Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Mechanisms of innate preconditioning towards ischemia/anoxia tolerance: lessons from mammalian hibernators
Time:2019-07-03
Number:10417
Author Affiliations

Conditioning Medicine, 2019. 2(3): 134-141.
Abstract
Hibernating mammals exhibit an innate physiological ability to withstand dramatic fluctuations in blood flow that occurs during hibernation and arousal or experimental models of ischemia reperfusion without significant damage. These innate adaptations are of significance particularly to organs that are highly susceptible to energy deprivation, such as the brain and the heart. Among vertebrates, the arctic ground squirrel (AGS) is a species that tolerates ischemic/anoxic insult. During the process of entering hibernation, a state of prolonged torpor, the AGS undergoes a profound decrease in respiratory rate, heart rate, blood flow, cerebral perfusion, and body temperature (Tb). The reduced level of blood flow during torpor resembles an ischemic state, albeit without energy deficit. During the process of arousal or emergence from torpor, however, when Tb, respiratory rate, heart rate, and blood flow rapidly returns to pre-torpid levels, the rapid return of cerebral blood flow mimics aspects of reperfusion such as is seen after stroke or cardiac arrest. This sublethal ischemic/reperfusion insult experienced by AGS during the process of arousal may precondition AGS to tolerate otherwise lethal ischemic/reperfusion injury induced in the laboratory. In this review, we will summarize some of the mechanisms implemented by mammalian hibernators to combat ischemia/anoxia tolerance.
Abstract
Hibernating mammals exhibit an innate physiological ability to withstand dramatic fluctuations in blood flow that occurs during hibernation and arousal or experimental models of ischemia reperfusion without significant damage. These innate adaptations are of significance particularly to organs that are highly susceptible to energy deprivation, such as the brain and the heart. Among vertebrates, the arctic ground squirrel (AGS) is a species that tolerates ischemic/anoxic insult. During the process of entering hibernation, a state of prolonged torpor, the AGS undergoes a profound decrease in respiratory rate, heart rate, blood flow, cerebral perfusion, and body temperature (Tb). The reduced level of blood flow during torpor resembles an ischemic state, albeit without energy deficit. During the process of arousal or emergence from torpor, however, when Tb, respiratory rate, heart rate, and blood flow rapidly returns to pre-torpid levels, the rapid return of cerebral blood flow mimics aspects of reperfusion such as is seen after stroke or cardiac arrest. This sublethal ischemic/reperfusion insult experienced by AGS during the process of arousal may precondition AGS to tolerate otherwise lethal ischemic/reperfusion injury induced in the laboratory. In this review, we will summarize some of the mechanisms implemented by mammalian hibernators to combat ischemia/anoxia tolerance.
1. Hibernation Physiology
Hibernation is a behavioral, physiological, and molecular adaptation exhibited by diverse mammalian species to withstand protracted periods or seasons of insufficient or unpredictable food availability. Hibernation is characterized by multiple bouts of torpor that are interrupted by brief periods of euthermia. During hibernation, there is a profound decrease in whole-body metabolic rate and body temperature (Tb), which last from days to several weeks, known as prolonged torpor. The arctic ground squirrel (AGS), a species native to the northern regions of Alaska and Canada, hibernate for approximately 7-8 months each year (Barnes, 1989). During hibernation, AGS and other species of ground squirrels enter a highly regulated and reversible state of prolonged torpor. The period of torpor is characterized by a profound decrease in respiratory rate, heart rate, blood flow, cerebral perfusion, and Tb. During torpor, Tb falls to within a few degrees of the ambient temperature (Carey et al., 2003a) and typically ranges from 2 to 10°C for most temperate-zone hibernators; however, in AGS, Tb can drop to as low as −2.9°C (Barnes, 1989) and metabolism can be reduced to 1–2% of resting metabolic rate. Torpor bouts are interrupted by arousal periods in which the AGS enters the state of interbout euthermy (Barnes, 1989). During an arousal in AGS, physiological changes that took place during torpor return to normothermic values for periods lasting approximately 24h (Daan et al., 1991). Species that hibernate differ in the depth and duration of torpor bouts, but when Tb falls below 30°C they all have in common the drive to interrupt torpor bouts with these periods of interbout arousal (Carey et al., 2003a; Dausmann et al., 2004).
2. Hibernation and mechanisms of neuroprotection
In mammals, the brain constitutes approximately 3% of the total body mass and utilizes 20% of the body’s O2. Much of the high-energy requirements of brain tissue are for maintenance of ionic gradients across excitable plasma membranes (Boveris and Chance, 1973). Within a few minutes of pathological conditions, such as anoxia and ischemia, the mammalian brain becomes isoelectric, suffers depletion of high energy intermediates, and produces an increase in lactate (Lowry et al., 1964; Heiss et al., 1976; Astrup et al., 1977). The loss of the ion gradient (depolarization) (Hansen, 1985) results in increased intracellular calcium concentrations, leading to activation of calcium-dependent processes as well as to a massive release of neurotransmitters, including the excitatory neurotransmitter glutamate (Lipton, 1999; Doyle et al., 2008). Increased glutamate concentration in synaptic clefts may activate N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5 methyl-4-isoxazolepropionic acid (AMPA) receptors, causing excitotoxic calcium influx (Lipton, 1999). Metabolic derangements with subsequent acidosis also activate pH sensitive ion channels, which contribute to calcium influx and acidotoxicity (Bhowmick et al., 2017b).
Although most mammalian brains are highly sensitive to anoxia, ischemia, and subsequent energy supply, not all mammals are equally susceptible. Hibernating mammals are natural models of tolerance to insults, such as ischemia, which would be injurious or lethal to non-hibernating species. Tolerance to hypoxia in hibernating species was first documented in the early 1800’s (Biorck et al., 1956). Although hibernating animals experience prolonged ischemic-like low levels in blood flow during torpor and the reperfusion-like return of blood flow during arousal, no neuronal damage ensues (Frerichs et al., 1994; Ma et al., 2005). Interestingly, evidence also suggests that the blood-brain barrier remains intact during torpor (Wells, 1972) in contrast to pathological conditions (Bhowmick et al., 2019b). As early as the 1960’s, mechanisms of hypoxia tolerance in hibernating species were proposed to be attributable to factors other than cold temperature (Bullard and Funkhouser, 1962). Mechanisms unrelated to temperature may include hypo-metabolism, differential modulation of NMDA receptors (NMDAR), immunosuppression, anticoagulant properties of the blood, and antioxidant defenses (Drew et al., 2001; Zhou et al., 2001). Subsequent in vitro studies in thirteen-lined ground squirrel explained that hibernation season (Lindell et al., 2005; Kurtz et al., 2006) or lowering temperature (Frerichs and Hallenbeck, 1998) in brain slices from active animals contributes towards tolerance to ischemia-reperfusion (I/R) injury. Later, in vivo studies demonstrated that tolerance is not just attributed to hibernation season or hypothermia since AGS and other species of ground squirrels tolerate hypoxia (D'Alecy et al., 1990) and ischemic-like conditions even when they are not hibernating (Dave et al., 2006; Dave et al., 2009; Bhowmick et al., 2017a). This suggests that resistance to I/R injury in hibernators is not just attributed to hibernating season or hypothermia but could also be due to tissue and circulating factors that persist in summer active hibernators. Investigation into the mechanisms in hibernating species that provide this protection from insults is a promising approach that could lead to effective therapeutics for stroke and cardiac arrest, where cerebral I/R injury produces death and long-term disability.
2.1 Energy metabolism
Reduction in metabolic rate is thought to be one of the key processes to decrease oxygen demand (Heldmaier et al., 2004). Several species of turtles suppress metabolism while overwintering in the mud at the bottom of a pond in a hibernation-like state. These turtles also tolerate hypoxia under laboratory conditions and have been studied in detail. The freshwater turtle (Trachemys scripta) is one of the most robust hypoxia-tolerant vertebrates (Milton, 2019). These animals can withstand a completely anoxic state for days at room temperature or weeks at colder temperatures (Jackson and Ultsch, 2010). Such profound tolerance to anoxia in turtles is accomplished by entering into a state of deep reversible hypo-metabolism. The state of hypo-metabolism, a type of hibernation that like mammalian hibernation reduces demand for ATP by 70 to 80 percent, helps the turtles survive on the ATP generated through anaerobic metabolism (Hochachka and Lutz, 2001), while lactic acidosis is buffered by CaCO3 stored in the animal’s shell. Likewise, in AGS, during hibernation, oxygen consumption decreases from ~1 to 0.01·ml·O2·g–1·h–1 (Buck and Barnes, 2000; Toien et al., 2001), electroencephalogram (EEG) remains isoelectric (Frerichs et al., 1994), and the heart and respiratory rates decrease by >10-fold (Ma et al., 2005). Suppressed O2 demand clearly contributes to anoxia tolerance during the hibernation state. Lowered tissue temperature in tissues isolated from torpid animals contributes to tolerance possibly by suppressing metabolism. Studies have shown that the hibernating state contributes to tolerance to oxygen and glucose deprivation in some species and that this state-dependent tolerance is additive with the protective influence of lower tissue temperatures (Frerichs and Hallenbeck, 1998). Reducing tissue temperature decreases cerebral metabolic rate by approximately 5 percent for every 1°C decrease in tissue temperature (Yenari et al., 2008). In AGS heart, a shift from carbohydrate to lipid metabolism during the hibernation season contributes to resistance to myocardial I/R (Quinones et al., 2016). Similarly, season is necessary for tolerance to I/R in liver and gut in 13-lined ground squirrels (Kurtz et al., 2006), and although metabolic rate and core TB decrease slightly during the hibernation season, even when animals are not torpid, it remains unclear how much metabolic suppression contributes to seasonal protection from I/R injury. In AGS brain tissue held at a normothermic temperature of 36°C, we have seen that tolerance to oxygen glucose deprivation (OGD) is the same during the summer and winter (hibernation) season. Indeed, in AGS, tolerance to cerebral I/R persists even in the absence of metabolic suppression when animals are not hibernating, at normothermic (36-37°C) tissue temperature and outside of the hibernation season. Such tolerance has been observed in vitro (Ross et al., 2006; Christian et al., 2008; Bhowmick and Drew, 2017; Bhowmick et al., 2017a) and in vivo (Dave et al., 2006; Dave et al., 2009).
Moreover, recent studies have demonstrated that resistance to OGD in vitro in AGS is not due to preservation of energy homeostasis. Depletion in ATP is a hallmark of ischemic injury (Sato et al., 1984). Although a coordinated balance between ATP-demand and ATP-supply prevails in other anoxia tolerant species, such as the painted turtle (Bickler and Buck, 2007), AGS display significant tolerance to ischemic injury despite ATP depletion (Figure 1 B, C) (Bhowmick et al., 2017a). This suggests that unlike hypoxia and anoxia tolerant species where tolerance is linked to preservation of ATP (Pamenter et al., 2012), AGS have evolved unique adaptations that cannot be explained by enhanced energy homeostasis, the hibernation state, or the hibernation season (Bhowmick et al., 2017a).
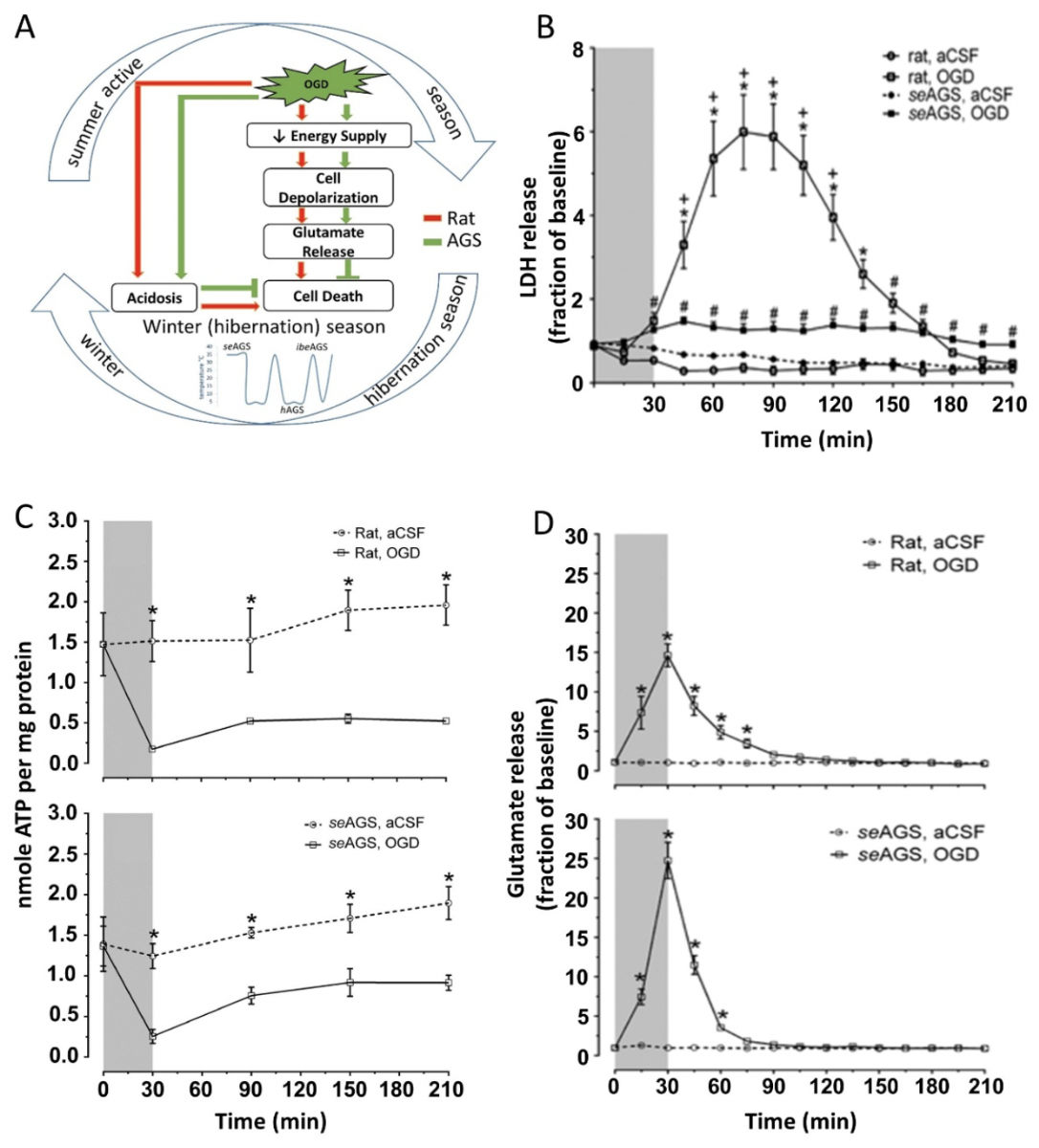
In a new window | Download PPT
Figure 1: AGS tolerate OGD injury despite ATP loss and glutamate efflux (A) Schematic representation of possible pathway in AGS tolerance to injury. (B) Lactic acid dehydrogenase (LDH) in perfusates collected every 15 min increases in rat hippocampal slices exposed to oxygen-glucose deprivation (OGD) (rat, OGD), but not in rat slices exposed to artificial cerebral spinal fluid (aCSF) (rat, aCSF), nor in slices harvested from summer euthermic AGS and exposed to aCSF (seAGS, aCSF). A small amount of LDH is released from slices collected from seAGS and exposed to OGD (seAGS, OGD). *p < 0.05 rat aCSF vs. rat OGD, +p < 0.05 rat OGD vs. seAGS OGD, #p < 0.05 seAGS aCSF vs. seAGS OGD. (C) Levels of whole tissue ATP were determined over time following bath application of treatment in slices from (a) rat (aCSF versus OGD) and (b) seAGS (aCSF versus OGD) subjected to either 30 min of aCSF or OGD followed by 3 h reperfusion. Gray bar indicates insult period. Data shown are means ± SEM, *p < 0.05 versus aCSF. (D) Time-dependent excitatory neurotransmitter (glutamate) efflux in rat and seAGS hippocampal slices induced by 30 min OGD insult. (n = 17 slices from six rats, n = 14 slices from five seAGS). Gray bar indicates insult period. *p < 0.05 for OGD versus aCSF group. Data shown are means ± SEM. [Reprinted with permission from Bhowmick et al. 2017a.]
2.2 Loss of ion homeostasis and effect of low pH in injury
Disruption in ion homeostasis has been tightly linked to cell death/injury (Bortner and Cidlowski, 2004). Ischemia or anoxia results in “ischemic” or “anoxic” depolarization and activation of acid-sensing ion channels, which in turn leads to brain damage (Kaminogo et al., 1998; Bhowmick et al., 2017b). One way of improving brain recovery from ischemic injury is to block or delay ischemic depolarization (ID) (Takeda et al., 2003; Anderson et al., 2005). Dave et al. (2009) showed that persistent ion homeostasis in AGS brain delays membrane depolarization. In both in vitro and in vivo models of cerebral ischemia, AGS delay onset of depolarization by 1.23 minutes when compared to I/R vulnerable species such as rats through activation of epsilon protein kinase C (εPKC) (Dave et al., 2009). Activation of εPKC, known to inhibit Na+/K+-ATPase and voltage-gated sodium channels, may contribute to ion channel arrest during experimental ischemia in AGS (Dave et al., 2009). Another crucial event during ischemia is the electrolyte shift that underlies anoxic depolarization and a subsequent decrease in extracellular pH that contributes to injury. Using an in vitro microperfusion approach that offered good temporal resolution throughout insult and reperfusion, we studied the role of electrolyte shift and low pH on I/R injury in AGS hippocampal slices at 36°C (Bhowmick et al., 2017b). Using an ischemic shift solution (ISS) sufficient to induce spreading depression in the cortex (Zhou et al., 2010), we found that mimicking ischemia-induced changes in the extracellular ionic microenvironment produces significant injury in hippocampal slices from non-hibernating mammals (rats), but not in AGS (Bhowmick et al., 2017a). The ISS mimics tissue acidosis sufficient to activate acid sensing ion channels, which increase calcium permeability and leads to cell death during cerebral I/R in rats and mice (Xiong et al., 2004; Bhowmick et al., 2017b). When we manipulated pH independently from other ions, low pH (~6.5) produced cell death in rats but not AGS slices (Bhowmick et al., 2017a) suggesting that AGS are immune to acid sensing ion channel activation as well as to depletion of tissue ATP. Previous studies have shown that painted turtle neurons also tolerate prolonged ISS treatment (Pamenter et al., 2012). Tolerance to low pH in AGS could be due to enhanced pH buffering capacity because in normothermic AGS, blood pH remains around 7.4 despite normal arterial PCO2 levels of 60 mmHg. Indeed, plasma HCO3−concentrations tend to be higher in AGS than in rat (Ma et al., 2005; Bogren et al., 2014b; Bogren et al., 2014a). Taken together, these findings suggest that persistent ion homeostasis and tolerance to deleterious effects of acid sensing ion channel activation may contribute to the innate tolerance to ischemic-like conditions in AGS.
2.3 Excitatory/inhibitory neurotransmitters
Neurotransmitters are broadly classified into two groups: excitatory and inhibitory. Excitatory neurotransmitters stimulate the activation of receptors and results in depolarization of the neuronal membrane thereby moving the membrane potential towards the action potential threshold. In contrast, inhibitory neurotransmitters activate receptors that cause hyperpolarization or directly decrease neuronal excitability. Previous studies have suggested that downregulation of ion channels, termed channel arrest, contributes to I/R tolerance in brain tissue from torpid ground squirrels (Doll et al., 1991). It is evident from previous studies that although AGS display delayed ischemic depolarization as compared to ischemic susceptible species, cells do eventually depolarize and release glutamate (Figure 1D) and aspartate in quantities similar to rat (Bhowmick et al., 2017a). Release of excitatory neurotransmitters in AGS contrasts with an absence of glutamate release in anoxia tolerant species such as fresh-water turtles and crucian carps (Nilsson, 1990). Moreover, addition of 500 μmol l−1 NMDA plus 20 mmol −1 KCl to activate NMDA receptors in a semi-acute slice preparation of AGS hippocampal slices fails to induce cell death as compared to significant cell death in rats (Ross et al., 2006). We found evidence of downregulation of NMDAR, specifically expression of NR1, an obligatory subunit of the NMDA receptor in AGS (both active and hibernating) compared to rat (Zhao et al., 2006). Further studies demonstrated that the Na+/K+ ATPase inhibitor ouabain enhanced survival of slices from hibernating arctic ground squirrel (hAGS). We interpreted enhanced survival in hAGS slices as evidence of channel arrest in hibernation (Ross et al., 2006). Another study using primary cortical neuronal cultures from Daurian ground squirrels (Spermophilus dauricus) found that ground squirrel neurons are better at maintaining calcium homeostasis than rat neurons, potentially through the activity of the ground squirrel Na+/Ca2+ exchanger 2 (Zhao et al., 2014). In AGS, a glutamate-induced increase in intracellular Ca2+ concentration does not exceed 400 nM in hippocampal slices prepared from both euthermic and hAGS. By contrast, the same stimulus in rat slices produces intracellular Ca2+ concentrations greater than 500 nM (Zhao et al., 2006). Similar observations of channel arrest were also observed in naked mole-rats (Peterson et al., 2012b; Peterson et al., 2012a) and other ischemia/anoxia-tolerant species such as turtles (Chrysemys picta) (Bickler et al., 2000). In previous studies, the concept of ion channel arrest has also been validated in non-hibernating species such as rats wherein knocking out the NR2A subunit results in ischemia tolerance (Morikawa et al., 1998). Taken together, the literature suggests that hibernating animals have an innate ability to preserve neuronal ion homeostasis and decrease excitotoxicity.
In the vertebrate brain, gamma aminobutyric acid (GABA) is a major inhibitory neurotransmitter that hyperpolarizes neurons through GABAA receptors. In the brain of anoxia-tolerant vertebrates such as crucian carp (Nilsson, 1990), freshwater turtles, and sea turtles (Nilsson et al., 1990; Nilsson and Lutz, 1991), a substantial increase in GABA has been documented during anoxia. We and others speculate that increased levels of GABA in the brain of anoxia tolerant animals suppress neural activity and brain energy consumption, which is an important part of these animal’s defense against anoxic brain damage (Nilsson and Lutz, 1993). Although GABA has not been measured to our knowledge during anoxia or OGD in AGS brain tissue, GABA measured with quantitative microdialysis surprisingly decreases in AGS striatum during hibernation (Osborne et al., 1999), while extracellular glutamate remains unchanged (Zhou et al., 2002).
A1 adenosine receptor (A1AR) activation during ischemic preconditioning induces ischemic tolerance in homeothermic species through activation of phospholipase C and subsequent activation of εPKC (Di-Capua et al., 2003; Raval et al., 2003; Lange-Asschenfeldt et al., 2004). Moreover, adenosine release during anoxia contributes to ion channel arrest (Perez-Pinzon et al., 1993; Pek and Lutz, 1997), including a decrease in NMDAR currents (Buck and Bickler, 1998), and dopamine release (Milton et al., 2002; Milton and Lutz, 2005), as well as cerebral blood flow (CBF) (Hylland et al., 1994). While evidence suggests that seasonal sensitization of A1AR signaling facilitates entrance into torpor in AGS (Jinka et al., 2011), ischemic tolerance is not attenuated by an A1AR antagonist despite an OGD-induced increase in adenosine release in brain slices harvested from AGS during the hibernation season (Bhowmick et al., 2017a). Overall, these studies suggest that something downstream of anoxic depolarization, tissue acidosis, and excitatory neurotransmitter release contributes to tolerance to I/R injury in AGS.
2.4 Oxidative stress in mammalian hibernators
Under normal physiological conditions, the redox environment of cells modulates signal transduction cascades that balance pro-death and pro-survival pathways (Crack and Taylor, 2005). However, during I/R injury, oxidative and nitrosative stress are traditionally I/R injury. These reactive species include hydroxyl radical, superoxide (O2•-), nitric oxide (NO), and peroxynitrite, that are highly reactive and damaging to multiple cellular components, leading to cell death. O2•- and NO react to form the potent oxidant peroxynitrite (ONOO-). In the brain, NO is formed by the NO synthase (NOS) isoforms, a family of enzymes (Guix et al., 2005; Calabrese et al., 2007). Under physiological conditions, NO is responsible for maintenance of basal CBF (Toda et al., 2009). However, during ischemia, cerebral NO rapidly increases to 2–4 μM, producing damaging levels of NO (Murphy, 1999). Concomitantly, a burst of free radicals such as O2•− (El Kossi and Zakhary, 2000; Cuzzocrea et al., 2001; Suh et al., 2008; Tang et al., 2012) is produced after the ischemic event. This scenario is deleterious because O2•− anion has a high affinity for NO, higher than for the superoxide dismutase (SOD) (Cudd and Fridovich, 1982; Huie and Padmaja, 1993). NO and O2•− react with each other in an equimolar stoichiometric ratio forming the potent oxidant, ONOO-, further contributing to injury (Beckman et al., 1990; Reiter et al., 2000; Zielonka et al., 2010).
During hibernation, entrance into hypometabolic states causes fluctuations in aerobic metabolism, which in turn alters reactive oxygen species (ROS) production and cause potential oxidative damage (Brown et al., 2012). In mammalian hibernators, intermittent arousals from torpor increase the rate of oxygen consumption to fuel thermogenesis and rewarm the body to euthermia. This process results in an increase in ROS generation, proportional to the oxygen consumed. Concomitantly, polyunsaturated fatty acid (PUFA) levels increase during hibernation and are highly susceptible to lipid peroxidation (Frank and Storey, 1995; Frank et al., 2008). Previous studies have demonstrated an increase in antioxidant defenses during anoxia/hypoxia including SOD, catalase, peroxiredoxin (PRX), thioredoxin (TRX), glutathione peroxidase, and other glutathione-linked enzymes (Hermes-Lima and Zenteno-Savin, 2002; Bickler and Buck, 2007; Hall et al., 2009) indicating that animals emerging from a hypometabolic state may generate large quantities of ROS, but be protected from oxidative stress. Expression of pro-inflammatory cytokines was also seen to be upregulated early in arousal (90 min), and normalized after 8h of arousal in Syrian hamsters that might serve to prevent or offset brain damage resulting from the substantial physiological changes accompanying torpor and their rapid change during early arousal (Cogut et al., 2018). Indeed, studies in AGS show no evidence of oxidative stress during hibernation or after arousal except in brown adipose tissue, which may be the most metabolically active tissue during arousal (Carey et al., 2000; Carey et al., 2003b; Ma et al., 2005). We found that in AGS acute hippocampal slices, using a novel microperfusion technique, that AGS resist up to 2h of OGD (Figure 1B) (Christian et al., 2008; Bhowmick et al., 2017a) despite previous evidence of anoxic depolarization (Dave et al., 2009) and concomitant demonstration of a loss of ATP (Bhowmick et al., 2017a). We also demonstrated that detoxification of ONOO- downstream of glutamate release contributes to ischemia tolerance in AGS (Figure 2 A). This conclusion is based on observations that resistance to OGD was not lost even when high concentrations of NO donor or O2•− donors were paired with OGD (Figure 2 B) (Bhowmick and Drew, 2017). ONOO- is identified as the damaging species since neither NO nor O2•− donors alone are sufficient to induce cell death in rats or AGS, however, when paired with OGD these radicals exacerbate cell death in rats. This suggests that OGD generates the endogenous NO and O2•− that when combined with exogenous NO or O2•− produces ONOO- and causes injury, and that AGS detoxifies ONOO- (Bhowmick and Drew, 2017).
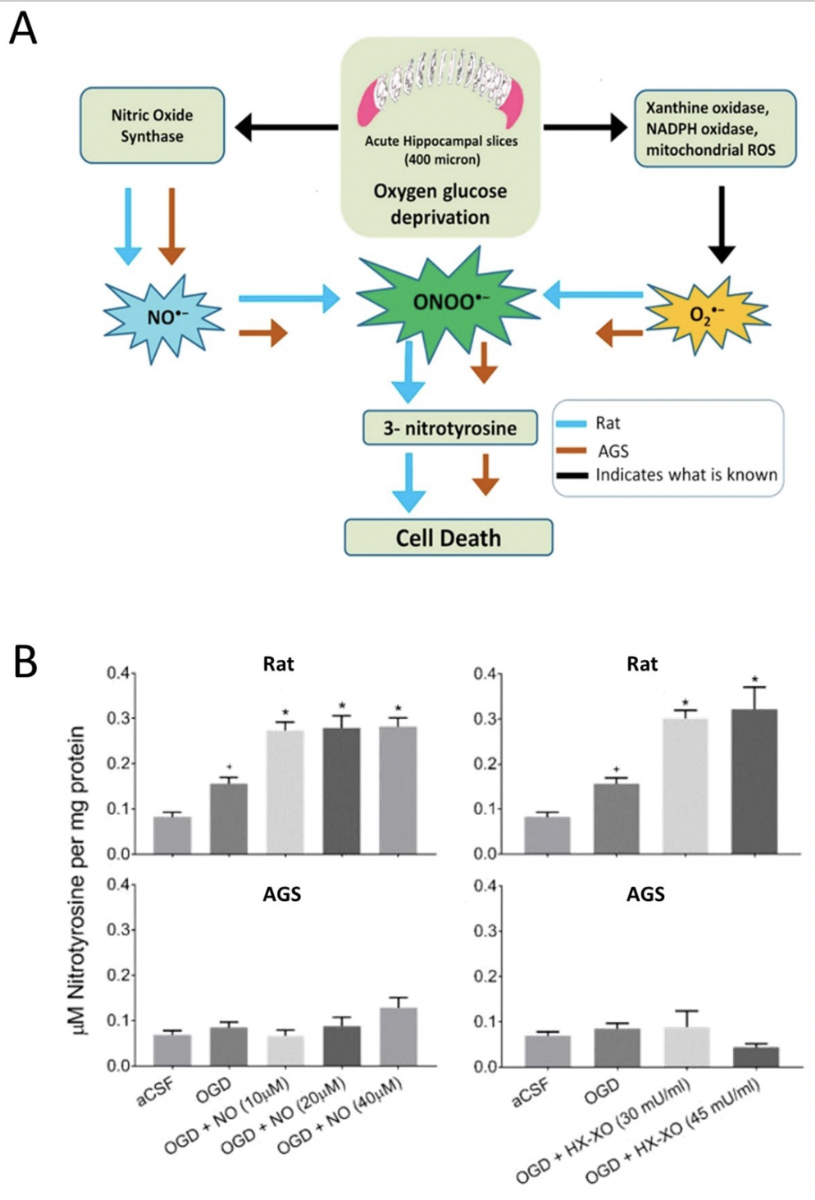
In a new window | Download PPT
Figure 2: (A) Graphical representation of peroxynitrite mediated oxygen-glucose deprivagtion (OGD) injury in ischemic susceptible rat and ischemic tolerant AGS (B) 3-Nitrotyrosine (3-NT) was measured in rat and AGS slices either subjected to NO donor along with OGD or subjected to O2•- donor along with OGD. *p< 0.05 vs. OGD, +p< 0.05 vs. aCSF. Data shown are means ± SEM. [Reprinted with permission from Bhowmick et al. 2017.]
2.5 Intracellular survival signals and neuroprotection
Although metabolic suppression has been considered as one of the key factors in providing tolerance in hibernating species, recent work has shown that a variety of protective mechanisms are activated at the molecular level in AGS. This includes mitogen activated protein (MAP) kinases, heat shock proteins, and hypoxia inducible factors (HIF). MAP kinases are serine/threonine protein kinases that play a critical role in signal transduction and are activated by phosphorylation in response to a variety of pathophysiological stimuli causing cell death (Bhowmick et al., 2019a). In AGS, tolerance to OGD, which occurs downstream of ATP depletion, is associated with decreased, but sustained baseline levels of ERK1/2 and JNK after OGD (Nilsson and Lutz, 1993). In contrast, euthermic and aroused AGS exhibit MAP kinase activation in accord with activation of a stress response. The degree of activation of ERK and JNK in euthermic or following arousal from hibernation correlates to increased HIF1α levels as a preconditioning like phenomena that may result from tissue hypoxia due to low arterial O2 and high arterial CO2 tension (Ma et al., 2005). HIF1α activates multiple mechanisms via increased expression of various downstream effectors known to contribute to ischemic preconditioning and adaptation (Ratan et al., 2004). Thus, the apparent lack of oxidative stress and pathology in aroused AGS brains after hibernation implies that some neuroprotective mechanisms are mobilized in the arousal phase and elevated levels of HIF1α could contribute to OGD tolerance (Ma et al., 2005). While the cellular stress of interbout arousals in hibernating rodents may precondition these species to resist I/R insult, this hypothesis has not yet been objectively tested.
3. Conclusion
Hibernating mammals possess a repertoire of neuroprotective adaptations that contribute to ischemic tolerance. Deciphering cellular mechanisms and unique adaptations to tolerate physiological extremes and subsequently cerebral I/R has become an active field of inquiry with new results and novel insights emerging at a steady pace. Influence of season or state of hibernation on cerebral ischemia tolerance is inconsequential in AGS's ability to resist ischemic injury. Tolerance to OGD in AGS occurs despite loss of ATP and glutamate release. Furthermore, this tolerance persists during conditions that mimic acidosis and ionic shifts characteristic of cerebral I/R. A better understanding of the cellular and molecular mechanisms that transform the cellular network into a neuroprotective state in the brain of the AGS could open new and different dimensions in translating the innate preconditioning feature displayed by mammalian hibernators for therapeutic approach.
Acknowledgements
This work was supported by the US Army Medical research and Material Command, No 0517800; the National Institute of Neurological Disorder and Stroke, Nos. NS041069-06 and R15NS070779; Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103395, and NSF IOS-1258179.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
References
Saurav Bhowmick1
1Laboratory of CNS Injury and Repair, JFK Neuroscience Institute, Hackensack Meridian Health JFK Medical Center, 65 James St., Edison, NJ 08820, USA.
Kelly L Drew2
2Department of Chemistry and Biochemistry, University of Alaska Fairbanks, Fairbanks, AK, USA; Institute of Arctic Biology, University of Alaska Fairbanks, Fairbanks, AK, USA.
Corresponding author:
Saurav Bhowmick
Email: sbhowmick@alaska.edu
In a new window | Download PPT
Figure 1: AGS tolerate OGD injury despite ATP loss and glutamate efflux (A) Schematic representation of possible pathway in AGS tolerance to injury. (B) Lactic acid dehydrogenase (LDH) in perfusates collected every 15 min increases in rat hippocampal slices exposed to oxygen-glucose deprivation (OGD) (rat, OGD), but not in rat slices exposed to artificial cerebral spinal fluid (aCSF) (rat, aCSF), nor in slices harvested from summer euthermic AGS and exposed to aCSF (seAGS, aCSF). A small amount of LDH is released from slices collected from seAGS and exposed to OGD (seAGS, OGD). *p < 0.05 rat aCSF vs. rat OGD, +p < 0.05 rat OGD vs. seAGS OGD, #p < 0.05 seAGS aCSF vs. seAGS OGD. (C) Levels of whole tissue ATP were determined over time following bath application of treatment in slices from (a) rat (aCSF versus OGD) and (b) seAGS (aCSF versus OGD) subjected to either 30 min of aCSF or OGD followed by 3 h reperfusion. Gray bar indicates insult period. Data shown are means ± SEM, *p < 0.05 versus aCSF. (D) Time-dependent excitatory neurotransmitter (glutamate) efflux in rat and seAGS hippocampal slices induced by 30 min OGD insult. (n = 17 slices from six rats, n = 14 slices from five seAGS). Gray bar indicates insult period. *p < 0.05 for OGD versus aCSF group. Data shown are means ± SEM. [Reprinted with permission from Bhowmick et al. 2017a.]
In a new window | Download PPT
Figure 2: (A) Graphical representation of peroxynitrite mediated oxygen-glucose deprivagtion (OGD) injury in ischemic susceptible rat and ischemic tolerant AGS (B) 3-Nitrotyrosine (3-NT) was measured in rat and AGS slices either subjected to NO donor along with OGD or subjected to O2•- donor along with OGD. *p< 0.05 vs. OGD, +p< 0.05 vs. aCSF. Data shown are means ± SEM. [Reprinted with permission from Bhowmick et al. 2017.]
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 10417 | 20 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA