Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Regulation of cellular oxidative metabolism by hypoxia-inducible factors
Time:2019-07-03
Number:13641
Author Affiliations

Conditioning Medicine, 2019. 2(3): 114-121.
Abstract
Ischemia-reperfusion episodes are characterized by pathological oxygen fluctuations that typically result in exacerbated oxidative stress, leading to cell death in the affected tissue. Cells respond to limited oxygen by triggering multiple adaptive mechanisms through the hypoxia-inducible factors (HIFs). A central action executed by the HIFs is a profound repression of mitochondrial cellular respiration. As a consequence, oxygen is salvaged in hypoxic/ischemic tissues, and the production of mitochondrial reactive oxygen species is attenuated, which limits ischemia-reperfusion damage. Here we discuss multiple actions simultaneously triggered by the HIF oxygen-sensing pathway to restrain oxidative metabolism and mitochondrial respiration when oxygen is limited.
Keywords: mitochondria; oxygen consumption; HIF; hypoxia-inducible factor; reactive oxygen species; TCA cycle
Abstract
Ischemia-reperfusion episodes are characterized by pathological oxygen fluctuations that typically result in exacerbated oxidative stress, leading to cell death in the affected tissue. Cells respond to limited oxygen by triggering multiple adaptive mechanisms through the hypoxia-inducible factors (HIFs). A central action executed by the HIFs is a profound repression of mitochondrial cellular respiration. As a consequence, oxygen is salvaged in hypoxic/ischemic tissues, and the production of mitochondrial reactive oxygen species is attenuated, which limits ischemia-reperfusion damage. Here we discuss multiple actions simultaneously triggered by the HIF oxygen-sensing pathway to restrain oxidative metabolism and mitochondrial respiration when oxygen is limited.
Keywords: mitochondria; oxygen consumption; HIF; hypoxia-inducible factor; reactive oxygen species; TCA cycle
1. Hypoxia-inducible factors, ischemia and oxidative stress
Insufficient oxygen supply can occur locally or systemically. A systemic reduced oxygen supply affects the whole organism, for example, upon exposure to high altitude environments or hypoxemia associated with obstructive lung disease. A local reduction in oxygen supply can be a consequence of vessel occlusion leading to areas of tissue ischemia, which can be reperfused to restore blood flow in the case of myocardial infarction or stroke. Cells exposed to an ischemic environment are characterized not only by insufficient oxygen supply, but also by a reduced availability of other nutrients such as glucose and amino acids, in addition to a compromised cellular waste removal, ultimately leading to acidosis of the ischemic tissue. This stressful event is also associated with profound mitochondrial dysfunction and the inefficient transfer of electrons to molecular oxygen in the respiratory chain, resulting in the generation of harmful mitochondrial reactive oxygen species (ROS). This is particularly manifest in the reperfusion phase, when oxygen/blood flow is re-established (Hausenloy et al., 2003; Murphy, 2009).
Against this background, the hypoxia-inducible factors (HIFs) have emerged as central factors that confer cellular tolerance against ischemia-reperfusion injury because of their ability to save oxygen and counter oxidative stress. The HIFs function as heterodimeric transcription factors composed of one alpha subunit (HIF1α, HIF2α or HIF3α) and one beta subunit (HIFβ), of the aryl hydrocarbon receptor nuclear translocator family. Whereas the HIFβ subunits are stable, the stability of the HIFα subunits fluctuates in response to changes in oxygen tension in a process controlled by the prolyl-4-hydroxylase (PHD) domain proteins (PHD1, PHD2 and PHD3), which are 2-oxoglutarate dependent Fe2+-dioxygenases (Figure 1) (Bruick and McKnight, 2001; Epstein et al., 2001). Under normoxic conditions, PHDs use oxygen to hydroxylate two conserved proline residues in the HIFα subunits (Figure 1). In turn, the hydroxylated prolyl residues are recognized by the von Hippel-Lindau (VHL)/E3 ubiquitin ligase complex, which targets the HIFα subunits for proteasomal degradation (Figure 1) (Safran and Kaelin, 2003; Schofield and Ratcliffe, 2004). By contrast, under hypoxic conditions there is insufficient oxygen for the PHDs to hydroxylate the HIFα subunits, preventing their recognition by VHL/E3 and resulting in their stabilization (Figure 1). Consequently, HIFα subunits can shuttle to the nucleus where they heterodimerize with HIFβ subunits and bind to DNA at hypoxia response elements (HREs) of target genes, to drive a HIF-dependent transcriptional program (Jiang et al., 1996; Ratcliffe et al., 1998; Ortiz-Barahona et al., 2010). Moreover, HIFα subunits can be stabilized constitutively in normoxia in a background of Vhl or PHD1–3 gene inactivation – as will be described in detail in some of the mouse models below – or in human clear cell renal cell carcinoma (ccRCC), which is characterized by loss of Vhl and subsequent constitutive HIF stabilization irrespective of the oxygen supply.
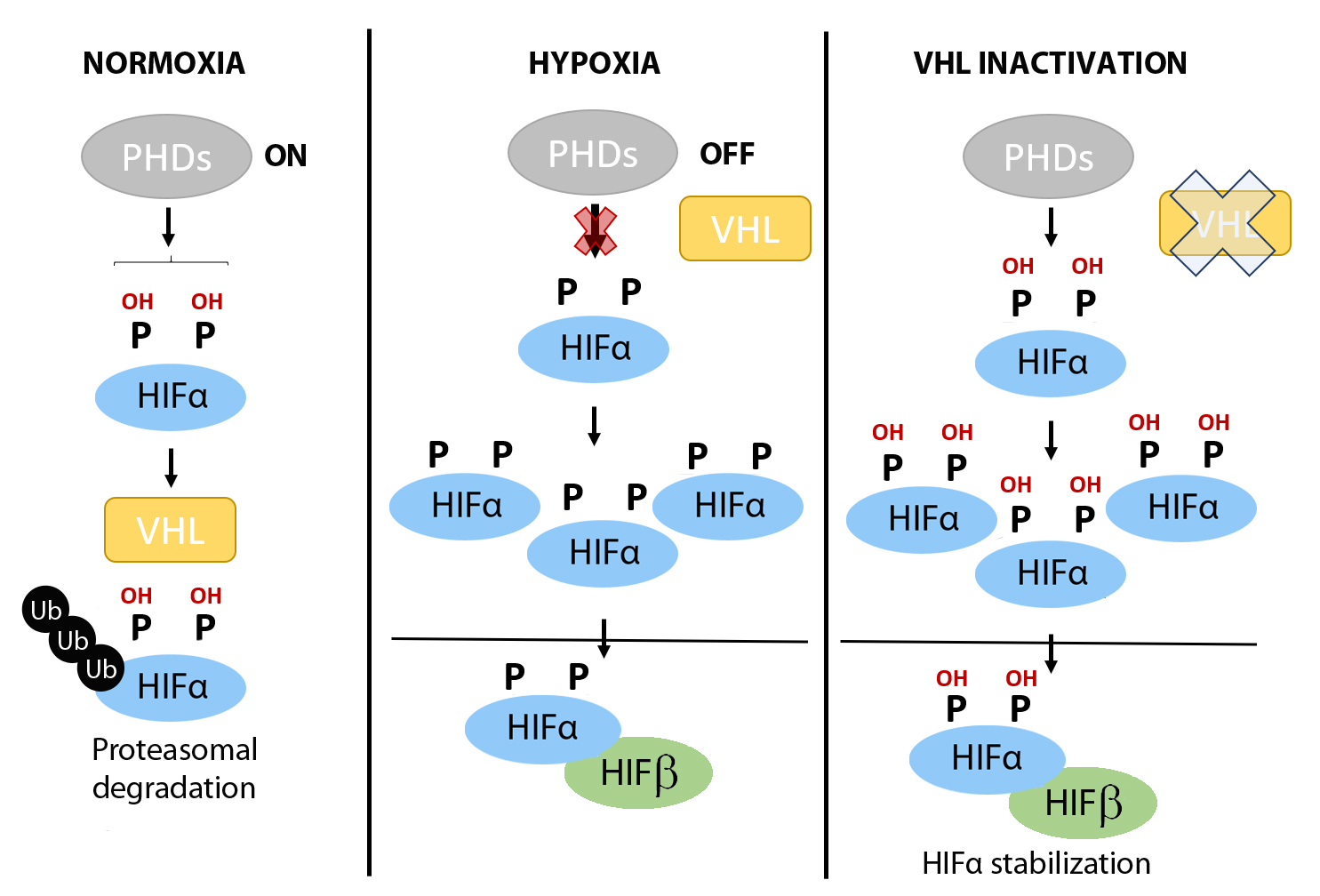
In a new window | Download PPT
Figure 1: Regulation of HIFα subunits stabilization by oxygen fluctuations and in VHL deficient tumor cells. When oxygen is available (left panel), prolyl-4-hydroxylases (PHDs) can hydroxylate HIFα subunits in two critical residues, which may be recognized by von Hippel Lindau (VHL) protein. Then, HIFα will be ubiquitinated and degraded by the proteasome. However, when oxygen becomes limited (middle panel), PHDs do not have enough oxygen to work, thus, HIFα subunits are not hydroxylated and VHL cannot recognize them. Therefore, HIFα subunits are accumulated and activated to promote gene transcription. In Vhl deficient cells, HIFα subunits can accumulate even under normoxic conditions (pseudohypoxic scenarios; right panel) because in the absence of VHL, HIFα subunits cannot be degraded despite an adequate oxygen supply, and therefore HIF-dependent pathways are constitutively active.
Two of the central biological functions mediated by the HIFs are (i) angiogenesis, to restore normal oxygen conditions in hypoxic environments through the induction of vascular endothelial growth factor A (VEGF-A) expression (Semenza et al., 1994; Forsythe et al., 1996), and (ii) erythropoiesis, to increase the oxygen-carrying capacity of the blood by inducing the expression of erythropoietin (EPO) (Rankin et al., 2007). A third biological function of the HIFs is the reprogramming of cellular metabolism by inducing a transition from aerobic to anaerobic metabolism. This metabolic switch suppresses cellular respiration and fosters energy generation through glycolysis, independently of oxygen. In this review, we will discuss the multiple mechanisms triggered by the HIFs to restrain mitochondrial activity, which save oxygen and quashes ROS production in low oxygen environments.
2. HIF pathway and mitochondrial content
Some, but not all, studies have reported that HIF activation can decrease the content of mitochondria in cells, which offers a plausible explanation for the known decrease in cellular respiration in response to hypoxia (Figure 2A). For example, the deficiency of Vhl or Phd2/3 in mouse hearts leads to HIF overactivation and reduced mitochondrial content (Lei et al., 2008; Minamishima et al., 2009; Moslehi et al., 2010; Menendez-Montes et al., 2016). Other studies have indicated that this suppression can be executed by the HIF1α isoform (Krishnan et al., 2009). Similarly, Vhl gene inactivation in mouse renal epithelial cells reduces mitochondrial mass and oxygen consumption in renal epithelium (Farsijani et al., 2016). Importantly, mitochondrial content was restored when HIF1α or HIF2α were simultaneously inactivated with Vhl, demonstrating that loss of mitochondrial abundance in Vhl-deficient renal epithelium can be entirely attributed to the overactivation of both HIF1α and HIF2α isoforms (Farsijani et al., 2016). In the setting of human renal cancer, ccRCC can originate from biallelic gene inactivation of VHL in the epithelium of renal tubules (Mandriota et al., 2002; Meléndez-Rodríguez et al., 2018). In line with the findings in Vhl-deficient mouse renal epithelium, RCC4 renal cancer cells, which lack VHL activity, show reduced mitochondrial content as a consequence of HIF1α and HIF2α overactivation (Zhang et al., 2007). In the context of metabolic disease, white adipose tissue expansion in obesity has been associated with an insufficient oxygen supply, leading to adipocyte hypoxia and subsequent activation of HIF1α. In this scenario Krishnan et al. (2012) found that in extreme obesity scenarios such as high-fat diet-fed mice, HIF1α is accumulated in white adipose tissue, leading to a reduced adipocyte mitochondrial content, which compromised fat oxidation and favored adipocyte intracellular lipid accumulation and obesity.
Regarding the molecular basis for the HIF-dependent reduction in mitochondrial abundance, several studies have described the participation of the peroxisome proliferator-activated receptor gamma (PPARγ) coactivators (PGCs), which are key transcriptional regulators involved in mitochondrial biogenesis (Puigserver et al., 1998). For example, Zhang et al. (2007) found that HIF1α and HIF2α activation can also represses PGC-1β-dependent mitochondrial biogenesis in RCC4 cells. At the molecular level, this study showed that HIF1α overactivation inhibits c-myc activity, which sustains PGC-1β gene expression in cells lacking VHL (Zhang et al., 2007). In addition, previous studies have found in different biological settings – including Vhl-deficient renal cancer cells – that HIF1α induces the expression of the basic-helix-loop-helix transcription factor Dec1 (Stra13/Bhlhe40) (Ivanova et al., 2001; Yun et al., 2002; LaGory et al., 2015), which acts as a transcriptional repressor of PGC-1α-dependent mitochondrial biogenesis (LaGory et al., 2015). Two conserved putative Dec1 binding sites have been identified in the PGC1α promoter, suggesting that Dec1 could directly repress PGC1α gene expression (LaGory et al., 2015). In addition, Krishnan et al. (2012) also found that HIF1α reduces the expression of sirtuin 2 (Sirt2) deacetylase, leading to increased PGC-1α acetylation in white adipose tissue. Interestingly, PGC-1α hyperacetylation has been associated with a weakening of its coactivator function (Rodgers et al., 2005).
In addition to its role in mitochondrial biogenesis, several studies have indicated that HIF1α can also reduce mitochondria abundance by promoting mitochondrial autophagy (mitophagy). Indeed, Zhang et al. (2008) proposed that HIF1α can stimulate mitophagy through the induction of Bcl2 interacting protein 3 (bNIP3) expression. At the molecular level, increased levels of bNIP3 compete with Beclin-1 for binding to Bcl2, which increases the levels of free Beclin-1, in turn triggering autophagy (Zhang et al., 2008). This HIF-dependent mitophagy pathway has been proposed to contribute to the reduced mitochondrial content in Vhl- or PHD-deficient hearts, which show damaged mitochondria and elevated levels of bNIP3 (Minamishima et al., 2009; Menendez-Montes et al., 2016). Accordingly, HIF factors have the ability to decrease mitochondrial content by suppressing mitochondrial biogenesis or increasing mitophagy (Figure 2A).
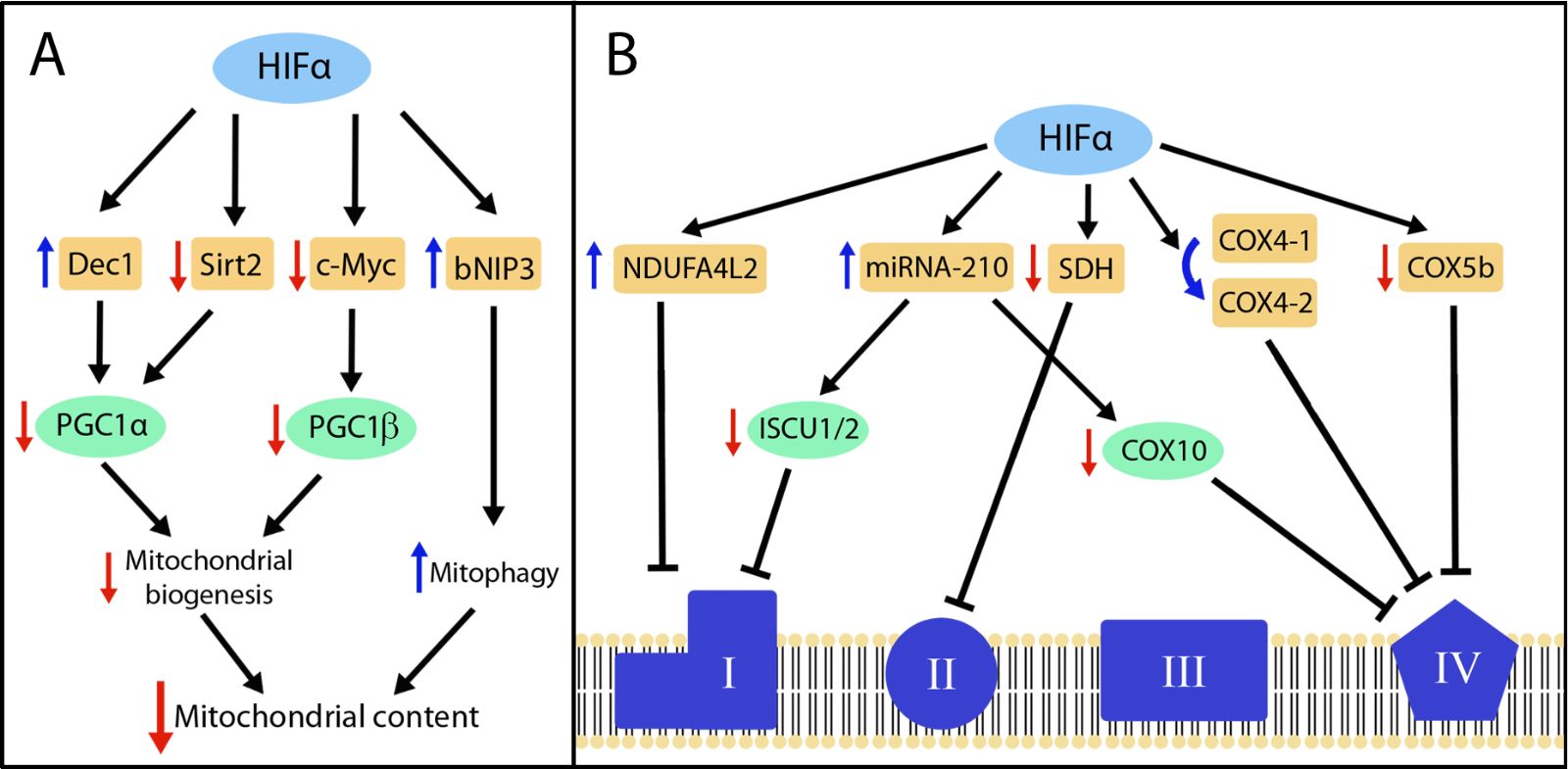
In a new window | Download PPT
Figure 2: HIF-dependent pathways involved in the suppression of mitochondrial content and mitochondrial respiratory chain activity. (A) The figure represents those genes shown to be controlled by HIFs (orange boxes) and involved in the suppression of peroxisome proliferator-activated receptor gamma coactivators (PGC)1α- and PGC1β-dependent mitochondrial biogenesis and in induction of mitophagy, which altogether can result in reduced mitochondrial content. (B) The figure represents those genes controlled by HIFs (orange boxes) that are involved in the suppression or reprograming of mitochondrial complex I, II and IV. In both panels, the blue and red arrows represent upregulation and downregulation respectively. The HIF isoform involved in each of these metabolic actions are detailed in the text.
Of note, in other biological settings, HIF activation is associated with a reduction in cellular respiration, but without the concurrent reduction in mitochondrial content. For instance, gain of HIF1α function in white adipocytes during aging does not lead to reduced mitochondrial abundance, but rather promotes the parallel decline of white adipose tissue respiration (Soro-Arnaiz et al., 2016). Likewise, HIF activation in PHD1-deficient skeletal muscle leads to a decrease in mitochondrial oxygen consumption but without the corresponding reduction in mitochondrial mass (Aragonés et al., 2008). Finally, tumor cells exposed to hypoxia can markedly reduce oxygen consumption, but without any form of mitochondria content decline (Papandreou et al., 2006). It is conceivable that the loss of mitochondria requires a robust (close to maximal) activation of HIF1α such as is observed in Vhl-deficient cells and PHD-deficient tissues, or is specifically operative only in some cell types. In this line, cardiac mitochondrial content decline is more clearly manifested in Phd2/Phd3 deficient mice than in Phd2-deficient mice, which activates HIF1α and HIF2α to a lesser extent (Moslehi et al., 2010). Of note, massive HIF overactivation in Vhl-deficient or Phd2/Phd3 deficient hearts can become pathologically more possible as a consequence of such profound mitochondrial decline. Therefore, cardiac tolerance as a consequence of HIF-dependent mitochondrial reprogramming can require submaximal HIF activation. Independently of these considerations, it is clear that HIF-dependent repression of cellular respiration cannot be attributed exclusively to the loss in mitochondria abundance, and that other HIF-dependent key mitochondrial players are involved for full suppression of mitochondrial activity in response to hypoxia, as described in the next sections.
3. HIF pathway and glucose/fatty acid-driven tricarboxylic acid cycle
The tricarboxylic acid (TCA) cycle is essential to sustain mitochondrial respiration since it generates the reducing equivalents NADH and FADH2, which are subsequently oxidized in the mitochondrial respiratory chain to fuel oxygen consumption and ATP generation. Replenishment of the TCA cycle can be dependent on glucose, fatty acids, and glutamine taken up from the extracellular media. Indeed, glucose entering into cells is converted to pyruvate through multiple steps of the glycolysis pathway. Subsequently, mitochondrial pyruvate dehydrogenase converts pyruvate to acetyl-coenzyme A (CoA), which condenses with oxaloacetate in a reaction catalyzed by citrate synthase to generate citrate. The activity of the mitochondrial pyruvate dehydrogenase complex is attenuated when it is phosphorylated by pyruvate dehydrogenase kinases (PDKs) (Holness and Sugden, 2003; Sugden and Holness, 2003). Key studies in this field have established that hypoxia induces PDK-1 gene expression through HIF1α activation (Kim et al., 2006; Papandreou et al., 2006). Indeed, a HIF1α binding site identified in the PDK-1 promoter is associated with PDK-1 mRNA elevation in response to hypoxia. Furthermore, several studies using 13C5-glucose tracer analysis have shown that HIF1α activation – but also HIF2α activation – leads to reduced glucose-dependent 13C5-citrate generation (Wise et al., 2011; Gameiro et al., 2013). The metabolic consequence of this HIF-dependent PDK-1 upregulation is the inhibition of the pyruvate dehydrogenase complex, which reduces glucose-dependent TCA cycle replenishment and subsequently attenuates glucose-driven oxygen consumption. Further studies showed that HIF signaling also induces the expression of PDK3 and PDK4 isoforms in different biological settings (Aragonés et al., 2008; Lu et al., 2008; Zhong et al., 2010). Together, these studies have established that augmented cellular PDK activity is a key feature of the HIF-dependent metabolic program to repress glucose-dependent oxygen consumption in hypoxic cells (Figure 3). As mentioned earlier, reduced oxygen consumption executed by HIF activity not only saves oxygen, but also reduces the formation of mitochondrial ROS leading to cellular tolerance to ischemia-reperfusion oxidative injury (Aragonés et al., 2009; Semenza, 2011). Therefore attenuation of glucose oxidation through PDK elevation is considered a HIF-dependent protective response for cells residing in hypoxic environments such as ischemic tissue (Aragonés et al., 2009; Semenza, 2011). This protection program seems to be also present in the inner core of solid tumors, which are also characterized by poor perfusion and oxygen/nutrient deficiency, mimicking the environment of ischemic regions (McFate et al., 2008). The relevance of this metabolic control is also highlighted by the fact that pyruvate that is not converted to acetyl-CoA – as a consequence of the HIF-PDK pathway – can be converted to lactate, which favors ATP generation through the glycolytic pathway in an oxygen-independent manner. In addition, HIF1α can also attenuate acetyl-CoA generation by reducing the expression of key players in fatty acid oxidation. Indeed, a previous study in hepatocellular carcinoma cells has shown that HIF1α-dependent repression of PGC-1β activity (see above) also leads to the reduced expression of PGC-1β-dependent fatty oxidation enzymes such as medium-chain (MCAD) and long-chain (LCAD) acyl-CoA dehydrogenases (Huang et al., 2014). Overall, simultaneous HIF1α-dependent suppression of pyruvate to acetyl-CoA reaction by inducing PDK activity, as well as reduced expression of acyl-CoA dehydrogenases leads to a remarkable suppression of acetyl-CoA-dependent TCA cycle activity upon HIF1α activation.

In a new window | Download PPT
Figure 3: HIF-dependent pathways that reprogram the TCA cycle. The figure represents those metabolic reactions that are regulated by HIF factors such as pyruvate dehydrogenase kinases (PDKs) elevation to suppress glucose-dependent oxidation, reduced expression of medium-chain (MCAD) and long-chain (LCAD) acyl-CoA dehydrogenases enzymes, inhibition of succinate dehydrogenase (SDH) and OGDH2 activities that can restrain glucose, fatty acid, and glutamine oxidative pathways, and finally the role of HIF on glutamine-dependent TCA replenishment by inducing GLS activity and favoring IDH-dependent glutamine reductive metabolism. In both panels, blue and red arrows represent upregulation and downregulation respectively. The HIF isoform involved in each of these metabolic actions are detailed in the text.
4. HIF pathway and glutamine-driven tricarboxylic acid pathway activity
The studies discussed above have predominately focused on glucose metabolism, but more recently the role of the HIF pathway in glutamine metabolism has become increasingly appreciated (Figure 3). Glutamine is also essential to replenish the TCA cycle. Glutamine entering into cells is first converted to glutamate by the enzyme glutaminase, and is subsequently converted to the TCA intermediate α-ketoglutarate (Altman et al., 2016). A recent study in PHD2-deficient murine periosteum-derived cells showed that HIF1α activity promotes glutamine uptake, which is accompanied by a concomitant elevation of glutaminase expression, leading to intracellular glutamate accumulation (Stegen et al., 2016). Moreover, increased intracellular glutamate accumulation has been found in hypoxic as well as in Vhl-null CD8+ T lymphocytes, and occurs through a mechanism dependent on HIF1α (Tyrakis et al., 2016). More recently, our own studies in tumor cells showed that restoration of HIF1α in tumor cells lacking HIF1α leads to increased glutamine-dependent glutamate generation (Meléndez-Rodríguez et al., 2019). Importantly intracellular glutamate is a precursor of glutathione synthesis and, accordingly, HIF1α-dependent intracellular glutamate accumulation can be used for glutathione biosynthesis, which can ultimately promote cell survival by counteracting oxidative stress (Stegen et al., 2016). These recent data suggest that HIF1α-dependent metabolic reprograming can neutralize oxidative stress not only by diminishing mitochondrial respiration, but also by rewiring glutamine metabolism to favor glutathione biosynthesis. Furthermore, it has been shown that glutamate secretion is induced in hypoxic human hepatocellular carcinoma Hep3B cells (Hu et al., 2014), suggesting that not all of the glutamate accumulated in response to HIF1α activation is used for glutathione generation. Of note, glutamate can be also subsequently converted to α-ketoglutarate, which can be oxidized through the TCA pathway to produce succinyl-CoA as a first step, and subsequently to other TCA intermediates such as succinate, fumarate, malate, oxaloacetate, and citrate. Our recent 13C5-glutamine metabolic tracing studies showed that HIF1α reduces glutamine oxidation (Meléndez-Rodríguez et al., 2019). In line with these data, constitutive endogenous HIF1α activation in Vhl-deficient renal carcinoma cells suppresses glutamine-dependent citrate generation through the oxidative pathway (Metallo et al., 2011; Wise et al., 2011; Gameiro et al., 2013; Okazaki et al., 2017), as well as glutamine-dependent respiration (Sun and Denko, 2014). At the molecular level, Sun and Denko showed that HIF1α activation compromises the activity of the α-ketoglutarate dehydrogenase (αKGDH) complex, which converts α-ketoglutarate to succynyl-CoA (Figure 3). Specifically, HIF1α promotes SIAH2-targeted ubiquitination and proteolysis of the 48 kDa splice variant of the E1 subunit of the α-KGDH complex, OGDH2 (Sun and Denko, 2014). Moreover, our own studies have shown that HIF1α represses the expression of subunit A of succinate dehydrogenase (SDHA), the TCA enzyme that directly converts succinate to fumarate and a subunit of mitochondrial complex II. This is consistent with another study showing that HIF1α decreases the expression of the mitochondrial SDHB subunit (Dahia et al., 2005), suggesting that HIF1α can also decrease other complex II subunits not directly involved in the conversion of succinate to fumarate. Therefore, HIF1α-dependent repression of glutamine oxidation can be a consequence of the simultaneous repression of α-KG dehydrogenase and SDH activities, which limits metabolite biosynthesis, particularly of those TCA metabolites generated after the SDH reaction.
Glutamine-derived α-ketoglutarate is not only generated by oxidative metabolism, but can be alternatively converted to citrate through reductive carboxylation (Metallo et al., 2011; Mullen et al., 2011; Wise et al., 2011; Gameiro et al., 2013; Okazaki et al., 2017). This requires as a first step the conversion of α-ketoglutarate to isocitrate through isocitrate dehydrogenase (IDH), which is followed by the conversion of isocitrate to citrate through aconitase. Previous studies have shown that silencing of the cytoplasmic IDH1 isoform reduces citrate generation through reductive carboxylation, indicating that this reaction takes place in the cytosol (Metallo et al., 2011). Further studies showed that silencing of the mitochondrial IDH2 isoform also compromises glutamine reductive carboxylation, suggesting that this pathway can also occur in the mitochondrial matrix, and that mitochondrial IDH2 can simultaneously function in oxidative and reductive metabolism (Mullen et al., 2011). Importantly, HIF1α favors this alternative pathway in an attempt to compensate for the suppression of citrate generation through oxidative pathways (Metallo et al., 2011; Wise et al., 2011; Fendt et al., 2013; Gameiro et al., 2013; Okazaki et al., 2017). It could be considered that these reactions acting in reverse consume rather than generate NADH, which therefore might also limit the transfer of electrons to mitochondria and contribute to some extent to reduced respiration upon HIF1α activation.
Overall, these findings serve to illustrate that HIF stimulates multiple and simultaneous mechanisms to attenuate glucose and glutamine oxidation, which compromise the biosynthesis of reducing equivalents such as NADH and FADH2 required for cellular respiration.
5. HIF pathway and the mitochondrial respiratory chain
Another key mechanism by which HIF1α suppresses cellular respiration is by inhibition of the activity of mitochondrial respiratory complexes (Figure 2B). For instance, HIF1α impedes iron-sulfur cluster assembly in key mitochondrial respiration proteins by reducing the expression of iron-sulfur cluster assembly proteins 1/2 (ISCU1/2) (Chan et al., 2009; Chen et al., 2010; Favaro et al., 2010). Iron-sulfur clusters are prosthetic groups required for critical cellular functions including electron transport and mitochondrial oxidation-reduction reactions. ISCU1/2 proteins serve as assembly scaffolds, obtaining ferrous iron and inorganic sulfide to build the iron-sulfur clusters (Rouault and Tong, 2008). Several studies have confirmed that HIF activation – both in vitro and in vivo – decreases the expression of ISCU1/2, leading to compromised mitochondrial complex I activity, which is particularly dependent on iron-sulfur assembly (Kucejova et al., 2011; White et al., 2015). Importantly, the decrease in expression of ISCU in response to hypoxia involves the HIF-dependent upregulation of miRNA-210, which targets ISCU1 and ISCU2 transcripts via a conserved binding site located in the 3’ untranslated region (Chan et al., 2009; Chen et al., 2010; Favaro et al., 2010). HIF-dependent miRNA-210 expression also negatively regulates aconitase, a key TCA enzyme that requires iron-sulfur assembly to convert citrate in isocitrate (Chan et al., 2009; Favaro et al., 2010). Moreover, miRNA-210 was also found to target cytochrome c oxidase 10 (COX10), a COX (complex IV) assembly protein (Chen et al., 2010). Accordingly, HIF-dependent miRNA-210 upregulation simultaneously attenuates not only mitochondrial complex I and IV, but also controls TCA cycle activity, which together can explain the reduced oxygen consumption observed when cellular levels of miRNA-210 are upregulated. Furthermore, studies have found that HIF1α-dependent miRNA-210 expression conferred cell tolerance by counteracting apoptosis under hypoxic conditions, whereas the opposite effect was found under normoxic conditions (Chan et al., 2009; Chen et al., 2010; Favaro et al., 2010). Interestingly, Chan et al. (2009) found that the HIF1α-miRNA-210 pathway serves to prevent ROS formation in human pulmonary arterial endothelial cells exposed to hypoxia, which might explain the protective effect of miRNA-210. By contrast, Favaro et al. (2010) found that miRNA-210 elevates ROS levels under hypoxic conditions in HCT116 and MCF7 cell lines. Therefore, it is still unclear whether mitochondrial reprograming executed by the HIF1α-miRNA210 pathway contributes in general to protect against oxidative damage in a large number of cell types.
We recently showed that HIF1α also induces the expression of NDUFA412, which has a HIF-binding site in its proximal promoter (Tello et al., 2011). NDUFA412 is strongly induced in numerous in vitro and in vivo biological settings in response to hypoxia or Vhl gene inactivation (Tello et al., 2011; Lai et al., 2016; Minton et al., 2016; Schönenberger et al., 2016). Elevated NDUFA412 expression in response to HIF1α reduces oxygen consumption and attenuates mitochondrial ROS generation, which confers cellular tolerance in hypoxia (Tello et al., 2011). Hypoxia-induced NDUFA412 was also found to attenuate mitochondrial complex I activity, suggesting that NDUFA412 upregulation as well as ISCU1/2 suppression can cooperate to assure complex I inhibition in response to HIF activation. As mentioned previously, HIF1α also reduces the expression of SDHA, which is simultaneously a key TCA enzyme and an essential subunit of complex II, and therefore HIF1α also has the capacity to repress mitochondrial complex II activity (Meléndez-Rodríguez et al., 2019). In addition to complex II activity, HIFs also control complex IV activity. Indeed, HIF1α induces a switch from COX4-1 to COX4-2 subunit via simultaneously inducing the expression of COX4-2 and LON, a protease that degrades COX4-1 (Fukuda et al., 2007). This switch optimizes mitochondrial respiration, which reduces mitochondrial ROS generation under hypoxic conditions. We have also shown in the context of age-dependent obesity that HIF1α can destabilize complex IV by decreasing the expression of the complex IV subunit COX5b (Soro-Arnaiz et al., 2016), which is sufficient to destabilize the entire complex, resulting in the reduced expression of other complex IV subunits such as Ndufa4 and COX-1 (Soro-Arnaiz et al., 2016). This age-dependent HIF1α activation in white adipose tissue compromised complex IV activity without affecting the activity of the other complexes. Accordingly, it is possible that mitochondrial complex IV is more vulnerable to moderate HIF1α accumulation, as in aged adipocytes, and more robust HIF1α accumulation (as in Vhl-deficient cells) is necessary to simultaneously suppress complex I, II and IV by the mechanisms described above.
6. Conclusions
In this review, we have summarized the responses that HIF1 and HIF2 factors trigger simultaneously to assure that mitochondrial respiration is repressed. To do this, HIFs decrease mitochondrial content and TCA replenishment, and suppress the assembly and activity of different mitochondrial respiratory complexes. These metabolic actions executed by HIFs have beneficial effects by saving oxygen and preventing oxidative stress in hypoxic, poorly perfused, or ischemic tissues. However, this HIF-dependent metabolic rewiring also impairs fatty acid oxidation, resulting in pathological lipid accumulation, therefore providing a HIF-dependent molecular basis for diseases characterized by fat accumulation such as liver steatosis or obesity. Because of the central role of HIF-dependent metabolic reprograming in several pathological scenarios, the identification of novel metabolic actions executed by the HIFs that could lead to the development of new therapeutic strategies will be important future challenges.
Conflicts of interest
Authors declare that there are no conflicts of interest.
Acknowledgments
We acknowledge funding support from the Spanish Ministerio de Economia y Competitividad (SAF2016-76815 and SAF2017-90794-REDT) and Fundació La Marató de TV3 (534/C/2016).
References
Claudia Mesa-Ciller1
1Research Unit, Hospital of Santa Cristina, Research Institute Princesa (IP), Autonomous University of Madrid, Madrid 28009, Spain.
Guillermo Turiel1
1Research Unit, Hospital of Santa Cristina, Research Institute Princesa (IP), Autonomous University of Madrid, Madrid 28009, Spain.
Julián Aragonés1,2
1Research Unit, Hospital of Santa Cristina, Research Institute Princesa (IP), Autonomous University of Madrid, Madrid 28009, Spain.
2CIBER de Enfermedades Cardiovasculares (CIBERCV), Carlos III Health Institute, Madrid, Spain.
Corresponding author:
Julián Aragonés
Email: julian.aragones@uam.es
In a new window | Download PPT
Figure 1: Regulation of HIFα subunits stabilization by oxygen fluctuations and in VHL deficient tumor cells. When oxygen is available (left panel), prolyl-4-hydroxylases (PHDs) can hydroxylate HIFα subunits in two critical residues, which may be recognized by von Hippel Lindau (VHL) protein. Then, HIFα will be ubiquitinated and degraded by the proteasome. However, when oxygen becomes limited (middle panel), PHDs do not have enough oxygen to work, thus, HIFα subunits are not hydroxylated and VHL cannot recognize them. Therefore, HIFα subunits are accumulated and activated to promote gene transcription. In Vhl deficient cells, HIFα subunits can accumulate even under normoxic conditions (pseudohypoxic scenarios; right panel) because in the absence of VHL, HIFα subunits cannot be degraded despite an adequate oxygen supply, and therefore HIF-dependent pathways are constitutively active.
In a new window | Download PPT
Figure 2: HIF-dependent pathways involved in the suppression of mitochondrial content and mitochondrial respiratory chain activity. (A) The figure represents those genes shown to be controlled by HIFs (orange boxes) and involved in the suppression of peroxisome proliferator-activated receptor gamma coactivators (PGC)1α- and PGC1β-dependent mitochondrial biogenesis and in induction of mitophagy, which altogether can result in reduced mitochondrial content. (B) The figure represents those genes controlled by HIFs (orange boxes) that are involved in the suppression or reprograming of mitochondrial complex I, II and IV. In both panels, the blue and red arrows represent upregulation and downregulation respectively. The HIF isoform involved in each of these metabolic actions are detailed in the text.
In a new window | Download PPT
Figure 3: HIF-dependent pathways that reprogram the TCA cycle. The figure represents those metabolic reactions that are regulated by HIF factors such as pyruvate dehydrogenase kinases (PDKs) elevation to suppress glucose-dependent oxidation, reduced expression of medium-chain (MCAD) and long-chain (LCAD) acyl-CoA dehydrogenases enzymes, inhibition of succinate dehydrogenase (SDH) and OGDH2 activities that can restrain glucose, fatty acid, and glutamine oxidative pathways, and finally the role of HIF on glutamine-dependent TCA replenishment by inducing GLS activity and favoring IDH-dependent glutamine reductive metabolism. In both panels, blue and red arrows represent upregulation and downregulation respectively. The HIF isoform involved in each of these metabolic actions are detailed in the text.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13641 | 42 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA