroprotection can be achieved by concerted activation of various mechanisms to achieve neuroprotection in vivo, including cells not directly affected by harmful insult. These observations are comparable to observations in the heart, where short occlusions of the coronary circumflex artery significantly decrease the size of the myocardial infarction following extended occlusion of the left anterior descending artery (Przyklenk et al., 1993). However, PC is not only capable of increasing tolerance of the unaffected tissue by applying insult to a distant locus of the same organ, but can also protect other organs as well. Protection of organs and tissues by applying stimuli to sites remote from them is known as “remote preconditioning”.
Remote preconditioning. It was suggested (Kirino, 2002) that the PC phenomenon is part of the universal stress response observed across species (Feder and Hofmann, 1999). After demonstration that femoral artery occlusion combined with gastrocnemius muscle stimulation (Birnbaum et al., 1997) is capable of decreasing myocardial infarction size, remote preconditioning attracted significant attention due to its simplicity (i.e. temporary limb ischemia) and potential efficacy. The presence of the remote PC phenomenon in which protection of remote organs is induced by making other organs (Gho et al., 1996; Song et al., 2007) or limbs (Wei et al., 2012b) ischemic, suggests the existence in complex organisms of innate protective mechanisms, which provide global defense against adverse conditions (Iadecola and Anrather, 2011a; Przyklenk and Whittaker, 2011).
Remotely preconditioned ischemic tolerance can be induced in heart (Eisen et al., 2004), brain (Hess et al., 2015), liver (Koti et al., 2003), intestine (Sileri et al., 2004), kidneys (Ogawa et al., 2000), skeletal muscles (Lee et al., 1996), and skin (Zahir et al., 1998). Due to the simplicity and relative safety of remote PC procedures, clinical trials are being carried out to explore the clinical efficacy of this method. While overall it seems that remote PC (applying tourniquet to legs or arms) is capable of exerting cardioprotective effects, the data are still not conclusive and further studies are required (Hong et al., 2010; Kottenberg et al., 2014; Hausenloy et al., 2015; Heusch and Gersh, 2016; Basalay et al., 2018; Chong et al., 2018). There are fewer clinical trials exploring the efficacy of pre-, peri or post-conditioning against brain damage compared to myocardial infarction (Meller and Simon, 2015; Basalay et al., 2018). Brain remote conditioning is a simple and well-tolerated therapy, which has been tested with different degrees of success in such conditions as ischemic stroke, transient ischemic attack, subarachnoid hemorrhage, cerebral small vessel disease, and severe carotid atherosclerotic stenosis (Koch et al., 2011; Hess et al., 2015; Meller and Simon, 2015; Basalay et al., 2018; Zhao et al., 2018; Zhou et al., 2018; Zhao et al., 2019).
Remote PC can only be implemented in complex organisms, implying the existence of systemic protective mechanisms that remains to be identified. A significant body of literature is devoted to analysis of the possible mechanisms of myocardial remote PC (Heusch et al., 2015; Meller and Simon, 2015; Basalay et al., 2018). It seems to include humoral and neural, including central, mechanisms. More scarce data on the mechanisms of remote PC in cases of ischemic stroke show that comparable mechanisms seem to be involved. Reversal of the neuroprotective effect of conditioning ischemia by transection of the femoral and sciatic nerves of ischemic hindlimbs (Yu and Liu, 2014) by administration of ganglion blocker hexamethonium or by nerve block with capsaicin (Dong et al., 2004; Ren et al., 2009; Malhotra et al., 2011; Wei et al., 2012a; Pignataro et al., 2013), strongly suggest the presence of a neural component in the mechanism of remote PC. These observations are supported by demonstration of the salvaging effect of peripheral nerve stimulation (Xiao et al., 2015). Humoral mechanisms also seems to be involved as indicated by reversal of remote PC by naloxone, an opioid receptor antagonist, by insulin antibodies, or by selective CGRP receptor blocker (Rehni et al., 2007; Zhou et al., 2011).
Neurogenic neuroprotection.
At the level of the organism, preconditioning can be triggered by signal(s) activating only specific “sensory” cells while other cells are not affected by the changing condition. Examples of such sensors are oxygen sensing neurons or astrocytes localized to different brain areas including medulla and cerebellar fastigial nucleus (Guyenet et al., 2010; Angelova et al., 2015). The function of these cells/systems is to anticipate upcoming potentially dangerous changes and take protective measures before harmful changes occur to other cells and organs. In summary, available data strongly suggest the existence of multi-level systems of endogenous mechanisms, which when activated protect cells and organs against the injurious effects of hypoxia, ischemia, and other damaging insults. These levels include individual cell protection, intercellular protection, organ protection, and last but not least organism level protective mechanisms. Coordinated interaction of these mechanisms at all levels, from cellular to organismal, provides robust protection allowing organism survival under various hostile conditions. The advantage of the systemic organismal response is that various cells and organs of the organism do not have to be severely affected by potentially damaging factors. Instead, existing “sensory” or “receptor” cells can trigger defensive changes, before cells are directly affected. The oxygen sensing neurons of the medulla are examples of such warning mechanisms (Reis et al., 1994). These neurons increase their activity to trigger processes counteracting hypoxia before the hypoxia sets in and other neurons become affected by it (Sun and Reis, 1994; Neubauer and Sunderram, 2004). Another example of a complex coordinated protective response is the “diving reflex”, which is triggered by trigeminal nerve stimulation and functions to promote survival during the period of anoxia (Panneton, 2013). Diving reflex includes coordinated activation of the sympathetic and parasympathetic systems (Chowdhury et al., 2015; Golanov, 2015; Chiluwal et al., 2017).
Endogenous neuroprotection. The existence of neurogenic neuroprotective mechanisms (i.e., systemic coordinated activation of various cellular to organismal mechanisms to provide neuroprotection triggered by activation of sensors) was suggested previously (Reis et al., 1997a; Golanov and Zhou, 2003; Schaller et al., 2009), and related phenomenon of endogenous neuroprotection have attracted significant attention as of late (Perez-Pinzon, 2007; Dirnagl et al., 2009; Iadecola and Anrather, 2011a; Kitagawa, 2012; Koch et al., 2012), especially in light of unsatisfactory outcome of numerous attempts to find therapy for stroke and other brain injuries.
Systemic neurogenic neuroprotection (Reis et al., 1997b; Golanov and Zhou, 2003) seems to result from the coordinated activation of endogenous mechanisms at different levels. Excitation of neurons of selected brain structures such as the cerebellar fastigial nucleus (FN) (Berger et al., 1990; Zhang and Iadecola, 1992a; Golanov et al., 1998; Reis et al., 1998b), dorsal periaqueductal grey matter (Glickstein et al., 2003), subthalamic vasodilator area (SVA) (Glickstein et al., 2001; Golanov and Zhou, 2003), rostral ventrolateral medulla (RVLM, S. Yamamoto, unpublished data), or the vagus nerve (Miyamoto et al., 2003; Mravec, 2010; Ay et al., 2011; Hiraki et al., 2012; Sun et al., 2012) protects brain tissue against global or focal ischemia. The neuroprotection triggered by brain stimulation was termed neurogenic neuroprotection (Reis et al., 1997a) to stress its neurogenic origin. Our research provided substantial evidence in support of the existence of intrinsic brain systems, which when activated offer acute and prolonged (up to three weeks) neuroprotection (Golanov and Zhou, 2003). Due to numerous commonalities shared by different types of PC we suggest that neurogenic neuroprotection integrates protective mechanisms of different levels to exert its neuroprotective effect and potentially participates in other forms of PC.
Phenomenology.
Electrical stimulation of FN globally increases cerebral blood flow (CBF) by decreasing cerebrovascular resistance (Doba and Reis, 1972; Iadecola and Reis, 1990; Talman et al., 1991; Golanov and Reis, 1995). FN-evoked increases in CBF are independent of cerebral glucose utilization (CGU) (Nakai et al., 1983) suggesting that CBF elevation is independent of non-specific general functional brain activation. FN-evoked increases in CBF represent so-called neurogenic cerebrovasodilation mediated by intrinsic brain circuitry (Iadecola et al., 1983; Golanov et al., 2001a, Golanov, unpublished data). These observations led to the hypothesis that stimulation of FN would be capable of improving CBF without changes in metabolism. This would result in the improvement of conditions in the stroke penumbral area, which is known to have increased metabolism when CBF is limited.
Indeed, in anesthetized rats, unilateral electrical stimulation of FN for one hour immediately after permanent MCA occlusion (MCAO) decreases contra- or ipsilateral infarction volume by ~40-50% as determined 24 hours after occlusion (Reis et al., 1989; Underwood et al., 1989; Reis et al., 1991; Yamamoto et al., 1993a; Golanov et al., 1996). Salvaged areas involve the periphery of the infarction and coincide with the penumbral zone surrounding the infarction core (Yamamoto et al., 1993a; Golanov et al., 1996). The effect of FN stimulation is strain independent and comparable in SHR, Wistar, Fisher, and Sprague-Dawley rats (Reis et al., 1989; Reis et al., 1991; Zhang and Iadecola, 1992b; Yamamoto et al., 1993a; Zhang and Iadecola, 1993; Golanov et al., 1996; Glickstein et al., 2001; Zhou et al., 2003).
In MCA occluded animals FN stimulation increases CBF in the non-ischemic areas of the ipsilateral hemisphere and in the whole contralateral hemisphere. However FN stimulation does not increase CBF in the underperfused penumbral zone – salvaged area - (Yamamoto et al., 1993a; Golanov et al., 1996) suggesting that mechanisms distinct from CBF elevation underlie salvaging of the penumbra, such as suppression of metabolism. However, further analysis revealed that salvaging of the penumbra by FN stimulation is not dependent on metabolism suppression (Golanov et al., 1996). In other words, the salvaging effect of FN stimulation is mediated by mechanisms other than CBF and CGU modifications.
Further exploration revealed that FN stimulation efficiently diminishes the volume of the lesion triggered by direct application of the excitotoxin – ibotenic acid (Schwarcz et al., 1979) – into striatum (Glickstein et al., 1999a). Delayed hippocampal neuronal death observed after >12 hours after the episode of global ischemia involves mostly apoptotic mechanisms (Macmanus et al., 1993; Honkaniemi et al., 1996; Petito et al., 1997; Ozawa et al., 1999; Back et al., 2004). FN stimulation preceding global ischemia decreases the number of damaged CA1 area neurons in hippocampus by ~60%, demonstrating that FN stimulation also effectively exerts neuroprotection against delayed neuronal death (Golanov et al., 1998).
The salvaging effect of FN stimulation requires at least 45 minutes of stimulation. Stimulation was shown to be effective if delivered immediately before (Yamamoto et al., 1993b; Glickstein et al., 1996) or after (He et al., 2014; Wang et al., 2019) the MCAO. The salvaging effect of electrical stimulation of the FN develops immediately after the stimulation. However, its maximum level of infarct volume attenuation of 50% occurs 72 hours after the stimulation and dissipates by 10 days (Reis et al., 1998a).
These findings raise the possibility that the FN may be a component of the system’s mechanisms that participates in the systemic defense reaction. This assumption is supported by the observation that transient global ischemic preconditioning-induced salvage of CA1 hippocampal neurons is completely reversed by preceding excitotoxic lesion of FN neurons. Moreover, sublethal preconditioning induced by transient global ischemia alone becomes lethal for CA1 neurons in animals with lesioned FN neurons, and drastically decreases the ability of animals to survive global sublethal ischemia (Rollins et al., 2003; Golanov et al., 2017). These observations are in-line with the earlier observations of a critical role of the FN in surviving hemorrhage (Lutherer et al., 1983) or myocardial infarction (Abulaiti et al., 2011). These findings strongly suggest that the FN plays a physiological role in the mechanisms of ischemic preconditioning.
FN-evoked neuroprotection is initiated by the excitation of neurons of the rostral-ventromedial part of the FN. Selective excitotoxic lesion (ibotenic acid) of these neurons reverses the neuroprotective effect of FN stimulation on infarct volume triggered by MCAO three days after the stimulation, while FN-evoked increases of CBF and arterial pressure are preserved (Glickstein et al., 1999b). These data allow the conclusion that excitation of FN neurons rather than fibers of passage produce neuroprotection, and that CBF increase and neuroprotection are independent and mediated by different circuitry.
Neuroprotective properties of FN are not unique suggesting that intrinsic neuroprotective circuitry within the brain may include multiple components. The SVA is the relay station for vasodilator signals generated in the medulla (RVLM) as well as in the FN (Golanov and Reis, 1998; Golanov et al., 2001b). Electrical stimulation of the SVA induces comparable metabolism-independent neuroprotection. Neuroprotective effects of stimulation of the SVA or the FN are independent: excitotoxic lesions of SVA or FN, respectively, do not affect salvaging effects of FN or SVA stimulation (Glickstein et al., 2001). Another known neuroprotective site is dorsal periaqueductal grey (PAG). One-hour electrical stimulation of dorsal PAG exerts robust neuroprotective effects (Glickstein et al., 2003).
Mechanisms.
Several possible mechanisms of neurogenic neuroprotection have been explored. Selective excitation of FN neurons by microinjection of excitatory amino acids decreases global CGU (Chida et al., 1989), which is indicative of suppression of functional activity (Sokoloff et al., 1977). In support of decreased functional activity is the appearance of synchronized slow high-amplitude EEG activity under electrical stimulation of the FN (Iadecola et al., 1986; Golanov et al., 2000). In line with the suggestion that excitation of FN neurons decreases functional activity are observations that stimulation of FN elevates seizure thresholds in experimental animals (Hablitz and Rea, 1976; Wang et al., 2008), reduces seizure susceptibility in man (Levy and Auchterlonie, 1979), and increases the threshold of spreading depression (Golanov and Reis, 1997).
Decrease in neuronal excitability evoked by FN stimulation counteracts peri-infarct depolarizing waves (PIDs). PIDs initiated by membrane depolarization resulting from stroke (Petzold et al., 2005; Hartings et al., 2009; Dreier, 2011; Lauritzen and Strong, 2017) aggravate ischemia-induced deep ionic disbalance (Giza and Hovda, 2001), and exacerbate energy depletion (Hartings et al., 2008) while CBF is compromised. In our experiments, stimulation of the FN increases latency and reduces the number of PIDs appearing after MCAO (Golanov and Reis, 1999a, b), which may have protective effects following brain ischemia.
Opening of potassium channels is known to decrease neuronal excitability (e.g. Lutz et al., 1996). Decreased neuronal excitability following FN neuroprotective stimulation is in line with possible opening of potassium channels. An increase of interstitial potassium levels during FN stimulation (Iadecola and Kraig, 1991) and reversal of the neuroprotective effect of FN stimulation by intracerebroventricular preferential KATP-channel blocker glibenclamide (Golanov et al., 1999; Golanov and Reis, 1999c), support the suggestion that opening of potassium channels may play a role in the neuroprotective effect of FN stimulation. This observation points to a commonality in cellular mechanisms between the FN evoked neuroprotection and neuroprotection evoked by ischemic or chemical preconditioning, which is also dependent upon KATP-channel opening (Heurteaux et al., 1993; Nakagama et al., 2002).
A substantial number of neurons after global (Macmanus et al., 1993; Honkaniemi and Sharp, 1996; Petito et al., 1997; Ozawa et al., 1999) or focal ischemia (Li et al., 1997; Velier et al., 1999) undergo apoptosis, where the mitochondria play a crucial role (Haeberlein, 2004). Inhibition of apoptosis is neuroprotective (e.g. Robertson et al., 2000; Wiessner et al., 2000). Staurosporine is known to induce cell death through an apoptosis-like mechanism: mitochondrial release of cytochrome c with subsequent activation of caspases-9 and -3 (Koh et al., 1995; Krohn et al., 1998; Velier et al., 1999; Strasser et al., 2000). In “ex vivo” brain slices obtained 72 hours after FN stimulation, we observed suppression of the release of cytochrome c by mitochondria induced by staurosporine, calcium overload, or by mastoparan, in addition to suppression of caspase-3 activity (Zhou et al., 2001). In these slices staurosporine-induced insertion of the pro-apoptotic protein Bax into mitochondria was significantly reduced. Following FN stimulation mitochondria exerted an increased capability of calcium sequestration and tolerance to depolarization. These results indicate that FN stimulation protects the mitochondria from calcium overload, and suppresses mitochondrial apoptotic pathways, suggesting a significant role of mitochondria in neuroprotection exerted by FN stimulation. The effect of the suppression of cytochrome c release is comparable to that observed in cultured cells in response to calcium-evoked release of cytochrome c by OGD preconditioned neurons (Zhou et al., 2001; Zhan et al., 2002; Zhou et al., 2005).
Mitochondria in brain slices obtained 72 hours after FN stimulation exert significant resistance to the depolarizing effect of the mitochondrial uncoupler, carbonyl cyanide-phenylhydrazone, indicating that FN stimulation stabilizes the mitochondrial membrane potential (Yamamoto and Golanov, 2004a). Increased mitochondrial tolerance against depolarization may be a component of the endogenous neuroprotective mechanism mediated by upregulation of uncoupling protein 4 (UCP4) in response to opening of potassium channels (Yamamoto and Golanov, 2004a, b; Yamamoto et al., 2011). Seventy-two hours after one-hour FN stimulation, protein and mRNA levels of UCP4 increased throughout the cortex. Following MCAO, mRNA and protein levels of UCP4 increased even more. These findings suggest that FN-evoked neuroprotection might involve modification of UCP4 expression, which can exert neuroprotective effects by rendering mitochondria more tolerant to ischemic insult (Shant et al., 2005b). Uncoupling proteins decrease production of reactive oxygen species (ROS) and can be protective against ischemic stroke (Mattiasson et al., 2003). In our experiments we observed participation of potassium channels in the early phase of FN stimulation. Opening of potassium channels, especially mitoK increases production of ROS and exerts neuroprotective effects (Shimizu et al., 2002; Andrukhiv et al., 2006). We hypothesized that this increase in ROS may trigger increased expression of UCP4. In cultured neurons, an observed increase in UCP4 expression in response to diazoxide exposure was reversed by superoxide dismutase (Golanov and Yamamoto, 2004; Shant et al., 2005a). These observations may explain the different action of the potassium channels blocker, glibenclamide, which effectively reversed FN-induced neuroprotection when injected at the time of stimulation, but was not effective in reversing the protective effect when applied 72 hours after stimulation, at the time of MCAO. This observation suggests that opening of potassium channels is necessary at the initial phase of FN-induced protection, thereby providing an acute phase of protection, and triggers long-term changes such as gene-expression that exert long term neuroprotection by increasing ROS levels.
Prohibitin is another mitochondrial protein whose expression increases 72 hours after FN stimulation. Prohibitin is also upregulated in neuronal cultures or hippocampal slices in response to hypoxia, and silencing of its expression increases neuronal loss. It seems that its effects are also associated with mitochondrial membrane potential and ROS production. It was suggested that prohibitin may stabilize the function of mitochondrial complex I (Zhou et al., 2012). These data are in line with the suggestion that FN stimulation-induced neuroprotection also involves mitochondria in conditioning processes (Pamenter, 2014; Prendes et al., 2014; Thompson et al., 2015; Silachev et al., 2016).
Excitotoxicity, which is accompanied by cellular calcium overload, is an important component of ischemic/hypoxic neuronal damage (Mergenthaler et al., 2004). Excessive calcium overload activates protein kinases, such as PKCγ and δ, and proteases, such as calpain, which are known to exert deleterious effects on neurons under conditions of brain ischemia/hypoxia (Yamashima, 2004; Chou and Messing, 2005; Zhao et al., 2016). FN stimulation at 1 to 7 days before the stroke decreased expression of these protein kinases (Yu et al., 2004) and inhibited calpain activity (Deng and Dong, 2003), decreasing stroke volume and improving recovery.
Peroxisome proliferator-activated receptor gamma (PPARγ) is known as a master regulator of numerous genes involved in neuroinflammation, energy metabolism, and redox equilibrium (Cai et al., 2018) and is neuroprotective when activated (Luo et al., 2006; Cai et al., 2018). FN stimulation increases expression of PPARγ, and reduces infarct volume, (He et al., 2014; Tang et al., 2015; Liu et al., 2017) while suppression of PPARγ expression using small hairpin RNA reverses the neuroprotective effect of FN stimulation (Liu et al., 2017).
Available data also indicate the possible participation of microRNA in the salvaging effects of FN stimulation. One-hour FN stimulation decreased expression of microRNA miR-29c in parallel with the decrease in infarct volume in a standard ischemia/reperfusion model. A control antagomir was not effective in reducing infarct volume. This microRNA directly binds to the predicted 3'-UTR target sites of Birc2 and Bak1 genes, suppressing their expression. Over-expression of miR-29c effectively reduced Birc2 (also Bak1) mRNA and protein levels, increased infarct volume and apoptosis, and worsened neurological outcomes (Huang et al., 2015). Further exploration of the possible involvement of microRNA in the salvaging effect of FN stimulation revealed over 9 microRNA whose expression increased following FN stimulation, and that may be involved in the salvaging effect. However, their specific targets remain to be established (Feng et al., 2015). One new specific microRNA, rno-miR-676-1, has been identified as participating in the salvaging effect of FN. However, it’s specific target has not been established (Pang et al., 2015).
Regeneration. Besides improving survival of brain cells, FN stimulation seems to be capable of stimulating axonal regeneration. FN stimulation 1 hour after MCAO led to upregulation of growth associated protein 43 (GAP43), which was accompanied by improvement of neurological recovery. The effect seems to be mediated by the protein kinase A (PKA) pathway, as an antagonist of PKA reversed the positive effect of FN stimulation (Wang et al., 2019).
A series of correlative studies also suggests that FN stimulation may improve axon growth. Thus growth arrest and DNA damage inducible gene β (Gadd45β), which may participate in axon growth (Liu et al., 2015), increased significantly in rats after FN stimulation demonstrating improvement in motor behavior (Liu et al., 2012). At the same time FN stimulation decreased expression of repulsive guidance molecule A (RGMa), which was accompanied by increased optical density of neurofilaments, indicating improved axon recovery (Jiang et al., 2012). Comparably, FN stimulation 2 hours after the ischemia decreased expression of Nogo receptor mRNA and protein, which are known to suppress axon regeneration (Zhang et al., 2008).
Electric stimulation of FN also exerts positive effects on neuronal stem cell proliferation and survival. It promotes the proliferation of bromodeoxyuridine (Brdu) positive cells after stroke (Huang and Luo, 2008) and improves survival and differentiation of neuronal stem cell transplanted into rats with MCAO (Jin et al., 2007; Huang et al., 2010).
Inflammation/Immune response (diencephalon/hypothalamus). Modulation of the immune/inflammatory response plays an important role in poststroke induced pathology (Helmy et al., 2011; Iadecola and Anrather, 2011b), and development of ischemic tolerance (Garcia-Bonilla et al., 2014). FN-evoked neuroprotection also seems to suppress the inflammatory response, which plays an important role in the protective effects. Expression of inducible nitric oxide synthase (iNOS) by cerebral microvessels and leukocytes is one of the components of the inflammatory reaction to ischemia (Iadecola et al., 1995a; Iadecola et al., 1995b; Nagafuji et al., 1995; Iadecola et al., 1996; Cobbs et al., 1997; Galea et al., 1998c; Cernak et al., 2001), which while important for reparative processes after brain damage, can also exacerbate it (see (Barone and Parsons, 2000; Iadecola and Alexander, 2001; Morganti-Kossmann et al., 2002)). FN stimulation 48 h prior to MCAO reduces induction of iNOS mRNA and expression of active iNOS in brain microvessels, and the infiltration of macrophages into the territory which is salvaged (Galea et al., 1998a). Moreover, stimulation of FN suppresses induction of intercellular adhesion molecule-1 and iNOS by interleukin-1β, a leading mediator of the inflammatory response (Liu et al., 1993; Barone and Parsons, 2000; Iadecola and Alexander, 2001; Morganti-Kossmann et al., 2002) in vivo in striatum and in vitro by cerebral microvessels obtained from rats 72 hours after FN stimulation (Galea et al., 1998b). These findings suggest that FN stimulation renders cerebral microvessels less sensitive to inflammatory stimuli and can be interpreted as evidence that suppression of the inflammatory reaction is one of the mechanisms of neurogenic neuroprotection.
Cerebellar FN has multiple connections with various hypothalamic nuclei (Del Bo and Rosina, 1986; Min et al., 1989; Haines et al., 1990; Çavdar et al., 2001; Soto-Tinoco et al., 2016; Li et al., 2017), including areas involved in immune control (Soto-Tinoco et al., 2016). Excitotoxic lesion of FN neurons resulted in increased mesenteric T lymphocyte proliferation and splenic NK cell cytotoxicity (Peng et al., 2005), which reflect deactivation of glutamatergic projections from the FN to the hypothalamus. Inhibition of glutamate synthesis in the FN decreased glutamate levels in the lateral hypothalamus and attenuated the percentage and cytotoxicity of natural killer cells, as a well as lowered the percentage of cytokine production by T lymphocytes (Cao et al., 2012; Cao et al., 2015). γ-Aminobutyric acid (GABA)-ergic projection from the FN to the hypothalamus exerted opposite effects: vigabatrin, an inhibitor of GABA-transaminase, significantly reduced concanavalin A (Con A)-induced lymphocyte proliferation, anti-sheep red blood cell (SRBC) IgM antibody levels, and splenic natural killer (NK) cell number and cytotoxicity (Cao et al., 2013).
Another intriguing regulatory function of the FN nucleus in the immune system is its potential regulation of the intestinal mucosa, and as a consequence, regulation of microbiome/host interaction. Lately it has become clear that the microbiome plays an important role in neurological disorders (see (Winek et al., 2016)). While currently no data is available on the effect of FN stimulation on the gut microbiome or gut wall alterations affecting the microbiome/organism interaction, there are potential consequences of FN stimulation on the organism/microbiome interaction. The effect of FN on gastrointestinal motility is known (Manchanda et al., 1972). Activation of FN GABAergic cells aggravated stress-induced gastric mucosa damage (Zhu et al., 2013). The effect seems to be mediated through the lateral hypothalamus and greater splanchnic nerve (Zhu et al., 2012), which plays and important role in the control of inflammation and the intestine (Martelli et al., 2014). It is conceivable that activation of the FN may also regulate organism/microbiota relations and affect the microbiome itself.
Conclusion
Cerebellar FN stimulation delivered before or after a brain-damaging event is capable of significantly attenuating the damage. Numerous molecular and systemic mechanisms are involved in the neurogenic neuroprotection induced by FN. Available data indicate that the FN is a part of an endogenous protective system and provides warning signaling for activation of neuroprotective mechanisms (Parsons et al., 2001; Nayak et al., 2016). Moreover, the FN is critical for survival during life threatening conditions (Lutherer et al., 1982; Lutherer et al., 1983; Golanov et al., 2017). Available data provide strong substantiation for the existence of an intrinsic neuroprotective system, which offers lasting neuroprotection when activated. An intrinsic protective system, which includes at least FN, SVA, and PAG, probably is activated by adverse conditions, such as ischemia, hypoxia, or traumatic brain injury, thereby protecting the brain (Fig. 2). We hypothesize that it is activated reflexively under normal physiological conditions in anticipation of the development of adverse situations, for instance as part of the coordinated diving response. “To tolerate and survive hypoxia, the mammalian nervous system must (a) reduce metabolism, (b) prevent cell death and injury, and (c) maintain functional integrity” (Ramirez et al., 2007). Data provided are in accord with these requirements and allow us to hypothesize that activation of the intrinsic neuroprotective system through activation of the naturally occurring response, mobilizes systemic (activity and metabolism suppression, suppression of inflammatory response) and innate cellular (changes of membrane properties of neurons and mitochondria, suppression of apoptosis) protective mechanisms. Endogenous neurogenic neuroprotection is mediated by a coordinated integrative response, which involves protective mechanisms at all levels, from cellular to organismal as we described. It is conceivable that the innate brain protective system, which includes the FN, may also play a role in other types of conditioning. Understanding the systemic protective mechanisms, their triggers, effectors, components, and their coordination will allow us to control and amplify naturally existing protective mechanisms.
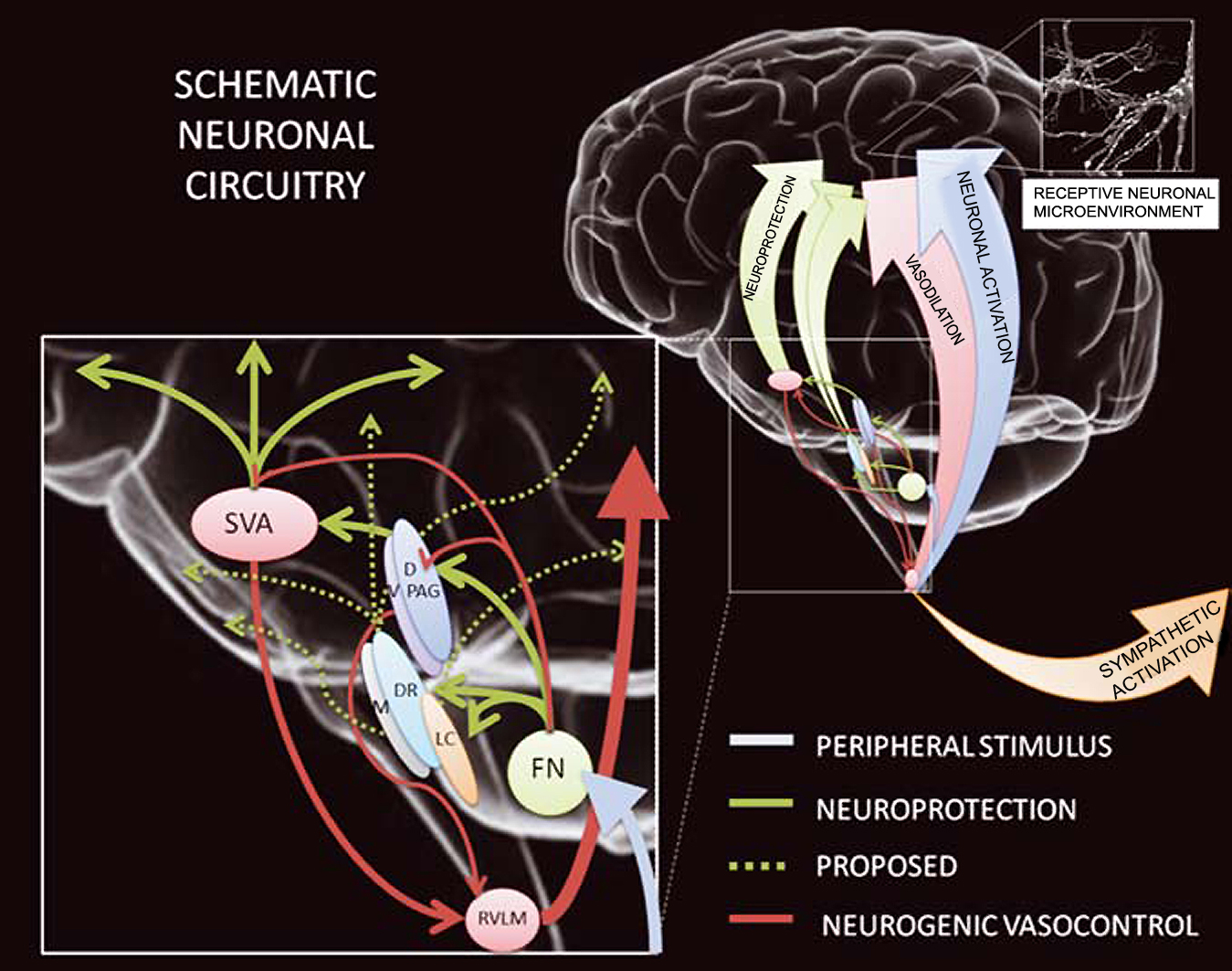
In a new window | Download PPT
Figure 2: The neuroprotection circuitry might encompass neuronal circuitry involved in the coupling between neuronal activation and its consequent energetic demands. (1) Excitation of neurons and/or fibers projecting through the subthalamic vasodilator area (SVA) reduces ischemic infarctions to the same degree as excitation of the fastigial nucleus (FN) neurons. (2) Conditioned neuroprotection is independent of increased cerebral blood flow (CBF). The effects are long-lasting and not attributable to changes in blood gases, brain temperature, or rat strain. (3) The neuroprotective effects of SVA and FN stimulation are mutually independent, and FN-evoked cerebrovasodilation is mediated by SVA neurons. (4) Both the systemic and cerebrovascular components of FN stimulation are abolished by bilateral lesions of the rostral ventrolateral medulla (RVLM). (5) The SVA also mediates the primary elevation of CBF elicited by hypoxic excitation of the sympathoexcitatory neurons of the RVLM. (6) Intrinsic neurons of dorsal- and ventral periaqueductal grey (D- and VPAG) differentially regulate CBF. (7) Neurons of DPAG mediate neuroprotective effects, independently of changes in CBF and/or arterial pressure. (From (Mandel et al., 2012)).
References
Eugene V. Golanov1
1Department of Neurosurgery, Houston Methodist Hospital, Houston, Texas, 77030.
Angelique S. Regnier-Golanov1
1Department of Neurosurgery, Houston Methodist Hospital, Houston, Texas, 77030.
Gavin W. Britz1
1Department of Neurosurgery, Houston Methodist Hospital, Houston, Texas, 77030.
Corresponding author:
Eugene V. Golanov
Email: evgolanov@houstonmethodist.org
In a new window | Download PPT
Figure 1: Different levels of innate self-defense. A. Innate self-defense can be triggered in a single cell by a strong but not lethal signal. In response to cellular changes triggered by the sublethal stimulus, the cell temporarily mobilizes protective mechanisms and becomes tolerant to the stimulus of lethal strength. B. The cell can be sensitive to the potentially dangerous stimulus and not only mount its own defense but also trigger changes in a neighbor cell making both cells tolerant to the subsequent lethal strength stimulus. C. In brain specific cells, receptors can sense changes in the environment before their own or their neighbor’s metabolism becomes affected and trigger a coordinated multicomponent response, which would make the brain or the whole organism protected against lethal stimulus.
In a new window | Download PPT
Figure 2: The neuroprotection circuitry might encompass neuronal circuitry involved in the coupling between neuronal activation and its consequent energetic demands. (1) Excitation of neurons and/or fibers projecting through the subthalamic vasodilator area (SVA) reduces ischemic infarctions to the same degree as excitation of the fastigial nucleus (FN) neurons. (2) Conditioned neuroprotection is independent of increased cerebral blood flow (CBF). The effects are long-lasting and not attributable to changes in blood gases, brain temperature, or rat strain. (3) The neuroprotective effects of SVA and FN stimulation are mutually independent, and FN-evoked cerebrovasodilation is mediated by SVA neurons. (4) Both the systemic and cerebrovascular components of FN stimulation are abolished by bilateral lesions of the rostral ventrolateral medulla (RVLM). (5) The SVA also mediates the primary elevation of CBF elicited by hypoxic excitation of the sympathoexcitatory neurons of the RVLM. (6) Intrinsic neurons of dorsal- and ventral periaqueductal grey (D- and VPAG) differentially regulate CBF. (7) Neurons of DPAG mediate neuroprotective effects, independently of changes in CBF and/or arterial pressure. (From (Mandel et al., 2012)).
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14069 | 31 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA