Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Molecular mediators of cardioprotective ischemic conditioning: focus on cytokines and chemokines
Time:2019-07-03
Number:13110
Author Affiliations

Conditioning Medicine, 2019. 2(3): 122-133.
Abstract
Ischemic conditioning is a promising treatment strategy to provide cardioprotection against ischemic heart disease (IHD), and remote ischemic preconditioning (RIPC) has been successfully demonstrated in numerous preclinical and clinical studies to protect from myocardial ischemia/reperfusion injury. However, large-scale multicenter clinical trials examining the efficacy of RIPC on clinical outcomes have been disappointing. Future strategies may encompass an altered clinical study design, the use of different anesthetics to avoid propofol, and specific molecular approaches that focus on the mediators that convey the RIPC signal from the remote organ to the heart. This review focuses on cytokines and chemokines that have been suggested to, at least partially, account for the remote cardioprotective RIPC signal cue. We discuss the classical chemokine CXCL12, the atypical cytokine/chemokine macrophage migration-inhibitory factor (MIF), and the anti-inflammatory cytokine interleukin-10 (IL-10), and touch upon other cytokine- and alarmin-like factors such as adipocytokines, myokines, and RNase1. The available evidence for these factors is weighed against their roles in cardiac ischemia and their suitability as RIPC cues, including their expression and release profiles and receptors. Some of these mediators may qualify as cardioprotective target molecules in IHD.
Abstract
Ischemic conditioning is a promising treatment strategy to provide cardioprotection against ischemic heart disease (IHD), and remote ischemic preconditioning (RIPC) has been successfully demonstrated in numerous preclinical and clinical studies to protect from myocardial ischemia/reperfusion injury. However, large-scale multicenter clinical trials examining the efficacy of RIPC on clinical outcomes have been disappointing. Future strategies may encompass an altered clinical study design, the use of different anesthetics to avoid propofol, and specific molecular approaches that focus on the mediators that convey the RIPC signal from the remote organ to the heart. This review focuses on cytokines and chemokines that have been suggested to, at least partially, account for the remote cardioprotective RIPC signal cue. We discuss the classical chemokine CXCL12, the atypical cytokine/chemokine macrophage migration-inhibitory factor (MIF), and the anti-inflammatory cytokine interleukin-10 (IL-10), and touch upon other cytokine- and alarmin-like factors such as adipocytokines, myokines, and RNase1. The available evidence for these factors is weighed against their roles in cardiac ischemia and their suitability as RIPC cues, including their expression and release profiles and receptors. Some of these mediators may qualify as cardioprotective target molecules in IHD.
Introduction
Cardiovascular diseases (CVDs) including ischemic heart disease (IHD, also termed coronary heart disease (CHD) or coronary artery disease (CAD)), stroke, and peripheral arterial disease (PAD), are the world’s leading cause of death, accounting for an estimated 18 million death (or 31%) of all global deaths in 2016 (Moran et al., 2014) (https://www.who.int-/cardiovascular_diseases/en/). Approximately 80% of these cases are due to IHD and ischemic stroke. Atherosclerosis is the main underlying cause of these diseases, which, in the heart, causes reduced blood flow to the coronary arteries. This can result in myocardial infarction (unstable angina, ST segment elevation myocardial infarction (STEMI), and non-ST segment elevation myocardial infarction (NSTEMI)) or sub-acute symptoms of reduced oxygen supply to the heart that may necessitate planned surgical intervention at a later time point (Ruff and Braunwald, 2011; Pasterkamp et al., 2017; Vogel et al., 2017).
The highest priority of any intervention is to rapidly re-open the occluded coronary vessel. Re-opening of the blocked vessel not only restores impaired blood flow, but the ‘reperfusion’ process itself damages the heart due to a surge in reactive oxygen species (ROS) that cause cardiomyocyte stress, mitochondrial permeability transition pore (mPTP) opening, and death. This phenomenon is termed myocardial ischemia-reperfusion (I/R) injury (IRI) (Hausenloy and Yellon, 2013, 2016). IRI is not only observed during acute intervention with primary percutaneous coronary intervention (PPCI) in STEMI patients, but also in the setting of planned cardiac surgery, i.e. coronary artery bypass grafting (CABG) (Head et al., 2018). Extensive efforts, encompassing both preclinical and clinical studies, have been made in the past decades to develop strategies to ameliorate cardiomyocyte damage incurred by IRI. A main focus has been on cardioprotective strategies based on ‘ischemic preconditioning’ (IPC), which is brief episodes of ‘sub-threshold’ ischemia and reperfusion prior to prolonged coronary artery occlusion. This procedure, which was first introduced 30 years ago by Murry and colleagues, is typically performed with a blood pressure cuff (Figure 1), and was found to potently limit myocardial infarct size Murry et al., 1986). Ischemic conditioning can follow a variety of modalities, but remote ischemic preconditioning (RIPC; often also abbreviated as RIC) and ischemic postconditioning (IPost) are considered to have the highest translational value. Cardioprotection by IPC has been impressively demonstrated in a variety of preclinical models and numerous smaller clinical trials, and there has been a consensus amongst these studies and a multitude of ex vivo and in vitro experiments that IPC is beneficial and reduces infarct size (Hausenloy and Yellon, 2016). Surprisingly, in 2015 two large-scale multicenter trials investigating the role of RIPC in more than 3,000 enrolled patients undergoing elective cardiac surgery were simultaneously published in the New England Journal of Medicine (RIPHeart and ERICCA study) (Hausenloy et al., 2015a; Meybohm et al., 2015). Both studies tested the efficacy of upper-limb RIPC in patients undergoing elective open-heart surgery using on-pump coronary artery bypass graft (CABG) with or without valve surgery. Anesthetic management was not fully standardized across both trials, but the majority of patients were under propofol-induced anesthesia (Hausenloy et al., 2015a; Meybohm et al., 2015). With similar primary endpoints, both studies demonstrated a ‘neutral’ outcome, overall shedding some doubt on the validity of the IPC/RIPC therapeutic concept in IHD. In fact, these results somewhat questioned whether RIPC is a powerful and practical cardioprotective strategy and if it has a cardioprotective effect in the setting of cardiac I/R. The reasons for the inability of these trials to reproduce the clear efficacy of the earlier smaller clinical trials are still debated in the community and still not yet fully understood. One explanation may be an improvement in surgical and anesthetic management protocols that has led to an overall improved cardiovascular morbidity and mortality (Bell et al., 2016; Stoppe et al., 2017). Indeed, innovations in surgical myocardial preservation techniques, such as combined ante- and retrograde perfusion during bypass, are associated with smaller per se peri-operative myocardial damage (Candilio et al., 2014). Recent explorative studies and meta-analyses may offer mechanistic explanations for the lack of effect of RIPC (Bell et al., 2016; Heusch and Gersh, 2016; Heusch and Rassaf, 2016; Benstoem et al., 2017; Stoppe et al., 2017; Ney et al., 2018). Accordingly, propofol has been suggested as a major confounding factor, as it interferes with the effects of RIPC (Kottenberg et al., 2014; Heusch and Rassaf, 2016; Stoppe et al., 2016), and RIPHeart failed to demonstrate beneficial effects on troponin release and clinical outcomes in propofol-anesthetized cardiac surgery patients (Meybohm et al., 2015).
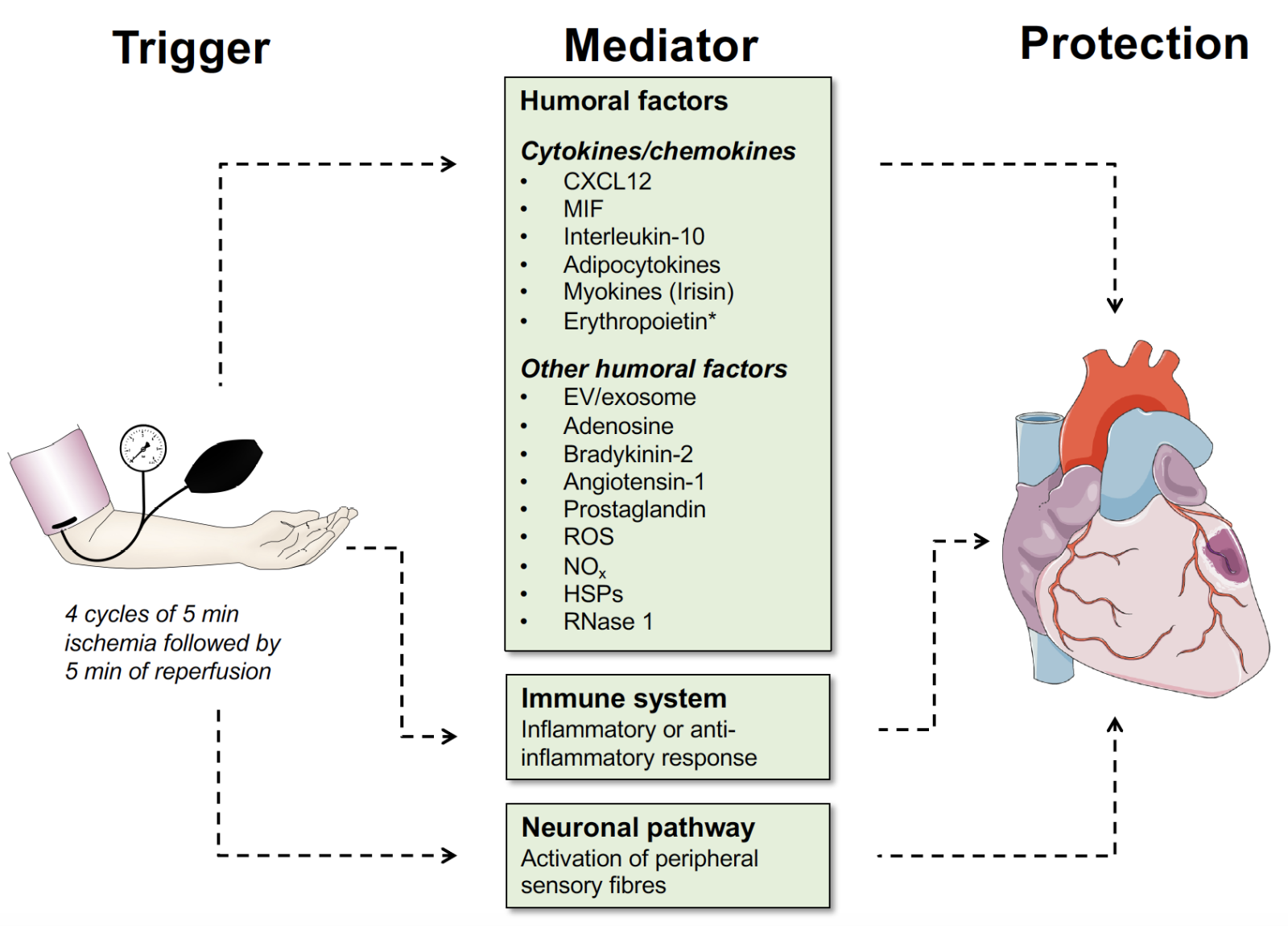
In a new window | Download PPT
Figure 1: Overview of mediators that can serve as a RIPC trigger in cardioprotection. Mediators can be humoral factors such as cytokines, chemokines, and other humoral factors as indicated, or neuronal pathways. The role of the inflammatory response, as it may generally contribute to ischemia-reperfusion stress of the heart, is indicated. In this article, we focus on the classical chemokine CXCL12, the atypical chemokine MIF, and the classical cytokine IL-10, while the evidence for adipocytokines, myokines, erythropoietin, RNase1, and extracellular vesicles/exosomes is briefly touched upon (see text). Abbreviations: CXCL12, CXC motif chemokine 12; EV, extracellular vesicle; MIF, macrophage migration-inhibitory factor; ROS, reactive oxygen species; NOx, reactive nitrogen species; HSPs, heat shock proteins.
Together, this has highlighted the challenges in translating IPC-based cardioprotection into clinical practice. Yet, while disappointing at first sight and contradicting the numerous previous smaller-scale trials, the results of the ERICCA and RIPHeart trials are not in contradiction to successful RIPC procedures used in elective or primary PCI, where surgery-associated inflammation and anesthesia-artifacts are not a confounding issue. To this end, it is of note that a multi-center trial is currently testing the hypothesis that RIPC protects from cardiac dysfunction in STEMI patients. The CONDI2/ERIC-PPCI trial is a randomized controlled clinical trial examining whether RIPC reduces cardiac death and hospitalization for heart failure one year after PPCI intervention in >5000 STEMI patients (Hausenloy et al., 2015b; Cabrera-Fuentes et al., 2016a; Chong et al., 2018). Results of this trial are expected in 2020 (https://clinicaltrials.gov/ct2/show/NCT02342522). Moreover, it has been suggested that the replacement of propofol, which specifically reverses the reduced troponin I release by RIPC in patients undergoing elective CABG, with other anesthetics, and/or altered clinical study design and patient selection might lead to a successful application of RIPC in IHD, at least for a sub-cohort of patients. After all, the procedure is simple, safe, non-invasive, and inexpensive, and the molecular and cellular mechanisms underlying RIPC-mediated cardiac protection are well understood on the effector (‘target organ’) side, involving cardiomyocyte signaling pathways such as the ‘reperfusion injury salvage kinase’ (RISK) and ‘survivor activating factor enhancement’ (SAFE) pathways (Heusch and Gersh, 2016; Rossello and Yellon, 2016). For the ‘trigger’ side, numerous humoral factors and neural pathways have been suggested and discussed (Figure 1), but the main RIPC stimulus, if there is such a key and decisive molecular player, has not been identified. Thus, while the factors on the ‘trigger’ side are less well characterized, a number of recent studies have investigated the causative roles of several cytokines, growth factors, and other humoral factors in cardiac RIPC, suggesting that some of them may be promising RIPC targets for cardioprotection. In fact, specific molecular strategies that mimic RIPC-based cardioprotection might be suitable to ‘replace’ the I/R conditioning cycles of the RIPC procedure, which likely leads to broad activation of several factors. Moreover, although no adverse effects of RIPC were reported in the ERICCA and RIPHeart trials, the ischemia per se or the repetitive clamping during CABG may bear risks ranging from embolic risks to complications during open heart surgery in aged patients.
Here, we focus specifically on the cytokines and chemokines that have been implicated as RIPC-mediating factors in cardioprotection that therefore may become potential targets that could mimic or replace the RIPC procedure of brief repetitive cycles of ischemia and reperfusion of a remote organ or limb. We will summarize the proteins implicated and discuss the evidence that may qualify them as future targets for cardioprotective strategies.
Mediators of remote ischemic preconditioning in cardioprotection: from specific humoral factors to neural pathways
Mediators that have been reported to convey the cardio-protective effect of RIPC range from specific humoral factors to immunological responses and neural pathways (Figure 1). The humoral factors may be classified into cytokines/chemokines, growth factors, and ‘other’ humoral factors (Tsibulnikov et al., 2019). The latter constitute a heterogeneous group of factors comprising released organelles such as extracellular vesicles (EVs) including exosomes, signaling-competent metabolites and lipids such as adenosine and prostaglandins, respectively, peptide hormones such as adrenomedullin, bradykinin-2 and angiotensin-1, danger-associated molecular patterns (DAMPs) or alarmins such as heat shock-proteins (HSPs), released endonucleases such as ribonuclease-1 (RNase1), as well as ROS and nitrogen (NOx) species. Inflammatory priming may encompass preconditioning with pattern recognition receptor (PRR) agonists such as the pathogen-associated molecular patterns (PAMPs) lipopolysaccharide or CpG-oligodeoxynucleotides (CpG-ODNs), which stimulate the Toll-like receptors (TLRs) TLR4 and TLR9, respectively (Knuefermann et al., 2008; Klinman et al., 2009; Eckle and Eltzschig, 2011; Eltzschig and Eckle, 2011). Neuronal pathways reported to contribute to cardioprotection may involve the activation of peripheral sensory fibers. These mediators and pathways have been extensively studied and their contributions to remote conditioning effects were discussed in several excellent recent reviews (Hausenloy and Yellon, 2009; Eckle and Eltzschig, 2011; Hausenloy et al., 2012; Hausenloy and Yellon, 2013; Heusch et al., 2015; Bell et al., 2016; Cabrera-Fuentes et al., 2016b; Davidson et al., 2017; Basalay et al., 2018; Tsibulnikov et al., 2019).
In the current article, we focus on the chemokines stromal cell-derived factor-1α (SDF-1α)/CXCL12 and macrophage migration-inhibitory factor (MIF), and the cytokine interleukin-10 (IL-10). We will also briefly mention adipocytokines as cytokine/chemokine-type humoral factors and include the growth factor erythropoietin (EPO) due to its cytokine-like properties. Although the focus in our review will be on ‘cardio’protective factors, we also include irisin, a recently identified myocyte-derived cytokine (‘myokine’) that has been reported to function as a protective RIPC signal for the lung and other organs affected by prolonged ischemia, as well as the heart. Moreover, we will cover the EV/exosome cardioprotection paradigm as these secreted cellular vesicles transport a variety of factors as their cargo including micro-RNAs (miRs) and cytokines/DAMPs, and mention the role of extracellular RNA (eRNA)/RNase1 system. While some of these factors have roles in both intracardiac signaling during IRI and remote signaling as ‘true’ RIPC cues (Heusch et al., 2015), we will confine our article to those factors for which an explicit role as remote RIPC signal has been suggested.
Cytokines and chemokines as mediators of cardioprotective remote ischemic pre-conditioning
Cytokines and chemokines that are predestined to serve as RIPC signaling cues (Figure 1) typically fulfill certain expression, release, and signaling properties. These include: i) rapid induction of their expression and/or secretion by cycles of ischemia and reperfusion, i.e. by the RIPC trigger, and/or by brief episodes of ischemia; and/or ii) expression in preformed intracellular stores (i.e. in secretion vesicles or in the cytosolic compartment); iii) substantial production from a remote tissue/organ that is well responsive to ischemic stimuli (the endothelium in limb muscle tissue would be an ideal tissue in this sense); reasonable stability for the mediator to reach the heart and engage in cardiac signal transduction at a prolonged time interval AFTER the RIPC trigger; iv) engagement of their cardiac-expressed signaling-competent receptors, which implies that the receptor(s) of the respective RIPC signal need to be expressed on cardiomyocytes at and/or before the time point of cardiac IRI (Table 1).
Table 1: Criteria of a remote signaling trigger that renders it suitable as a cardioprotective factor in remote ischemic conditioning of the heart.
|
|
Category |
Criteria |
|
1 |
Expression |
Rapid induction of expression and/or secretion by cycles of ischemia and reperfusion, or by brief episodes of ischemia |
|
2 |
Expression |
Expression in preformed intracellular stores |
|
3 |
Concentration and stability |
Production from a remote tissue/organ in substantial amounts of a mediator with appreciable stability in plasma |
|
4 |
Stimulus |
Remote tissue is readily responsive to ischemic stimulus; promoter of trigger substance may be HIF-1a-responsive |
|
5 |
Receptor / target organ |
Target organ/cell (here: heart/cardiomyocyte) expresses signaling-competent receptor(s) of remote trigger |
|
6 |
Receptor / target organ |
Receptor needs to be expressed at and/or before the time point of cardiac IRI |
Stromal cell-derived factor-1α (SDF-1α)/CXCL12
Chemokines are small 8-14 kDa cytokines that are structurally characterized by N-terminal signature cysteine residues and a so-called chemokine fold (Clark-Lewis et al., 1995; Mantovani, 1999; Murphy et al., 2000; Mackay, 2001; Fernandez and Lolis, 2002; Charo and Taubman, 2004). They are best known for their chemotactic capacity and their role in orchestrating the trafficking and homing of immune cells, but they have numerous other functions in homeostasis and disease. The chemokine family of ‘chemo’tactic cyto’kines’ consists of 49 chemokines that interact with 18 signaling-competent receptors, indicating a redundancy on the side of the ligands. Chemokines are classified according to the number and positioning of the signature cysteine residues into CC-, CXC-, CXXXC-, and C-type chemokines (Murphy et al., 2000). Their interacting receptors are classified accordingly. Complexity in the chemokine network is further enhanced by five atypical chemokine receptors (ACKRs) and more than 10 atypical chemokines (ACKs), an emerging and structurally heterogeneous ‘functional’ chemokine sub-family that encompasses mediators such as MIF and human β-defensins that can engage in high-affinity binding with classical chemokine receptors albeit lacking the signature cysteines and the chemokine-fold (see below) (Tillmann et al., 2013; Pawig et al., 2015; Kapurniotu et al., 2019).
Stromal cell-derived factor-1α (SDF-1α) is a member of the sub-category of CXC-type chemokines. Among these, it belongs to the ELR - sub-class (Murphy et al., 2000), and accordingly is generally considered a homeostatic chemokine with major roles in development and differentiation, stem cell recruitment, and immune cell homing (Zernecke et al., 2005; Zernecke et al., 2008a; Zernecke et al., 2009; Doring et al., 2017). However, CXCL12 also has tissue- and context-specific pro-inflammatory, tumorigenic, and pro-atherogenic roles (Doring et al., 2014; Weber et al., 2016). The many roles of CXCL12 in homeostasis, development/differentiation, cell trafficking, inflammation, and cancer have been extensively reviewed elsewhere (Burger and Kipps, 2006; Doring et al., 2014; Pawig et al., 2015; Pozzobon et al., 2016; Janssens et al., 2018). CXCL12 signals through the CXC-type receptor CXCR4, which mediates most of its functions in homeostasis, cell recruitment, and disease. For a long time, the CXCR4/CXCL12 axis was considered to be one of the few specific ligand/receptor pairs in the chemokine network. However, it is now clear that CXCL12 also binds to the chemokine receptor CXCR7/ACKR3, which (mostly) serves as a scavenger receptor to ‘shape’ CXCL12 gradients (Bachelerie et al., 2014; Koenen et al., 2019). Inversely, CXCR4 is engaged by the atypical chemokine MIF (see next chapter), human β-defensin 3 (HBD3), and a heterodimeric complex of the alarmin HMGB1 and CXCL12, as well as extracellular ubiquitin and HIV gp120 in stress and infections, respectively. The cardio-protective role of CXCL12 in RIPC thus also needs to be viewed in the context of CXCR4 interactions and functions of these alternative chemokine-like ligands (Pawig et al., 2015).
In the heart and in cardiac I/R, CXCL12 has been ascribed dual roles with both ameliorating and disease-exacerbating functions (Liehn et al., 2011b; Doring et al., 2014; Pawig et al., 2015). One of the major regulatory sites in the CXCL12 gene promoter is the hypoxia-inducible factor (HIF)-1α responsive element (HRE) (Ceradini et al., 2004). Accordingly, CXCL12 expression and secretion in endothelial cells is prominently driven by hypoxic stress and has been linked to progenitor cell recruitment during ischemia (Ceradini et al., 2004; Ceradini and Gurtner, 2005). These properties render CXCL12 a potential candidate to serve as a RIPC signaling cue. In fact, Yellon, Davidson and colleagues found that CXCL12 is profoundly elevated in a rat model of RIPC in cardiac IRI (Davidson et al., 2013). Strikingly, they observed that RIPC-mediated decreased myocardial infarct size and functional recovery of cardiac papillary muscle was partially reversed by AMD3100, a pharmacological inhibitor of the CXCR4/CXCL12 axis. Moreover, mimicking a therapeutic application of CXCL12, administration of the recombinant chemokine protein confirmed its protective role in this model, which was again blocked by AMD3100 (Davidson et al., 2013; Bromage et al., 2014). They also studied dipeptidyl peptidase 4 (DPP4), a circulating enzyme that rapidly inactivates CXCL12 by N-terminal cleavage (Noels and Bernhagen, 2016), and the inhibition of which is protective during myocardial infarction (Noels et al., 2018; Xie et al., 2018; Ziff et al., 2018). Interestingly, although DPP4 also has other substrates, such as the incretins, that play a role in metabolic and cardiovascular diseases (Matheeussen et al., 2012) and although a counter-intuitive association between post-operative DPP4 activity and worse patient outcome was noted in a cardiac surgery cohort (Noels et al., 2018), a potential role for full-length CXCL12 in the setting of cardiac RIPC could be uncovered capitalizing on a novel anti-CXCL12 antibody (Bromage et al., 2017). The role of DPP4-processed versus full-length CXCL12 in cardiac RIPC will have to be addressed further by future mechanistic studies.
While in this review article we focus on the role of ‘remote’ RIPC cues, the numerous protective activities of the intracardiac CXCL12/CXCR4 axis will be briefly mentioned. Two main activity types can be differentiated: i) recruitment of CXCR4-expressing stem cells with angiogenic and vasculogenic capacity; and ii) local loops involving cardioprotective signaling via cardiomyocyte expressed CXCR4 pathways that contribute to protection through the RISK and SAFE routes. For example, these activities involve the release of cardiac CXCL12 by the ischemic myocardium, which then serves as a major recruitment signal for e.g. CXCR4+ endothelial progenitor cells. Additionally, autocrine/paracrine action of cardiomyocyte-, cardiac fibroblast-, or myocardial endothelial cell-derived CXCL12 may act to enhance protective AKT and ERK1/2 signaling pathways in ischemic stressed cardiomyocytes. These activities and the origin and source of secreted CXCL12 cannot always be distinguished from ‘remote’ CXCL12 signals as it may derive following brief RIPC episodes of ischemia/reperfusion in peripheral organs or limbs. They have been reviewed extensively (Farouk et al., 2010; Liehn et al., 2011a; Penn et al., 2012; van der Vorst et al., 2015) and will not be further discussed herein.
Macrophage migration-inhibitory factor (MIF)
MIF is a long-known (David, 1966; Weiser et al., 1989; Calandra and Roger, 2003) evolutionarily highly conserved chemokine-like inflammatory cytokine that has well-known pro-inflammatory roles in many diseases (Bernhagen et al., 1993; Donnelly et al., 1997; Calandra et al., 2000; Calandra and Roger, 2003; Morand et al., 2006). In cardiovascular pathologies, a double-edged sword role has been suggested for MIF (Zernecke et al., 2008b; Rassaf et al., 2014; Sinitski et al., 2019).
MIF is a small 12.5 kDa protein that crystallizes as a trimer and shares some structural features with bacterial enzymes, the cytokine IL-1β, and the CXC chemokine CXCL8, but does not belong to any of the known cytokine or chemokine classes, if pro forma structural criteria are followed (Sun et al., 1996; Murphy et al., 2000). MIF is well known to be secreted from immune cells such as monocytes/macrophages and T cells, but it is widely expressed and can be secreted from preformed intracellular stores by non-conventional secretion from a variety of cell types such as endothelial and parenchymal cells, including cardiomyocytes following inflammatory, hypoxic, or other cell stress triggers (Calandra and Roger, 2003; Kapurniotu et al., 2019). As such, MIF ‘fulfills’ several of the above-discussed criteria that a RIPC signaling cue should have.
Having been discovered as the first cytokine over 50 years ago (David, 1966), MIF is considered an extracellular-acting cytokine with chemokine-like properties, but intracellular MIF has more recently been suggested to contribute to some of its functions (Kapurniotu et al., 2019). The activities of MIF are mediated by high-affinity interaction with its cognate receptor CD74, the surface-expressed form of class II invariant chain Ii (Leng et al., 2003), as well as by non-cognate engagement of the CXC chemokine receptors CXCR2 or CXCR4. As mentioned above, MIF thus ‘shares’ CXCR4 with CXCL12. Preliminary structural evidence suggests that MIF and CXCL12 cover distinct, though overlapping, binding sites on CXCR4 that make specific targeting approaches possible (Crump et al., 1997; Wu et al., 2010; Rajasekaran et al., 2016; Lacy et al., 2018). Of note, the MIF receptors, in particular CXCR4, can also be upregulated on endothelial and tissue cells upon inflammation and hypoxia (Kanzler et al., 2013). While CD74 is primarily considered a pro-proliferative, inflammatory, and metabolic receptor of MIF, the MIF chemokine receptor primarily controls inflammation, atherogenesis, and leukocyte recruitment (Bernhagen et al., 2007; Sinitski et al., 2019).
While MIF is an upstream regulator of innate immunity and generally exhibits pro-inflammatory disease-exacerbating effects in various inflammatory and autoimmune pathologies including atherosclerosis, it has also been found to protect from hepatic fibrosis and has pivotal protective activities in IHD. It thus displays a complex role in cardiovascular diseases, dependent on the stage (chronic versus acute), vessel type, and disease context. MIF promotes atherosclerosis through enhancing atherogenic monocyte and T-cell infiltration via CXCR2- and CXCR4-based pathways, respectively, and also fuels plaque inflammation and destabilization. MIF’s pro-atherogenic properties in atherogenesis and atheroprogression have been unanimously observed in various experimental models; this role is further supported by clinical correlations of MIF protein plasma levels and MIF’s polymorphic promoter in human atherosclerotic disease (Rassaf et al., 2014; Tilstam et al., 2017). However, MIF’s role in the ischemic heart and in cardiac IRI is bivalent. Brief (15-30 min) cardiac ischemia followed by up to a 4 h period of reperfusion in murine in vivo models suggests that MIF potently protects from IRI. Mechanistically, this cardioprotective effect is mediated by CD74/AMP kinase (AMPK)-mediated metabolic signaling in I/R-challenged cardiomyocytes. It is further enhanced by a MIF-based redox mechanism that also includes post-translational nitrosylation of MIF (Miller et al., 2008; Qi et al., 2009; Koga et al., 2011; Luedike et al., 2012; Rassaf et al., 2014). That this cardioprotective effect of MIF might be therapeutically exploitable was first shown in an elegant study by Young, Bucala, and colleagues. They identified an intriguing mechanism called ‘pharmacologic augmentation’ that is based on a small molecule agonist of MIF, a compound termed MIF20, which binds to endogenous MIF and induces a conformational change that enhances CD74 binding to foster the cardioprotective capacity of MIF by stimulating CD74/AMPK signaling (Wang et al., 2013). Curiously, augmentation capitalizes on MIF20 binding into the conserved N-terminal tautomerase cavity of MIF, indicating that this evolutionarily conserved enzyme activity of MIF, for which to date no role in mammalians has been found, could be harnessed for protection of the heart. However, the role of MIF in cardiac ischemia is probably more complicated. When longer periods of ischemia were applied and later time points after the onset of reperfusion were analyzed, cardioprotection was observed in Mif–/– mice, suggesting that under these conditions, MIF has disease-exacerbating inflammatory effects. In fact, it appears that MIF-triggered CXCR2- or CXCR4-mediated inflammatory activities dominate in this delayed post-reperfusion window (Gao et al., 2011; Dayawansa et al., 2014). We suggest a ‘wave’ or ‘phase-dependent’ model for MIF’s contribution in IRI, following up on an earlier suggestion by Dayawansa et al. (2014). Accordingly, MIF is protective in the early phase of IRI. The various data posit that it is cardiac MIF (most likely and predominantly derived from cardiomyocytes) that is released in the ischemic and/or early reperfusion phase that is responsible for the cardioprotective effect seen in this phase (‘1st wave cardioprotective MIF’) (Miller et al., 2008; Qi et al., 2009; Luedike et al., 2012; Rassaf et al., 2014; Pohl et al., 2016). In contrast, MIF produced in later stages of the reperfusion and post-reperfusion inflammatory phases is predominantly ‘inflammatory’, exacerbating the inflammatory cascade that prevails in this phase. ‘Second wave inflammatory MIF’ is mainly produced by the infiltrating myeloid cells, but local cardiac MIF may still contribute to this 2nd wave effect of MIF. It has been suggested that MIF-triggered inflammatory CXCR2/4 pathways ‘outcompete’ the protective CD74 pathway in this phase (Liehn et al., 2011b; Liehn et al., 2013; Bernhagen, 2019). In fact, it is obvious that there is a continuous transition between both phases, but although redox modifications have been suspected to play a role, the molecular switch converting ‘good MIF’ into ‘bad MIF’ has remained elusive (Schindler et al., 2018). Exacerbating inflammatory effects of MIF may also prevail under conditions of more profound (elongated) ischemia, although reports have been partially contradictory (Gao et al., 2011; Koga et al., 2011).
The MIF ligand family was recently ‘doubled’, when it became clear that the structural homolog of MIF, D-dopachrome tautomerase (D-DT)/MIF-2, partially phenocopies MIF activities (Merk et al., 2011; Merk et al., 2012). Evidence from an experimental model of cardiac IRI capitalizing on conditional cardiomyocyte-specific Mif-2-knockout mice suggests that this also holds true for the cardioprotective CD74/AMPK pathway, which is also addressed by MIF-2 (Qi et al., 2014). Although clinical correlations between MIF-2 plasma levels and outcome parameters from cardiac surgery patients suggest that the role of MIF-2 in IRI may differ from that of MIF (Stoppe et al., 2015), it has been speculated that MIF-2 could contribute to the phase-specific switch regulating the contribution of MIF family proteins in different phases of the IRI process. Thus overall, the evidence from the various experimental IRI models is in line with clinical data suggesting a correlation between high admission MIF levels in STEMI patients and adverse outcomes (Deng et al., 2018), as well as high MIF, inflammation markers, and unstable IHD.
Can this knowledge be exploited for MIF-based cardioprotection in the context of RIPC? A number of recent studies argue that this could be the case. Ruze et al. (2019) demonstrated in a mouse model that Mif deficiency counteracted the protective effect of RIPC on myocardial IRI. They first induced I/R in a Langendorff-perfused heart comparing wildtype and Mif-deficient (Mif–/–) mice with or without preceding cycles of ischemia and reperfusion. The protective effect of RIPC in wildtype hearts was lost in hearts from Mif–/– knockout mice. The same was found in an in vivo IRI model with evidence for a strongly reduced infarct size and cardiac dysfunction. Moreover, RIPC-induced increased cardioprotective signaling via the RISK and AMPK pathways and improved cardiomyocyte glucose uptake were blunted in hearts from Mif–/– mice (Bernhagen, 2019; Ruze et al., 2019). Of note, the reversal effect on RIPC-based reduction in infarct size noted in Mif-deficient hearts was marked (i.e. >30%), implying that MIF could indeed be one of the critical cardioprotective factors released in RIPC. A study by Wang et al. (2019) on the role of MIF in IPost confirms this conclusion. Wang and colleagues studied the causal effect of MIF in IPost in a rat model (Wang et al., 2019). They applied 4 cycles of 5 min I/R on the lower limbs immediately after reperfusion. IPost led to a significant elevation of plasma MIF. Moreover, femoral occlusion blocked the rise in plasma MIF, arguing that MIF behaved as a true remote conditioning signaling cue. To test for causality, the administration of the established pharmacological MIF inhibitor (S,R)-3-(4-hydroxy-phenyl)-4,5-dihydro-5-isoxazoleacetic acid methyl ester (ISO-1) was compared to a vehicle control. Of note, ISO-1 but not a vehicle control prevented cardioprotection and enhanced cardiomyocyte apoptosis in the hearts of IPost-treated rats. The study also provided initial insight into the mechanism. Confirming the earlier MIF/IRI studies, the cardioprotective effect of MIF after IPost was correlated with elevated myocardial phospho-AMPK levels in IPost-treated but not IPost+ISO-1-treated rats. Interestingly, inhibition of HIF-1α by a small molecule blocker led to decreased plasma MIF levels in IPost, in line with the notion that the remote production of MIF in limb tissue is triggered by an ischemia/HIF-1α-dependent mechanism (Wang et al., 2019). A possible role for MIF in cardioprotective conditioning is further confirmed by an in vitro study by Goetzenich and colleagues (2014), who studied MIF in anesthetic-induced myocardial preconditioning (AIPC). Although the stimulating trigger is a different one compared to RIPC (i.e. an anesthetic such as sevoflurane rather than cycles of hypoxia and hyperoxia), the AIPC phenomenon follows a comparable mechanistic principle as RIPC. In contrast, a cardiac RIPC study using exogenous recombinant MIF questioned whether MIF-based strategies may have practical translational potential, as exogenously administered recombinant MIF was unable to exert cardioprotection in a Langendorff-perfused heart, when administered before (RIPC model) the ischemic insult or at reperfusion (IPost model) (Rossello et al., 2016). In conjunction with the other studies discussed above, this may indicate the general complexities of using recombinant proteins on the one hand and specifically the MIF protein on the other hand. MIF is known to be susceptible to redox-modulation (Schindler et al., 2018), to oligomerize, and has a relatively high hydrophobic index (Sun et al., 1996; Mischke et al., 1998), which together may render it tricky to control its activity in I/R-based experimental set-ups. Pharmacologic augmentation by MIF20 may represent a solution to this problem, as MIF20 can act on endogenous MIF protein (Wang et al., 2013), and could thus be a valid RIPC target.
MIF was also included in one of the smaller follow-up studies of RIPHeart and ERICCA, aimed at elucidating the confounding factors and the potential role of propofol (Ney et al., 2018). The data demonstrated comparable perioperative kinetics of MIF and the cognate CXCR4 ligand CXCL12 in the RIPC and control group, sharing characteristics that overlap with the signaling mechanisms of RIPC, i.e. activation of ERK1/2, AKT, and PKCε (Heusch, 2015). In contrast, in the intra-operatively collected right atrial tissue specimens, MIF was decreased after RIPC, whereas in turn RIPC did not lead to an increase in MIF and CXCL12 serum levels, indicating that the RIPC stimulus itself limits cardiac MIF expression. This may be a preliminary hint that the release of these two CXCR4 ligands may have been inhibited by propofol in the RIPHeart cohort, while the classical inflammatory markers IL-6, CXCL8, and IL-10 were not different in the propofol-anesthetized patients (Ney et al., 2018).
Lastly, further -indirect- evidence for a potential utility of MIF or MIF-related target structures in cardiac RIPC came from a recent RIPC model of hepatoprotection after liver transplantation. Remote ischemic conditioning (RIC) was investigated in a rat model of orthotopic liver transplantation (OLT), using both RIPC and IPost settings. Graft micro- and macrocirculation and liver damage were the main readouts (Emontzpohl et al., 2018). Plasma MIF levels were down-regulated in this model following RIPC and inversely correlated with hepatoprotection, a notion that may be in line with hepatic translocation of remotely produced MIF.
Interleukin-10 (IL-10)
Interleukin-10 (IL-10) is a pivotal anti-inflammatory cytokine that affects both the innate and adaptive immune systems. IL-10 is produced by a wide range of cell types in an NFκ B-dependent manner following delayed kinetics compared to pro-inflammatory NFκB-triggered cytokines such as TNF-a or IL-6. It serves to dampen the inflammatory response as a prerequisite to transition into resolution and regeneration. The anti-inflammatory properties of IL-10 in the context of numerous diseases have been extensively reviewed and will not be covered further here (Renauld, 2003; O'Garra and Vieira, 2007; Saraiva and O'Garra, 2010; Ng et al., 2013; Hotchkiss et al., 2016; Comi et al., 2018). Instead, we will focus on a handful of recent studies reporting on a specific role of IL-10 in cardiac RIPC.
Since IL-10 is supposed to be a ‘delayed’ cytokine, Cai et al. (2012) used a mouse model of myocardial IRI and tested the hypothesis that ischemic conditioning may confer late protection against IRI through IL-10. In a setting of lower limb RIPC followed by 30-min ischemia and 120-min reperfusion, RIPC increased plasma and cardiac IL-10 protein levels. Of note, anti-IL-10 antibodies fully blocked the protective effect of RIPC. Similarly, IL-10 gene knockout led to a loss of RIPC cardioprotection, whereas recombinant exogenous IL-10 mimicked the protective RIPC effect. In a Langendorff heart model, IL-10 increased phospho-Akt levels, suggesting that RIPC-triggered IL-10 activates cardioprotective pathways such as RISK signaling. The study implied that RIPC induces protection against myocardial IRI by triggering the expression of IL-10 in remote muscle tissue. Muscle-derived IL-10 is then released into the circulation to promote protective signaling in the heart. The role of IL-10 as a remote signal in RIPC cardioprotection is underpinned by two studies in which preconditioning was achieved by TLR agonists such as CpG-oligonucleotides (CpG-ODNs) in a model in which IRI was applied 16 h after the conditioning trigger (in this case CpG-ODNs instead of RIPC cycles) (Markowski et al., 2013; Hilbert et al., 2018). In addition to pro-inflammatory cytokines, conditioning with CpG-ODNs caused a pronounced increase in circulating IL-10 levels that correlated with long-lasting protection from cardiac IRI. Moreover, inhibition of IL-10 increased the infarct size and counteracted the beneficial influence of CpG-ODN conditioning (Markowski et al., 2013). The conclusion from this study that IL-10 is a key remote protection signal is further strengthened by the notion that a closed-chest model of myocardial IRI was used, which circumvents a surge in peri-operative local inflammatory reactions. Hilbert and colleagues further confirmed these findings by combining CpG-ODN-mediated conditioning and IRI with a genetic profiling approach. The profiling showed that the expected induction of cardiomyocyte survival genes correlated with a decrease in inflammatory pathways that in turn were suppressed by IL-10 (Hilbert et al., 2018). Thus, the up-regulation of protective pathways and the down-regulation of inflammatory pathways represent a genetic correlate of the cardioprotective effects of ODN preconditioning, with the pro-inflammatory arm blocked by IL-10. The confirmation of this concept by clinical studies is yet pending.
Nederlof and colleagues (2017) performed a smaller randomized trial of RIPC and control treatment for cardioprotection in sevoflurane-anesthetized CABG patients. Their initial goal was to further probe the ‘propofol confounder’ hypothesis by restricting perioperative anesthesia regimens to sevoflurane and fentanyl in their CABG patients, while avoiding propofol. While the study remained underpowered and had to be halted regarding its initial inclusion target, it could be used to study inflammatory mediators such as IL-6, TNF-α, and MIF, as well as IL-10. RIPC was without effect on these mediators obtained before and immediately after RIPC. An interesting study links IL-10 to cardioprotective EVs/exosomes. Cambier et al. (2017) demonstrated that Y RNA fragment present in EVs/exosomes confers cardioprotection via modulation of IL-10 expression and secretion from cardiac macrophages (see also next chapter).
Other cytokines, alarmins, and extracellular vesicles/exosomes
It is beyond the scope of this review article to discuss the numerous humoral factors that have been implicated in RIPC cardioprotection in detail. Nevertheless, Table 2 summarizes selected references that have provided appreciable evidence (or contra-indicated data) on the role of cytokine- and alarmin-like mediators of RIPC cardioprotection, including adipocytokines and myokines, the cytokine-like growth factor erythropoietin (EPO), the eRNA/RNase1 system, and EVs/exosomes, which may among other cardioprotective cargo such as microRNAs (mIRs) carry cytokines or chemokines.
Table 2: Overview of the role of ‘other’ cytokines, alarmins, and extracellular vesicles/exosomes in cardioprotection by remote ischemic preconditioning.
|
Mediator |
Molecule class |
Selected references |
|
Adiponectin, leptin, resistin |
Adipocytokine |
(Smith et al., 2010; Smith et al., 2011; Wang et al., 2017a) |
|
Irisin |
Myokines |
(Chen et al., 2017; Wang et al., 2017b; Bernhagen, 2018; Wang et al., 2018) |
|
Erythropoietin |
Cytokine / growth factor |
(Baker, 2005; Hausenloy and Yellon, 2007; Diwan et al., 2008b; Diwan et al., 2008a; Riksen et al., 2008; Tsibulnikov et al., 2019) |
|
Extracellular vesicle/Exosome |
Secreted organelle |
(Giricz et al., 2014; Yellon and Davidson, 2014; Vicencio et al., 2015; Barile et al., 2017; Cambier et al., 2017; Davidson et al., 2017; de Couto et al., 2017; Davidson et al., 2018; Minghua et al., 2018; Hou et al., 2019; Nazari-Shafti et al., 2019; Spannbauer et al., 2019) |
|
RNase1 |
Alarmin / endonuclease |
(Cabrera-Fuentes et al., 2014; Cabrera-Fuentes et al., 2015; Cabrera-Fuentes et al., 2016b; Stieger et al., 2017) |
We will briefly discuss two prominent examples. Vicencio and colleagues (2015) demonstrated that EVs/exosomes deliver protective signals to the myocardium and that this occurs via the HSP70/TLR4 axis, expressed on the surface of EVs/exosomes and cardiomyocytes, respectively (Vicencio et al., 2015; Davidson et al., 2017). They also demonstrated that conditioning-competent EVs/exosomes derive from endothelium and that the conditioning effect on cardiomyocytes involves ERK1/2 signaling (Davidson et al., 2018). Cabrera-Fuentes, Preissner, Sedding, and colleagues (2014) identified a critical role for the eRNA/RNase1 system, which has emerged to have a significant clinical impact on the development and progress of cardiovascular diseases. Extracellular RNA (eRNA) is a cellular alarm signal for tissue damage and has been associated with increasing TNF-α levels and may trigger the progress of atherosclerosis. It also negatively impacts on the consequences of myocardial I/R injury (Cabrera-Fuentes et al., 2014; Simsekyilmaz et al., 2014; Zernecke and Preissner, 2016). The ubiquitous endonuclease RNase1 decreases damaging eRNA and TNF-α levels and RNase1 treatment was shown to significantly reduce infarct size (Stieger et al., 2017). Of note, RNase1 could be directly linked to RIPC. Patients undergoing RIPC exhibited increased cardioprotective RNase1 activity and decreased eRNA serum levels (Cabrera-Fuentes et al., 2015), while the exact mechanism of RNase1-induced cardioprotection remains to be explored.
Conclusions
Cytokines and chemokines such as CXCL12, MIF, and IL-10 have been implicated as remote triggers during RIPC-mediated cardioprotection. Moreover, there is appreciable evidence from experimental models that they may have a causal role and that they may, at least partially, mimic RIPC-based cardioprotection. While evidence from clinical trials is not yet available to predict whether they may eventually qualify as cardioprotective targets they fulfill several of the criteria that an effective RIPC signaling cue should have.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grants BE 1977/9-1 and SFB1123-A03 to J.B. and DFG grant STO1099/2-1 to C.S. It was co-supported by DZHK grant B 18-001 Extern/81X2600248 to J.B. and by DFG under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) to J.B. We also acknowledge support by the DFG-funded LMU excellence (LMUexc) program "Strategic cooperation Munich-Singapore.
References
Christian Stoppe1
1Department of Intensive Care Medicine, RWTH Aachen University, Universitätsklinikum 52074 Aachen, Germany
Sandra Kraemer1
1Department of Intensive Care Medicine, RWTH Aachen University, Universitätsklinikum 52074 Aachen, Germany
Jürgen Bernhagen2,3,4
2Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), Klinikum der Universität München (KUM), Ludwig-Maximilians-University (LMU), 81377 Munich, Germany
3Munich Heart Alliance, 80802 Munich, Germany
4Munich Cluster for Systems Neurology (SyNergy), 81377 Munich, Germany.
Corresponding author:
Jürgen Bernhagen
Email: juergen.bernhagen@med.uni-muenchen.de
In a new window | Download PPT
Figure 1: Overview of mediators that can serve as a RIPC trigger in cardioprotection. Mediators can be humoral factors such as cytokines, chemokines, and other humoral factors as indicated, or neuronal pathways. The role of the inflammatory response, as it may generally contribute to ischemia-reperfusion stress of the heart, is indicated. In this article, we focus on the classical chemokine CXCL12, the atypical chemokine MIF, and the classical cytokine IL-10, while the evidence for adipocytokines, myokines, erythropoietin, RNase1, and extracellular vesicles/exosomes is briefly touched upon (see text). Abbreviations: CXCL12, CXC motif chemokine 12; EV, extracellular vesicle; MIF, macrophage migration-inhibitory factor; ROS, reactive oxygen species; NOx, reactive nitrogen species; HSPs, heat shock proteins.
Table 1: Criteria of a remote signaling trigger that renders it suitable as a cardioprotective factor in remote ischemic conditioning of the heart.
|
|
Category |
Criteria |
|
1 |
Expression |
Rapid induction of expression and/or secretion by cycles of ischemia and reperfusion, or by brief episodes of ischemia |
|
2 |
Expression |
Expression in preformed intracellular stores |
|
3 |
Concentration and stability |
Production from a remote tissue/organ in substantial amounts of a mediator with appreciable stability in plasma |
|
4 |
Stimulus |
Remote tissue is readily responsive to ischemic stimulus; promoter of trigger substance may be HIF-1a-responsive |
|
5 |
Receptor / target organ |
Target organ/cell (here: heart/cardiomyocyte) expresses signaling-competent receptor(s) of remote trigger |
|
6 |
Receptor / target organ |
Receptor needs to be expressed at and/or before the time point of cardiac IRI |
Table 2: Overview of the role of ‘other’ cytokines, alarmins, and extracellular vesicles/exosomes in cardioprotection by remote ischemic preconditioning.
|
Mediator |
Molecule class |
Selected references |
|
Adiponectin, leptin, resistin |
Adipocytokine |
(Smith et al., 2010; Smith et al., 2011; Wang et al., 2017a) |
|
Irisin |
Myokines |
(Chen et al., 2017; Wang et al., 2017b; Bernhagen, 2018; Wang et al., 2018) |
|
Erythropoietin |
Cytokine / growth factor |
(Baker, 2005; Hausenloy and Yellon, 2007; Diwan et al., 2008b; Diwan et al., 2008a; Riksen et al., 2008; Tsibulnikov et al., 2019) |
|
Extracellular vesicle/Exosome |
Secreted organelle |
(Giricz et al., 2014; Yellon and Davidson, 2014; Vicencio et al., 2015; Barile et al., 2017; Cambier et al., 2017; Davidson et al., 2017; de Couto et al., 2017; Davidson et al., 2018; Minghua et al., 2018; Hou et al., 2019; Nazari-Shafti et al., 2019; Spannbauer et al., 2019) |
|
RNase1 |
Alarmin / endonuclease |
(Cabrera-Fuentes et al., 2014; Cabrera-Fuentes et al., 2015; Cabrera-Fuentes et al., 2016b; Stieger et al., 2017) |
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13110 | 33 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA