Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Neuroprotection in Parkinson’s disease: hypoxia inducible factor-1α, exercise, and preconditioning through hypoxia
Time:2019-07-02
Number:15381
Author Affiliations

Conditioning Medicine, 2019. 2(3):142-154.
Abstract
Exercise has been shown to be beneficial to neurological health. In fact, recent preclinical and clinical studies demonstrate that exercise, in particular vigorous aerobic exercise, is neuroprotective in Parkinson’s disease. One key protein activated by changes in cellular oxygen levels is Hypoxia Inducible Factor (HIF). HIF is a transcription factor that activates the expression of numerous genes, including those involved in glycolysis, angiogenesis, metabolism, and cell proliferation. In this review, we discuss exercise-induced neuroprotection and the necessity for the generation of hypoxia as a means to initiate neuroprotection within the dopaminergic neurons of the substantia nigra pars compacta. The role of HIF1α and its interacting partners (including the parkinsonian gene DJ-1) in the promotion of exercise-induced neuroprotection are also considered.
Keywords: hypoxia inducible factor; oxidative stress; dopaminergic neuron; substantia nigra
Abstract
Exercise has been shown to be beneficial to neurological health. In fact, recent preclinical and clinical studies demonstrate that exercise, in particular vigorous aerobic exercise, is neuroprotective in Parkinson’s disease. One key protein activated by changes in cellular oxygen levels is Hypoxia Inducible Factor (HIF). HIF is a transcription factor that activates the expression of numerous genes, including those involved in glycolysis, angiogenesis, metabolism, and cell proliferation. In this review, we discuss exercise-induced neuroprotection and the necessity for the generation of hypoxia as a means to initiate neuroprotection within the dopaminergic neurons of the substantia nigra pars compacta. The role of HIF1α and its interacting partners (including the parkinsonian gene DJ-1) in the promotion of exercise-induced neuroprotection are also considered.
Keywords: hypoxia inducible factor; oxidative stress; dopaminergic neuron; substantia nigra
Introduction
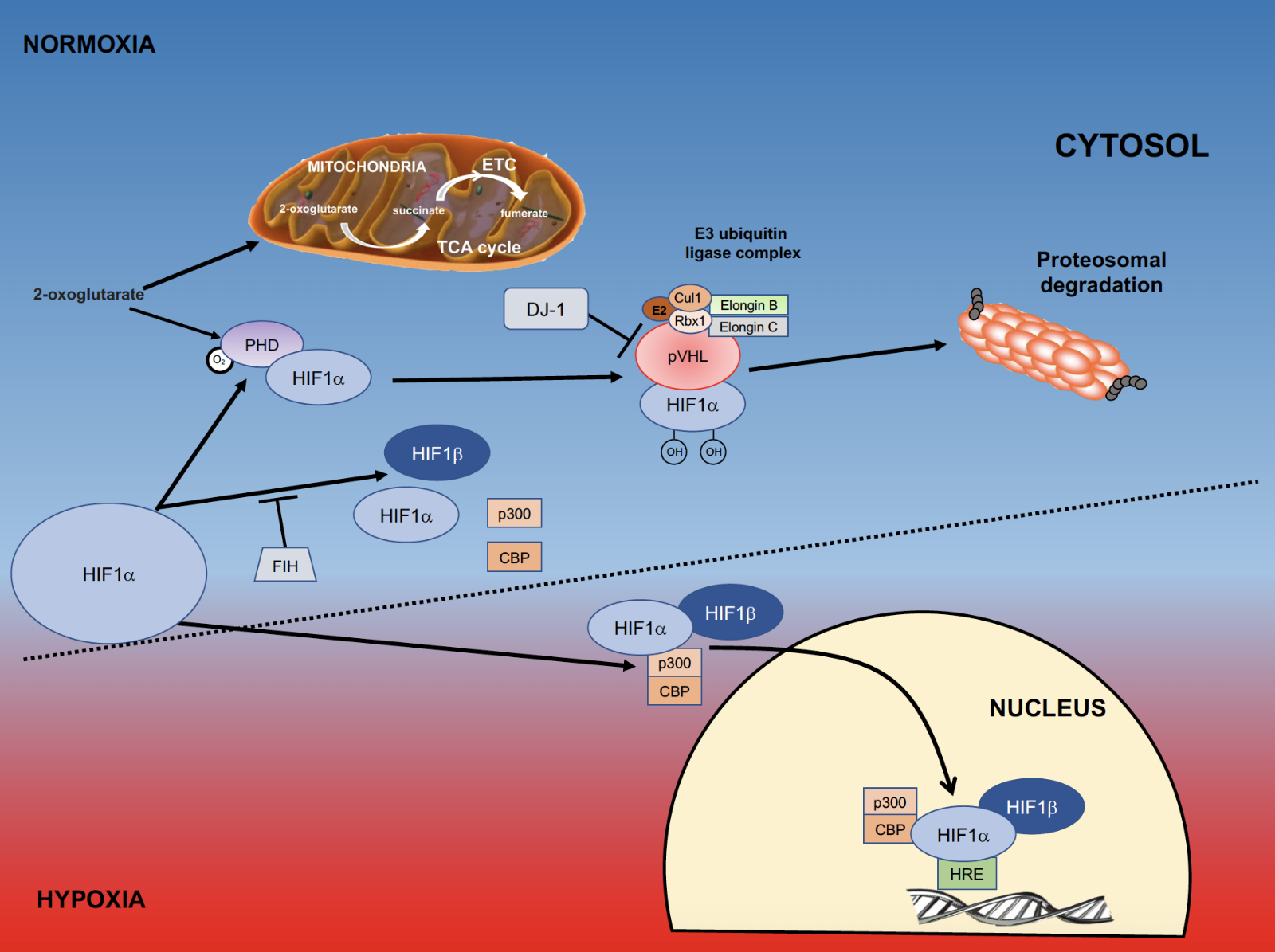
Mammalian cells require both oxygen and a source of energy to survive. Cellular energy, provided in the form of adenosine triphosphate (ATP) is predominantly produced by the electron transport chain (ETC) in the inner mitochondrial membrane. The production of ATP involves oxygen, which generates a small amount of reactive oxygen species (ROS) (Murphy, 2009). In addition to energy production, cellular metabolism also depends on the regulation of oxygen and glucose for their homeostasis. In the brain, neurons are particularly sensitive to changes in these substrates. A decline in molecular oxygen levels results in cellular hypoxia. Hypoxia inducible factor (HIF) is a transcription factor that is sensitive to cellular oxygen levels, and activates the expression of numerous genes, some of which are involved in glycolysis, angiogenesis, metabolism, and cell proliferation (Wang and Semenza, 1993a; Semenza et al., 1994; Sharp and Bernaudin, 2004). HIF1α, as well as the products of many of these genes, has been implicated in preconditioning cells, as well as in the process of neuroprotection itself (Figure 1). In this review, we will discuss the role of HIF and exercise-induced neuroprotection in experimental Parkinson’s disease.
In a new window | Download PPT
Figure 1: Role of HIFα in hypoxia. Within the cell molecular oxygen is tightly regulated, and a key component of this oxygen-sensing pathway is the transcription factor hypoxia inducible factor (HIF). During periods of normoxia, HIF1α is continually made and degraded. The degradation of HIF1α requires interaction with prolyl hydroxylases (PHDs), which are activated by molecular oxygen and 2-oxoglutarate, an intermediate of the TCA cycle that also functions as a substrate for formation of ATP in the electron transport chain (ETC). When HIF1α is hydroxylated by PHDs it changes conformation so that it can be recognized by von Hippel Lindau (VHL) factor. This complex interacts with the E3 ubiquitin ligase complex and is targeted for proteosomal degradation. Another means to prevent HIF-induced gene transcription is through the action of factor inhibiting hypoxia (FIH), which inhibits the formation of the HIF1α/HIF1β/p300/CBP complex. When cells are under oxygen stress they become hypoxic; a process where PHDs are inhibited and HIF1α accumulates. DJ-1 is a negative regulator of pVHL and also allows HIF1αaccumulation. Once HIF1α is stabilized and accumulates, it binds to HIF1β, complexes with p300 and CBP, and translocates to the nucleus where it binds to the Hypoxia Responsive Elements responsive elements (HRE’s) on DNA. (Adapted from Jochmanova et al., 2013).
Exercise and Parkinson's disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting 1-2% of adults over 60 years of age (Tysnes and Storstein, 2017). PD results in neuronal degeneration throughout the neuraxis, however the most notable cell loss is seen in the dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpc) (Kalia and Lang, 2015). The SNpc dopaminergic neurons are extremely sensitive to damage from oxidative stress, likely due to the fact that they contain high levels of iron and neuromelanin as well as free DA that, when not sequestered in vesicles, is readily nitrosylated (Gerlach et al., 2003; Sian-Hulsmann et al., 2011; Garcia-Garcia et al., 2012). The DA neurons of the SNpc project via the medial forebrain bundle to synapse primarily on D1 or D2-expressing medium spiny neurons (MSNs) in the striatum. Functionally, balanced output from this pathway is responsible for both the normal initiation and control of voluntary movement (Ledonne and Mercuri, 2017). When these SNpc DA neurons degenerate in PD, this system becomes unbalanced leading to its classical motor symptoms of tremor, bradykinesia, and rigidity (Kuno, 1997).
Exercise has been shown in epidemiological, preclinical, and human clinical studies to be beneficial to overall neurological health. In addition to Parkinson’s disease (Smith and Zigmond, 2003; Chen et al., 2005; Xu et al., 2010; Yang et al., 2015; Muller and Myers, 2018), which will be the main focus of this paper, examples of the neuroprotective properties of exercise can be seen after stroke (Austin et al., 2014), in reducing the risk of both vascular and Alzheimer’s dementia (Obisesan et al., 2012; Paillard et al., 2015), and in reducing the decline of function in multiple sclerosis (Motl and Sandroff, 2018), Huntington's Disease (Fritzet al., 2017) and Amyotropic Lateral Sclerosis (ALS)(McCrate and Kaspar, 2008).
Epidemiological studies have shown lower incidence of PD later in life for people who participated in moderate to vigorous physical activity (cycling, running, aerobic exercise, hockey, and basketball) at earlier stages in life (Sasco et al., 1992b; Chen et al., 2005; Thacker et al., 2008; Xu et al., 2010; Saaksjarvi et al., 2014; Yang et al., 2015; Shih et al., 2016; Muller and Myers, 2018). Recently, the positive impact of exercise on PD progression has been experimentally demonstrated. Shenkman et al. (2018) showed in a phase 2 randomized clinical trial of 128 participants that patients who exercised at least 4 times/week for 6 months and reached 85% maximum heart rate during their exercise period had a negligible progression in their UPDRS motor scores. In contrast, those individuals who performed exercise reaching only 65% maximum heart rate had a score increase of 2.0, and those who did not exercise or meet these parameters had a progression of 3.2, which was not significantly different from the 65% exercise group. Studies using forced lower extremity exercise by cycling have shown global improvement in PD patient symptoms (Ridgel et al., 2009; Rosenfeldt et al., 2015; Alberts et al., 2016; Shah et al., 2016). Forced exercise (FE) involves cycling at rates 30% faster than voluntary exercise (VE), and both groups were compared to an inactive control group. Functional MRI scans of patients in the FE and VE groups indicated improved thalamo-cortical connectivity in the FE group (Shah et al., 2016). When a FE group that was not currently receiving PD medication was compared to a control group not receiving PD medication, the FE group had a 50% improvement in UPDRS scores (Alberts et al., 2016). These studies and other trials of high-intensity exercise (Moore et al., 2013; Schenkman et al., 2018) advocate high intensity or aerobic physical activity as a viable preventative measure and possible therapy for PD. Literature reviews and meta-analyses of published studies also support vigorous or aerobic physical activity to reduce the risk of PD, lessen the severity of PD symptoms, and also to improve cognitive function during the course of the disease (Lauze et al., 2016; Flach et al., 2017; Ahlskog, 2018; da Silva et al., 2018; Ellis and Rochester, 2018; Fang et al., 2018). These studies also substantiate previous preclinical findings that demonstrated the neuroprotective effects of exercise (Hirsch and Farley, 2009; Zigmond et al., 2009; Radak et al., 2016). One caveat to these studies is the possibility that a predisposition to PD promotes lower levels of physical activity (Chen et al., 2005; Thacker et al., 2008). Contrary to the above evidence, Logroscino et al. (2006) reported the absence of an association between physical activity and the risk of PD. Also, Sasco et al. (1992a) suggested moderate physical activity reduces the risk of PD while vigorous activity provides less benefit.
The positive effect(s) of exercise on neurological health are likely due to the interactions of a multitude of factors. Exercise has been shown to increase the expression of neurotrophic factors (Cotman et al., 2007) including brain derived neurotrophic factor (BDNF) (Faherty et al., 2005; Wu et al., 2011; Gerecke et al., 2012), vascular endothelial growth factor (VEGF) (Storkebaum et al., 2004; Yasuhara et al., 2004; Tang et al., 2009; Villar-Cheda et al., 2009; Munoz et al., 2018; Rezaei et al., 2018), glial cell line derived neurotrophic factor (GDNF) (Smith et al., 2003; Sajadi et al., 2006; Zigmond et al., 2009; Tajiri et al., 2010), and insulin-like growth factor (IGF) (Carro et al., 2000). Exercise has also been shown to induce anatomical and chemical neuroplasticity in the brain. Fisher et al. (2004) showed that exercise after MPTP-induced parkinsonism can normalize changes in DA receptor expression, reduce levels of the DA transporter (DAT), and lead to behavioral improvement. In another study, treadmill exercise reduced the MPTP-induced loss of spines in striatal MSNs and increased the expression of two synaptic proteins, PSD-95 and synaptophysin (Toy et al., 2014). In a clinical study, eight weeks of treadmill exercise by PD patients resulted in increased striatal D2 receptor binding, measured by PET scan, as well as increased postural control; neither of which were seen in PD patients that did not exercise (Fisher et al., 2013).
Exercise effects on cellular plasticity in the brain
Interactions between living organisms and their environment often have a physical impact on that organism. Some adaptations may be brought about indirectly, through activity that induces change in the biochemical signaling pathways of cells. Studies in mammals have demonstrated that the environment- and the animal's response to it- affects the brain. Enriched environments, that allow an animal’s exposure to social interaction, mazes, toys, and running wheels, have been shown to increase the capacity for memory or learning by increasing dendritic length (Faherty et al., 2003), dendritic spine density (Leggio et al., 2005), and induction of neurogenesis in the hippocampus (van Praag et al., 1999b; van Praag et al., 1999a; Brown et al., 2003; Kronenberg et al., 2006). These changes are both brain region-specific and depend on the task performed. For example, rats executing acrobatic tasks such as training on an obstacle course have increased cerebellar molecular layer volume, increased Purkinje cell mitochondria number, and increased Purkinje cell synapse number (Black et al., 1990), while rats exercised on a treadmill (involuntary) or running wheel (voluntary) do not (Isaacs et al., 1992). However, rats in all active conditions show increased cerebellar capillary density compared to inactive animals (Isaacs et al., 1992). Similarly, young rats housed in a more complex environment (with toys and more social interaction), as well as rats in standard caging, (with other rats) have greater branching and surface area of capillaries in visual cortex than rats housed individually (Sirevaag et al., 1988). Exercise has also been shown to affect neurotransmitter levels in the brain (Gerecke et al., 2010; Klempin et al., 2013; Waters et al., 2013), however see (Gorton et al., 2010). Therefore, physical activity has the potential to alter anatomical and chemical substrates in the brain.
A number of studies have examined biochemical and molecular changes in the brain that are induced by exercise. Kinni et al. (2011) showed that forced exercise resulted in increased cerebral metabolic parameters indicated by higher levels of glucose transporters 1 and 3 (GLUT 1 and 3), phosphorylated 5'-AMP-activated protein kinase (AMPK) activity, and HIF1α in cortex, that were significantly higher in rats exercised on a treadmill compared to voluntary wheel running or sedentary rats. In this study, voluntary runners ran a greater distance (4432 m) than those on a treadmill (90 m), which suggests that the intensity of the running effort, rather than distance alone may affect neuroprotection.
Running exercise has also been shown to promote increases in VEGF (Ding et al., 2006a; Gao et al., 2014) and angiogenesis (Black et al., 1990; Isaacs et al., 1992; Ding et al., 2004). VEGF is protective in models of PD (Yasuhara et al., 2004; Yasuhara et al., 2005; Villar-Cheda et al., 2009) and an age dependent decrease in VEGF expression and microvascular density in rat SN is reversed with exercise (Villar-Cheda et al., 2009). There are also indications of angiogenesis in PD patients (Faucheux et al., 1999; Desai Bradaric et al., 2012) suggesting a possible neurorestorative functional relationship between the vascular system and VEGF in PD. VEGF is a target gene of the transcription factor HIF (Liu et al., 1995; Forsythe et al., 1996; Semenza, 2011b; Ahluwalia and Tarnawski, 2012), and in addition to the protective effects of VEGF on DA neurons, there are several other reasons to explore HIF as a modulator of DA neuron survival.
HIF as a modulator of SNpc DA neuron survival
HIF is a ubiquitous protein that is responsive to low oxygen levels in cells and is necessary for cell survival (Iyer et al., 1998). Several lines of evidence support the involvement of HIF in the survival and maintenance of DA neurons in the SN. HIF1α improves the viability of midbrain or dopaminergic precursor cells, while reduction of HIF1α expression in vitro or in vivo reduces the number of tyrosine hydroxylase (TH) positive neurons (Milosevic et al., 2007; Kim et al., 2008; Johansen et al., 2010). Conditional knockout of HIF1α from midbrain neural precursor cells reduces VEGF expression, TH expression, and the number of TH+ neurons in young adult mice, while neuronal precursors in frontal cortex are not significantly affected (Milosevic et al., 2007). A number of studies have shown that the stabilization of HIF1α is protective to neurons (Siddiq et al., 2005; Siddiq et al., 2009; Lushnikova et al., 2011). In particular, DA neurons are protected from MPTP toxicity (Lee et al., 2009) or 6-OHDA (Johansen et al., 2010) by inhibition of prolyl hydroxylases (PHDs), enzymes responsible for tagging HIF1α for proteosomal destruction (Mole et al., 2001; Huang et al., 2002). Inhibition of PHD allows the accumulation of HIF1α and the activation of downstream gene targets (Lee et al., 2009; Johansen et al., 2010). The expression of VEGF, shown to be a protective factor for DA neurons (Lee et al., 2009; Villar-Cheda et al., 2009) is regulated by HIF1α and HIF2α (Damert et al., 1997; Spinella et al., 2014). HIF2α is necessary for survival of DA neurons in the adult SNpc (Smeyne et al., 2015), for catecholamine homeostasis (Tian et al., 1998; Favier et al., 1999), as well as embryonic survival (Scortegagna et al., 2003). HIF2α is also stabilized with PHD inhibition under control conditions as well as with administration of MPTP (Lee et al., 2009). Although HIF1α and HIF2α have distinct and sometimes contrasting functions (Yuan et al., 2013), they may serve as surrogates for one another, but that remains to be tested (Lee et al., 2009; Vasseur et al., 2009; Johansen et al., 2010; Parsanejad et al., 2014; Smeyne et al., 2015). Complex I inhibitors such as rotenone and MPTP, which are known to be toxic to DA neurons (Giordano et al., 2012), attenuate HIF1α stabilization during hypoxia (Agani et al., 2000; Agani et al., 2002) while increasing cellular ROS (Przedborski and Ischiropoulos, 2005). Aerobic exercise that has been shown to be neuroprotective and may cause transient, intermittent hypoxia, stabilizes HIF1α (Lindholm and Rundqvist, 2016). In addition, PD patients have reduced HIF1α gene expression pathways in SN neurons isolated from postmortem brains (Elstner et al., 2011).
HIFs are master regulators of (cellular) oxygen homeostasis and adaptive responses
HIF was first described as a DNA binding complex, sensitive to cellular hypoxia (Wang and Semenza, 1993b). HIF is a heterodimer composed of an oxygen sensitive alpha subunit (HIF1α, HIF2α, or HIF3α) and constitutively expressed HIF1β (also called the aryl hydrocarbon receptor nuclear translocator, ARNT) (Hu et al., 2003), which are part of the basic Helix-Loop-Helix Per-Arnt-Sim (bHLH PAS) family of transcription factors (). The bHLH domain modulates DNA binding and subunit dimerization (Jiang et al., 1996). Distinct genes encode HIF1α (hif1α), HIF2α (epas1), and HIF3α (hif3α), and while part of their amino acid sequences are highly conserved, their functions and tissue distributions are different (O'Rourke et al., 1999; Semenza, 2000; Hu et al., 2003). HIF1α is ubiquitously expressed and mediates transcription of genes whose protein products are involved in angiogenesis, vascular remodeling, energy metabolism including glycolysis, erythropoiesis, and cell proliferation and viability (Sharp and Bernaudin, 2004; Semenza, 2011b, a). HIF2α and HIF3α have a more discreet expression pattern, with HIF2α being found in vascular tissue, endothelial, and smooth muscle cells, as well as in liver, kidney, lung, pancreatic progenitor cells, and catecholaminergic producing tissues (Jain et al., 1998; Tian et al., 1998; Favier et al., 1999; Chen et al., 2010). HIF2α plays a critical role in expression of genes encoding antioxidant enzymes (Scortegagna et al., 2003), factors that modulate vascular tone (Tian et al., 1997), and cardiac function (Tian et al., 1998; Sharp and Bernaudin, 2004). HIF3α is expressed at low levels in the cerebral cortex and hypothalamus (Heidbreder et al., 2000), as well as in the heart, lung, kidney (Makino et al., 2002; Heidbreder et al., 2007; Pasanen et al., 2010), pancreas, skeletal muscle (Pasanen et al., 2010), and adipose tissue (Pfeiffer et al., 2016).
HIF1α and HIF2α (HIFα) possess an oxygen-dependent degradation domain (ODD) (Huang et al., 1998), an N-terminal transactivation domain (aa 531-575), and a C-terminal transactivation domain (aa 786-826) (Jiang et al., 1996; Semenza, 2000). Under normal oxygen conditions, HIFα is a short-lived protein, with a half-life of less than 5 minutes and a low steady state level (Huang et al., 1998) (room air is 21% oxygen (160 mmHg), although oxygen levels in the brain are lower (1-5%) (Sharp and Bernaudin, 2004) and normal oxygenation of brain tissue occurs at ~pO2=35 mmHg (Carreau et al., 2011)). Under normoxic conditions, HIF1α and HIF2α are hydroxylated on 1 or 2 conserved proline residues (Pro402 and Pro564 in humans) within the ODD by PHDs (Tian et al., 2011; Snell et al., 2014). PHDs are dioxygenases that require O2, iron, and 2-oxoglutarate as cofactors (Epstein et al., 2001; Appelhoff et al., 2004; Berra et al., 2006). Hydroxylation of these residues changes the conformation of HIFα allowing the von Hippel Lindau protein (pVHL) to recognize and bind to it (Masoud and Li, 2015). Other factors bind to pVHL (elongin B and C, Cullin, and RBX1) (Kibel et al., 1995; Cockman et al., 2000; Sharp and Bernaudin, 2004) resulting in an E3 ubiquitin ligase complex, targeting HIF for ubiquitylation and proteasomal degradation (Maxwell et al., 1999; Sharp and Bernaudin, 2004; Samanta and Semenza, 2017) (Figure 1). HIFα protein is constantly produced but immediately degraded when sufficient oxygen is present, while the HIF1β subunit that is necessary for dimerizing with HIFα and translocating the HIF1α/β complex to the nucleus for DNA binding, is constitutively produced in normoxic and hypoxic conditions (Wang et al., 1995; Samanta and Semenza, 2017). There are differing reports pertaining to changes in Hif1α RNA with changes in oxygen tension. In one report hypoxia (1% O2) results in increased levels of HIF1α RNA and protein (Wang et al., 1995) while in another HIF1α protein, but not RNA, is increased with exposure to reduced oxygen (1%) (Huang et al., 1996). Cellular hypoxia prevents O2 activation of PHD and degradation of HIFα, allowing stabilization and accumulation of HIFα and the induction of its target genes. A family of 3 PHD genes has been described: egln2 (encodes PHD1), egln1 (encodes PHD2), and egln3 (encodes PHD3) (Epstein et al., 2001). Each of these have specificity for different hydroxylation sites of HIFα, and different expression levels and patterns (Appelhoff et al., 2004). PHD2 is able to hydroxylate both proline residues of HIF1α (human) (Huang et al., 2002; Berra et al., 2006) and has relatively more influence on the degradation of HIF1α than HIF2α (Appelhoff et al., 2004). Factor Inhibiting HIF (FIH) also hydroxylates HIF1α in the presence of oxygen, preventing the HIF1α/β complex from binding to the coactivator p300 and CREB-binding protein (CBP), which bind to and activate HIF target genes (Kallio et al., 1998; Lee et al., 2004; Ruas et al., 2010) (Figure 1).
In addition to hypoxia, HIF1α is stabilized by the presence of ROS (Chandel et al., 2000; Brunelle et al., 2005). The mitochondrial ETC is composed of Complexes I through V, and inhibition at several different points has been shown to interfere with the stabilization of HIF1α during hypoxia. Pharmacological inhibitors of Complex I , MPP+ and rotenone (Agani et al., 2000; Agani et al., 2002), or genetic deletion of Complex I (DeHaan et al., 2004) block HIF1α stabilization. Mitochondrial DNA-depleted ρo cells, which do not have a functional mitochondrial respiratory chain, do not increase ROS generation or stabilize HIF1α under hypoxic conditions (Chandel et al., 1998; Chandel et al., 2000). Further studies have demonstrated that the Qo site of Complex III is necessary for the generation of ROS and the stabilization of HIF1α during hypoxia (Bell et al., 2007). The addition of antioxidant enzymes to wild type or ρo cells decreases the stabilization of HIF1α during hypoxia (Chandel et al., 1998; Brunelle et al., 2005; Bell et al., 2007). The generation of ROS by the ETC during oxidative phosphorylation promotes HIF stabilization, however oxygen consumption or ATP production are not necessary for HIF accumulation. The addition of antioxidants reduces HIF accumulation, while hydrogen peroxide or free radical exposure promotes HIF stabilization (Brunelle et al., 2005). However, antioxidant administration does not affect HIF1α stabilization when mitochondrial respiration is inhibited. One possible explanation is that the inhibition of mitochondrial respiration may redistribute oxygen during hypoxic conditions, allowing the PHDs to function and reduce HIF1α stabilization (Hagen et al., 2003). Pharmacological inhibition of PHD by iron chelation or reduction of 2-oxoglutarate also promotes the stabilization of HIF during normoxic as well as hypoxic exposure (Lee et al., 2009; Siddiq et al., 2009). The regulation of HIF1α stabilization is complex, and ultimately depends on the availability of O2 and the activity of PHDs.
Hypoxia induces changes in the brain through HIF
Hypoxia in the brain is of significant importance because the brain utilizes a disproportionate amount of oxygen and glucose relative to its size and to other organs (Raichle and Gusnard, 2002; Mergenthaler et al., 2013). Further, neurons are particularly sensitive to changes in metabolism (Magistretti and Allaman, 2015). Oxygen pressure (pO2) is regulated by constriction or dilation of arterioles and by changes in capillary density (LaManna et al., 2004). Microvessel density has been shown to increase with hypoxia, by hypobaric control (LaManna et al., 2004; Benderro and Lamanna, 2011), induced ischemia (Pichiule et al., 2003), and by physical activity (Isaacs et al., 1992; Swain et al., 2003). Conversely, increases in capillary density may be reversed by reoxygenation (Benderro and LaManna, 2014) or when hyperoxia occurs (Benderro et al., 2012; Benderro and LaManna, 2013). Increased expression and accumulation of the transcription factor HIF1α, and subsequent transcription of some of its target genes, including erythropoetin (EPO), VEGF, and Angiopoetin 2 (ANG2), are activated when oxygen levels are reduced in the brain (Chavez et al., 2000) but return to baseline with chronic hypoxia (14-21 days) (Chavez et al., 2000; LaManna et al., 2004) or reoxygenation (Chavez and LaManna, 2002; Benderro and LaManna, 2014). In aged rodent brain or under hyperoxic conditions dysregulation of HIF1α also occurs. For example, 24 month old C57BL/6 mice exhibit HIF1α activation and accumulation that are attenuated and delayed, compared to young (4 month C57BL/6) mice (Ndubuizu et al., 2009; Ndubuizu et al., 2010; Benderro and Lamanna, 2011). This may be attributed to an increase in its degradation due to higher PHD protein in older animals (Ndubuizu et al., 2009). The reduction of HIF1α stabilization and accumulation that occurs with reoxygenation or chronic hypoxia does not occur under hyperoxic conditions (Benderro et al., 2012; Benderro and LaManna, 2013).
Preconditioning in the brain by hypoxia, oxidative stress and exercise-induced hypoxia
Controlled hypoxia in the brain affords preconditioning, a protective mechanism where a sublethal exposure to hypoxia prevents neuronal death from a later, normally detrimental bout of ischemia (Liu et al., 1992; Perez-Pinzon et al., 1997; Bernaudin et al., 2002; Ratan et al., 2007). Molecules activated by hypoxia have been demonstrated to be important for neuroprotection by ischemic preconditioning (Neumann et al., 2015; Morris-Blanco et al., 2016; Koronowski et al., 2018). Pharmacological preconditioning has been shown to protect DA neurons from agents that induce oxidative stress (see Leak, 2018, for review). In one study, dopaminergic cells treated with a sublethal concentration of 6-OHDA, used to produce lesions in the striatum, were later spared when administered a toxic dose of the same compound (Leak et al., 2006; Leak, 2018). Several factors are necessary to provide this protective conditioning; mild oxidative stress (activation of the ARE by 6-OHDA), increases in phosphorylated ERK1/2, AKT, JNK and Bcl2, and the ability to increase gene expression (Leak et al., 2006).
Exercise has been demonstrated to act as preconditioning protection from ischemic incidents through a number of factors (Ding et al., 2006b; Liebelt et al., 2010; Iadecola and Anrather, 2011; Zhang et al., 2011; Dornbos et al., 2013; Rezaei et al., 2018)(and reviewed in (Islam et al., 2017)). Increases in HIF1α, AMPK, and VEGF have been demonstrated with exercise and coincide with protection from post-exercise ischemic insults (Dornbos et al., 2013; Rezaei et al., 2018).
Vigorous exercise provides preconditioning via HIF1α for DA neuron survival
Preclinical studies support the neuroprotective role of exercise on DA neurons in the SNpc and describe the type or intensity of the exercise (Gerecke et al., 2010) necessary to provide protection, as well as possible mechanisms or contributions to the effect (Gerecke et al., 2012). Voluntary wheel running protects the DA neurons of the SNpc in an acute MPTP mouse model of PD. Administration of the mitochondrial complex I inhibitor, MPTP, leads to oxidative stress (Nicklas et al., 1987; Sriram et al., 1997; Jackson-Lewis et al., 2015) and dopaminergic neuron death in the SNpc in some strains of mice (Sundstrom et al., 1987; Muthane et al., 1994; Hamre et al., 1999; Sedelis et al., 2000). This neurotoxin is used to model parkinsonism since it was first discovered to rapidly produce symptoms such as rigidity, "pill-rolling" tremor, flexed posture, shuffling gait, bradykinesia, and hallucinations (Langston et al., 1983) in patients who had self-administered incorrectly synthesized heroin, which resulted in toxic MPTP. Brains from these patients at autopsy revealed ongoing (chronic) nigrostriatal damage years after the exposure to MPTP (Langston et al., 1999). Acute administration of MPTP (20 mg/kg in 4 doses 2 hours apart) to C57BL/6 mice results in a 58%-62% DA neuron loss in the SNpc, identified by cell counts of TH immunostained neurons (Jackson-Lewis et al., 2015; Hamre et al., 1999; Smeyne et al., 2015; Munoz-Manchado et al., 2016; Smeyne et al., 2016). The amount of wheel running necessary to afford protection was further elucidated to be 3 months of voluntary running at 18,000 revolutions per night (9.2% cell loss), while mice allowed to run only 12,000 revolutions per night for 3 months (30.7% cell loss), or 18,000 revolutions for 2 months (16.6% cell loss) were partially protected compared to mice in standard housing (SH). Mice allowed to run 6000 revolutions/night (35.6% loss) or unrestricted running for 1 month (41.5% cell loss) did not show a difference in cell loss compared to MPTP treated mice in SH conditions (Gerecke et al., 2010). The high threshold for voluntary running to protect SNpc DA neurons from the toxic insult may be comparable to the vigorous or forced exercise in human studies shown to reduce the risk of developing PD or improve motor symptoms once the disease has been diagnosed.
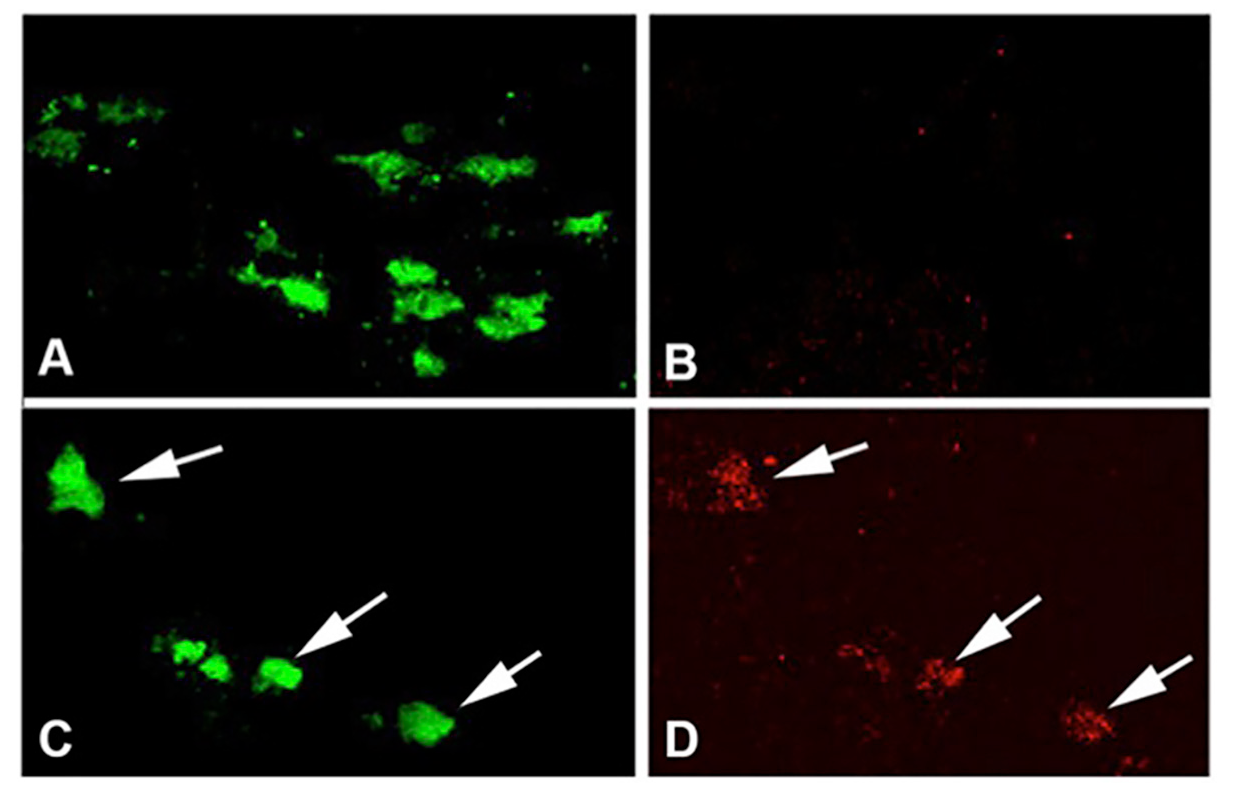
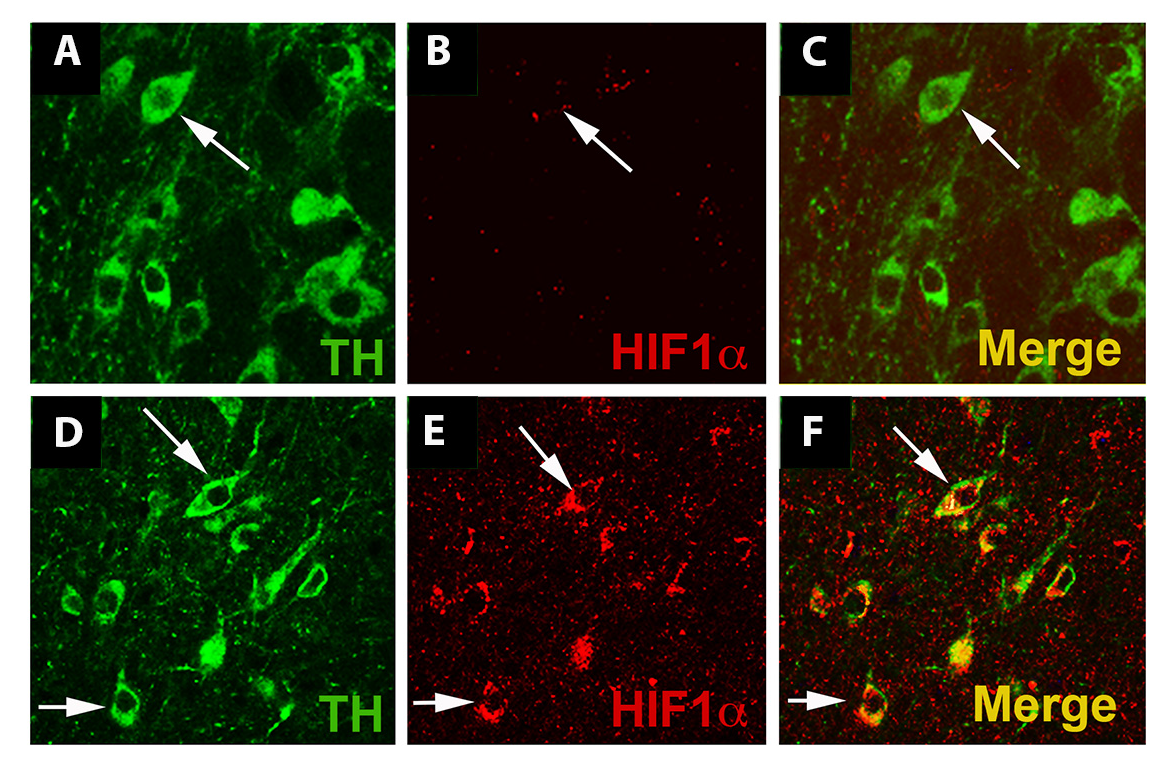
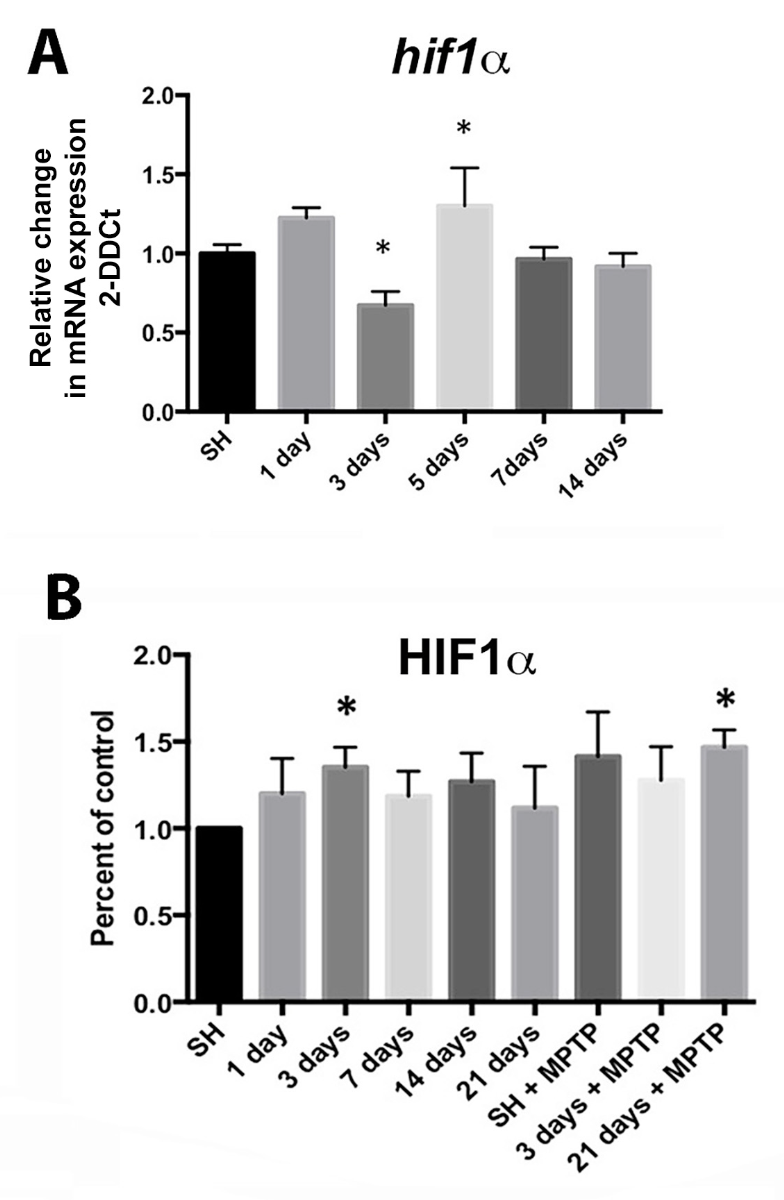
Preconditioning by hypoxia generated with exercise through HIF1α plays a role in the protection of SNpc DA neurons. In the MPTP toxin model of PD discussed above, where voluntary running is protective, it was hypothesized that the vigorous running necessary to provide DA neuron protection may involve HIF due to its role as a mediator of genes involved in cellular stress such as hypoxia, oxidative stress, and energy metabolism (Smeyne et al., 2015). In this paradigm, the inhibition of complex I and oxidative stress caused by administration of MPTP is the lethal insult that reduces DA neuron survival. Vigorous running is proposed as the means to produce a sublethal preconditioning insult, in the form of hypoxia and mild oxidative stress, which is later protective to the neurons. In fact, the DA neurons of the SNpc become hypoxic with intense running early in the exercise protocol (Smeyne et al., 2015) at 2-3 days of running (Figure 2). In sedentary conditions, HIF1α is not expressed at detectable levels by immunofluorescence, but after 90 days of voluntary exercise, the protein expression is enhanced (Figure 3). In addition, HIF1α protein is increased in the SN at 3 days of running, while hif1α mRNA is reduced at 3 days and rebounds at 5 days after running begins (Figure 4). HIF1α protein and RNA levels return to baseline at 7 days of running exercise, and remain at basal levels for 2-3 weeks. This is similar to the return to baseline of HIF1α with chronic hypoxia (Chavez et al., 2000). Administration of MPTP at 3 weeks of voluntary running also results in an increase of HIFα protein. Reduction of Hif1α in postnatal DA neurons by conditional knockout abrogates the protective effect of running with MPTP administration, and in addition, vigorous running alone (>16,000 average daily revolutions) reduces the number of SNpc DA neurons (Smeyne et al., 2015). It may be that the initial aerobic running and resulting hypoxia in neurons initiates a process where, with continued and regular running, there is a switch in metabolic pathways that is sustained and shields the neuron from later oxidative or toxic insult.
In a new window | Download PPT
Figure 2: Exercise induces hypoxia in SNpc DA neurons. (A, B) C57BL/6 mice in the standard housing condition taken during the evening active period show DA neurons labeled with TH (A) but not Elk3-51, an indicator of hypoxic conditions in the cell (Bergeron et al., 1999)(B). (C, D) SNpc from C57BL/6 mice in wheel cages taken during day 2 in the active period (running) show DA neurons co-labeled with TH (C) and Elk3-51 (D). Reprinted from Smeyne et al. (2015) with permission from Elsevier.
In a new window | Download PPT
Figure 3: Exercise enhances HIF1α immunostaining in dopaminergic cells. (A-C): SNpc from WT mice housed under standard conditions. An arrow identifies a TH+ dopaminergic neuron (A, C, green). (D-F): SNpc from WT mice housed in wheel cages and allowed voluntary access to running for 3 months. Exercise does not alter the appearance of TH+ neurons in the SNpc (D, F) (arrows), but HIF1α (E) immunostaining is more evident in SNpc DA neurons (arrows). Each image is taken from a single 1 micrometer z-plane to insure that any co-localization of immunofluorescence is from the same cell. Reprinted from Smeyne et al. (2015) with permission from Elsevier.
In a new window | Download PPT
Figure 4: Expression of hypoxia sensitive molecules in the SN is modulated by wheel running exercise. (A) hif1α expression from SN mRNA isolated at 1, 3, 5, 7, and 14 days of running show significant reduction at 3 days and increase at 5 days of running compared to controls in standard housing (SH). * = p ≤ 0.05, n = 17-18 for SH, 5-8 for each exercise condition. (B) HIF1α protein from the SN is significantly increased at running day 3 and after 21 days of running plus MPTP administration (SN taken at 2 hrs. after the last MPTP injection). * = p ≤ 0.05. Error bars = ± SEM. Values are expressed as percent of control relative to SH after normalization to β-actin n = 4 SN for each condition. Reprinted from Smeyne et al. (2015) with permission from Elsevier.
DJ-1 regulates HIF1α stability and affects dopamine neurons
Another point of regulation of HIF1α is DJ-1 (Figure 1), the product of the PARK7 gene, that functions as an antioxidant (Andres-Mateos et al., 2007; Cookson, 2012) and a regulator of pVHL (Parsanejad et al., 2014). PARK7 was shown to be important to DA neuron health when an increase in risk for the development of PD was linked to families with a PARK7 mutation (Bonifati et al., 2003). DJ-1 is protective to SNpc DA neurons under oxidative stress conditions (Kim et al., 2005; Oh et al., 2018). Reduction of DJ-1 results in DA neuron loss with oxidative insult, while in normal conditions, it's decrease does not affect the number of SNpc DA neurons (Zhou and Freed, 2005).
DJ-1 expression is necessary for the phosphorylation of AKT and ensuing mTOR function that helps to stabilize HIF1α. Reduction of DJ-1 in tumor cells also reduces phosphorylation of AMPK, a metabolic sensor that responds to hypoxia and glucose deprivation (Vasseur et al., 2009). Knockdown of DJ-1 in mouse embryonic fibroblasts (MEFs) reduces the amount of ATP, compared to wild-type MEFs, and the AMP:ATP ratio is also important for cell survival, especially under metabolically stressful conditions (Vasseur et al., 2009). SNpc neurons of DJ-1(-/-) mutant mice are hypersensitive to MPTP administration, and in vitro DJ-1 deficient cortical neurons have increased sensitivity to oxidative stress, both of which can be reversed by expression of DJ-1 (Kim et al., 2005). Modulation of AKT activation by DJ-1 has also been shown to be important in the protection of SNpc DA neurons and striatal DA terminals with in vivo administration of MPTP (Aleyasin et al., 2010). DJ-1 also negatively regulates pVHL, leading to increased stabilization of HIF1α (Parsanejad et al., 2014). Loss of DJ-1 reduces HIF1α levels in hypoxic or oxidative stress conditions, while accumulation of HIF1α rescues DJ-1 deficient DA neurons from MPTP toxicity (Parsanejad et al., 2014). DJ-1 deficiency also promotes mitochondrial fragmentation in vitro and increased ROS production in mitochondria isolated from brain (Irrcher et al., 2010). The antioxidant N-acetylcysteine (NAC) as well as DJ-1 expression in DJ-1 -/- neurons rescues the mitochondria morphology defect, although mutant DJ-1 expression, a C106A point mutation which is critical for DJ-1 antioxidant effects (Canet-Aviles et al., 2004), does not rescue the defect. In addition, lymphoblasts from PD patients with a DJ-1 (Park7) mutation show diminished HIF1α stabilization in the presence of oxidative stress (Parsanejad et al., 2014) and fragmented mitochondria (Irrcher, 2010). Neuroprotection by exercise is not afforded to SNpc DA neurons in DJ-1KO mice. Three months of exercise that protects WT mice after administration of MPTP, does not spare DA neurons in the SNpc of DJ-1 KO mice even though the MPTP dose necessary to produce the lesion is lower (Smeyne, unpublished research).
Conclusions
Numerous studies provide evidence that regular (vigorous) physical activity can reduce the risk of developing PD, as well as improve motor and cognitive function when used as therapy post-diagnosis. Regular physical activity is also a means to promote preconditioning in the brain; a phenomenon that has been shown to increase neuron survival in the event of later insult(s). Preconditioning afforded to neurons by pharmacological means and neuroprotection by preischemic sublethal hypoxia have been thoroughly demonstrated. Related to PD, DA neurons located in the SNpc benefit from vigorous aerobic exercise that generates hypoxia, inducing alterations in HIF1α-mediated gene expression and modifying neuronal metabolism. These alterations have been described in skeletal (White and Schenk, 2012; Kent and Fitzgerald, 2016) and cardiac muscle (Zhou et al., 2006), but remain to be fully elucidated in neurons and glia. The type and amount of physical activity necessary to promote metabolic change varies with the brain region, and it is possible that the threshold for preconditioning to acquire neuroprotection is unique for individuals depending on prior health, physical fitness, and genetic profile. Determining the molecular pathways and the particular substrates that confer neuroprotection through exercise may make it possible to find and provide treatment for individuals who are not able to participate in physical activities.
References
Michelle Smeyne1
1Department of Neuroscience, Thomas Jefferson University, 900 Walnut Street, Philadelphia, PA 19107.
Richard Jay Smeyne1
1Department of Neuroscience, Thomas Jefferson University, 900 Walnut Street, Philadelphia, PA 19107.
Corresponding author:
Michelle Smeyne
Email: michelle.smeyne@jefferson.edu
In a new window | Download PPT
Figure 1: Role of HIFα in hypoxia. Within the cell molecular oxygen is tightly regulated, and a key component of this oxygen-sensing pathway is the transcription factor hypoxia inducible factor (HIF). During periods of normoxia, HIF1α is continually made and degraded. The degradation of HIF1α requires interaction with prolyl hydroxylases (PHDs), which are activated by molecular oxygen and 2-oxoglutarate, an intermediate of the TCA cycle that also functions as a substrate for formation of ATP in the electron transport chain (ETC). When HIF1α is hydroxylated by PHDs it changes conformation so that it can be recognized by von Hippel Lindau (VHL) factor. This complex interacts with the E3 ubiquitin ligase complex and is targeted for proteosomal degradation. Another means to prevent HIF-induced gene transcription is through the action of factor inhibiting hypoxia (FIH), which inhibits the formation of the HIF1α/HIF1β/p300/CBP complex. When cells are under oxygen stress they become hypoxic; a process where PHDs are inhibited and HIF1α accumulates. DJ-1 is a negative regulator of pVHL and also allows HIF1αaccumulation. Once HIF1α is stabilized and accumulates, it binds to HIF1β, complexes with p300 and CBP, and translocates to the nucleus where it binds to the Hypoxia Responsive Elements responsive elements (HRE’s) on DNA. (Adapted from Jochmanova et al., 2013).
In a new window | Download PPT
Figure 2: Exercise induces hypoxia in SNpc DA neurons. (A, B) C57BL/6 mice in the standard housing condition taken during the evening active period show DA neurons labeled with TH (A) but not Elk3-51, an indicator of hypoxic conditions in the cell (Bergeron et al., 1999)(B). (C, D) SNpc from C57BL/6 mice in wheel cages taken during day 2 in the active period (running) show DA neurons co-labeled with TH (C) and Elk3-51 (D). Reprinted from Smeyne et al. (2015) with permission from Elsevier.
In a new window | Download PPT
Figure 3: Exercise enhances HIF1α immunostaining in dopaminergic cells. (A-C): SNpc from WT mice housed under standard conditions. An arrow identifies a TH+ dopaminergic neuron (A, C, green). (D-F): SNpc from WT mice housed in wheel cages and allowed voluntary access to running for 3 months. Exercise does not alter the appearance of TH+ neurons in the SNpc (D, F) (arrows), but HIF1α (E) immunostaining is more evident in SNpc DA neurons (arrows). Each image is taken from a single 1 micrometer z-plane to insure that any co-localization of immunofluorescence is from the same cell. Reprinted from Smeyne et al. (2015) with permission from Elsevier.
In a new window | Download PPT
Figure 4: Expression of hypoxia sensitive molecules in the SN is modulated by wheel running exercise. (A) hif1α expression from SN mRNA isolated at 1, 3, 5, 7, and 14 days of running show significant reduction at 3 days and increase at 5 days of running compared to controls in standard housing (SH). * = p ≤ 0.05, n = 17-18 for SH, 5-8 for each exercise condition. (B) HIF1α protein from the SN is significantly increased at running day 3 and after 21 days of running plus MPTP administration (SN taken at 2 hrs. after the last MPTP injection). * = p ≤ 0.05. Error bars = ± SEM. Values are expressed as percent of control relative to SH after normalization to β-actin n = 4 SN for each condition. Reprinted from Smeyne et al. (2015) with permission from Elsevier.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 15381 | 45 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA