Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Estrogen preconditioning: a promising strategy to reduce inflammation in the ischemic brain
Time:2019-07-03
Number:14055
Author Affiliations

Conditioning Medicine, 2019. 2(3): 106-113.
Abstract
During the premenopausal phase of a woman’s life, estrogen naturally protects against ischemic brain damage and its debilitating consequence of cognitive decline. However, the decline in estrogen at menopause exponentially increases a women’s risk for cerebral ischemia and its severity. Supplementation of estrogen during menopause is the most logical solution to abate this increased risk for cerebral ischemia; however, continuous therapy has proven to be contraindicative. Studies from our laboratory over the past decade have shown that a single bolus or long-term periodic 17β-estradiol treatment(s) two days prior to ischemia mimics ischemic preconditioning-conferred protection of the brain in ovariectomized or reproductively senescent female rats. These studies also demonstrated that 17β-estradiol-induced preconditioning (EPC) requires estrogen receptor (ER)-subtype beta (ER-β) activation. ER-β is expressed throughout the brain, including in the hippocampus, which plays a key role in learning and memory. Because periodic activation of ER-β mitigates post-ischemic cognitive decline in ovariectomized female rats, it can be surmised that EPC has the potential to reduce post-ischemic damage and cognitive decline in females. Estrogens are key anti-inflammatory agents; therefore this review discusses the effects of EPC on the inflammasome. Furthermore, as we now clearly know, the brain acts differently in males and females. Indeed, neurodegenerative diseases, including cerebral ischemia, and pharmacological drugs affect males and females in different ways. Thus, inasmuch as the National Institutes of Health and the Stroke Treatment Academic Industry Roundtable (STAIR) consortium mandate inclusion of female experimental animals, this review also discusses the need to close the gap in our knowledge in future studies of EPC in female animal models of cerebral ischemia.
Keywords: Reproductively senescent, Menopause, Inflammasome, NOD-like receptors (NLRs), Extracellular vesicles
Abstract
During the premenopausal phase of a woman’s life, estrogen naturally protects against ischemic brain damage and its debilitating consequence of cognitive decline. However, the decline in estrogen at menopause exponentially increases a women’s risk for cerebral ischemia and its severity. Supplementation of estrogen during menopause is the most logical solution to abate this increased risk for cerebral ischemia; however, continuous therapy has proven to be contraindicative. Studies from our laboratory over the past decade have shown that a single bolus or long-term periodic 17β-estradiol treatment(s) two days prior to ischemia mimics ischemic preconditioning-conferred protection of the brain in ovariectomized or reproductively senescent female rats. These studies also demonstrated that 17β-estradiol-induced preconditioning (EPC) requires estrogen receptor (ER)-subtype beta (ER-β) activation. ER-β is expressed throughout the brain, including in the hippocampus, which plays a key role in learning and memory. Because periodic activation of ER-β mitigates post-ischemic cognitive decline in ovariectomized female rats, it can be surmised that EPC has the potential to reduce post-ischemic damage and cognitive decline in females. Estrogens are key anti-inflammatory agents; therefore this review discusses the effects of EPC on the inflammasome. Furthermore, as we now clearly know, the brain acts differently in males and females. Indeed, neurodegenerative diseases, including cerebral ischemia, and pharmacological drugs affect males and females in different ways. Thus, inasmuch as the National Institutes of Health and the Stroke Treatment Academic Industry Roundtable (STAIR) consortium mandate inclusion of female experimental animals, this review also discusses the need to close the gap in our knowledge in future studies of EPC in female animal models of cerebral ischemia.
Keywords: Reproductively senescent, Menopause, Inflammasome, NOD-like receptors (NLRs), Extracellular vesicles
Introduction
Cerebral ischemia, resulting from cardiac arrest or stroke, is one of the leading causes of mortality and morbidity in the world, and strategies to prevent this disease or to mitigate its consequences are greatly needed. Ischemic preconditioning (IPC)/tolerance is emerging as an innovative and potent neuroprotective strategy to counter ischemic brain damage (Moncayo et al., 2000; Koch et al., 2012; Narayanan et al., 2013; Koch et al., 2014; Hausenloy et al., 2016; Esposito E, 2018; Tauskela JS, 2018). Clinical observations suggest that transient ischemic attacks (TIAs) before ischemic stroke in the same vascular territory are associated with milder initial clinical symptoms and more favorable outcomes in stroke (“protective TIA”) (Weih et al., 1999; Moncayo et al., 2000; Wegener et al., 2004). Multiple laboratories across the globe, including ours, have also proven that a sub-lethal ischemic episode increases the brain's ability to withstand a subsequent injurious ischemic insult, using in vitro and animal models of global or focal ischemia (Dowden and Corbett, 1999; Perez-Pinzon et al., 1999; Raval et al., 2003; Stenzel-Poore et al., 2003; Kim et al., 2010; Brand and Ratan, 2013; Bell et al., 2017; Li et al., 2017). Although the phenomenon of IPC is well documented, studies demonstrating the efficacy of ischemic tolerance have been conducted mainly on male experimental animals, and experiments on female animals have been neglected with the assumption that results from male studies would apply to females (Beery and Zucker, 2011). In addition, studies on female counterparts are commonly dismissed owing to their normal cycles of endogenous ovarian hormone fluctuations (Beery and Zucker, 2011). An amendment to the Stroke Treatment Academic Industry Roundtable (STAIR) consortium recommends that “efficacy studies should be performed in both male and female animals” (Fisher et al., 2009). In general, the National Institutes of Health also requests biomedical investigators to include female animals to avoid skewing data (Clayton and Collins, 2014). We already know that neurodegenerative diseases, including stroke, and pharmacological drugs affect males and females in different ways - a phenomenon that gives sex-specific therapy a promising future (Siegel and McCullough, 2013; Manwani et al., 2015; Chisholm and Sohrabji, 2016; Chauhan et al., 2017; Kim et al., 2019). If we are to translate IPC from bench to bedside, we must identify possible sex difference/similarities in IPC-conferred neuroprotection and understand the physiological changes in male and female brains, especially the latter, as it is often neglected by researchers. A better understanding of the cellular and molecular mechanisms that regulate differences between female and male brain activities could have future therapeutic implications. What we know thus far is that sex hormones play a major role in normal and abnormal brain development, maturation, and aging (Brown et al., 1994; Solum and Handa, 2002; Srivastava et al., 2013; Duarte-Guterman et al., 2015; Marrocco and McEwen, 2016). In fact, one of the ovarian hormones - 17β- estradiol (E2) - is a potent neuromodulator and a neuroprotective agent (Raval et al., 2006; Arevalo et al., 2015; Engler-Chiurazzi et al., 2017). Studies from our laboratory demonstrated that a single bolus of E2 48h prior to global ischemia mimics IPC-conferred protection of the selectively vulnerable CA1 region of the hippocampus (Raval et al., 2006; Raval et al., 2013; Cue et al., 2015; de Rivero Vaccari et al., 2016). It is important to note that the paradigm of E2 treatment hours prior to cerebral ischemia is similar to that of IPC/pharmacological preconditioning and therefore, we call this phenomenon E2 preconditioning (EPC). In a subsequent study, we demonstrated that EPC at 48h intervals for ten treatments reduced post-ischemic hippocampal neuronal death and improved hippocampal-dependent cognition in female rats (Raval et al., 2009; Raval et al., 2013). Cognitive decline remains the most significant problem among cardiac arrest (Frisch et al., 2017) and stroke survivors (Tatemichi et al., 1993; Barba et al., 2000; Stephens et al., 2004; Levine et al., 2015), and the aforementioned data suggest that EPC has the potential to reduce post-ischemic cognitive decline in females and emphasizes the need to investigate EPC for the prevention of incidents/impact of cerebral ischemia in peri- and post-menopausal women. The current review discusses the available literature on EPC and identifies the gap in our knowledge for future studies to test EPC in female animal models of cerebral ischemia.
Estrogen preconditioning (EPC) and ischemic neuroprotection
Decades of research have established that neuronal death and brain injury are prevented with E2 treatment in both in vitro focal and global ischemia models (Simpkins et al., 1997; Dubal et al., 1998; Pelligrino et al., 1998; Rusa et al., 1999; Alkayed et al., 2000; Wise et al., 2001; Jover et al., 2002; Kofler et al., 2005; Gulinello et al., 2006; Arevalo et al., 2015; Thakkar et al., 2016; Engler-Chiurazzi et al., 2017). Strong evidence from experimental animal models suggests that chronic continuous E2 treatment provides both potent and long-lasting improvements in pathophysiological outcome after focal and global cerebral ischemia (Alkayed et al., 2000; Wise et al., 2001; Jover et al., 2002). Although there are strong pre-clinical data supporting E2 treatment for prevention of ischemic brain damage, concern regarding the safety of chronic E2 has prohibited translation of this phenomenon to the clinic (Viscoli et al., 2001; Utian, 2007). Therefore, it is essential to identify an alternative E2 regimen that avoids known side effects of continuous E2 replacement, and the appropriate time window in which E2 treatment is efficacious. One possible solution to overcome this concern is to mimic endogenous estrogen release and develop a regimen to give periodic estrogen therapy that mimics IPC for ischemic neuroprotection. In this regard, we investigated whether endogenous hormonal variations during the estrous cycle have any effect on ischemic outcome in normal young rats (Raval et al., 2009). Our study showed that increasing titers of endogenous E2 during the transition from diestrus to proestrus of the estrous cycle conferred protection to the hippocampus against global cerebral ischemia (Raval et al., 2009). Ischemic tolerance at the proestrus and estrus stages correlated with increased immunoreactivity of the phosphorylated cAMP response element-binding protein (pCREB (Bourtchuladze et al., 1994)), a cellular transcription factor in the hippocampus. On the contrary, when circulating gonadal hormone concentrations were lowest at diestrus, the pCREB protein content of hippocampus was reduced and showed the least number of normal neurons after ischemia compared to other stages of the estrous cycle. These results suggest that endogenous intermittent rising levels of E2 induce pCREB-mediated priming in hippocampal neurons and are pivotal for neuronal survival against ischemia/stress (Raval et al., 2009). Further, mimicking this natural intermittent increase in ovariectomized rats by a single exogenous administration of E2 initiated the pCREB pathway, primed the hippocampus, and protected against cerebral ischemia. Similarly, a study by the Scharfman laboratory simulated the pre-ovulatory estrogen surge in ovariectomized rats after sequential low doses of E2, and confirmed that physiological E2 levels were sufficient to profoundly affect hippocampal function (Scharfman et al., 2007).
The aforementioned studies supporting the protective role of EPC were conducted in young animals evincing a normal estrous cycle, and in those animals the condition of menopause was simulated by surgical removal of the ovaries. The incidence of stroke/cardiac arrest in women increases after the onset of menopause, and menopause is a highly complex process that takes about 5-7 years in women (Harlow et al., 2012). Therefore, simulating menopause by surgical removal of the ovaries adopted in pre-clinical studies may not provide a clear picture, and future studies testing the efficacy of EPC for ischemic protection need to be conducted using reproductively senescent (mimicking peri-menopause) and aged (mimicking post-menopause) female animals. Targeting both peri- and post-menopause ages are critical as the systemic ovarian hormonal milieu varies during those stages of menopause, with a progressive decline in circulating estrogen. This circulating estrogen decline consequently reduces the availability of ER-α and ER-β in the brain, which is essential to E2 neuroprotective signaling (Nilsson et al., 2011; Waters et al., 2011). Also, long-term E2 deprivation after menopause was identified as an important issue by several clinical trials on estrogen, and the outcome of these trials suggested that there is a “critical period” beyond which E2 might not be effective. Identifying the critical period for EPC efficacy will be interesting and crucial for future translation (Sun, 2019). Studies have established that E2 mediates ischemic neuroprotection through activation of ER-β (Noppens et al., 2009; Raval et al., 2013). Therefore, preconditioning using ER-β receptor agonists could be more beneficial. The critical time window of ER-β agonist-mediated neuroprotection after onset of reproductive senescence in female rats needs further investigation.
Pharmacological preconditioning induced by ER-β activation reduces inflammation in the brain
The aging process is associated with changes in the composition and function of the immune system and these changes occur at an accelerated rate after onset of menopause in women. In contrast, an accelerated rate of immune and inflammation system dysfunction may not occur as rapidly in elderly men. Estrogen has been suggested to function as a potent anti-inflammatory factor (Cushman et al., 1999; Vegeto et al., 2008; Edwards and Li, 2013). Thus, the depletion of estrogen as a result of menopause activates systemic adaptive and innate immune responses (Giannoni et al., 2011). Menopause and the associated lack of steroidal hormones coincide with higher levels of circulating interleukins (IL-6, IL-4, IL-2) and tumor necrosis factor (TNF) in postmenopausal women, and the increase in these factors have been shown to be reversed by hormone therapy (Giuliani et al., 2001; Pfeilschifter et al., 2002; Yasui et al., 2007).
On the other hand, it is also well established that cerebral ischemia activates the innate immune response, and a key component of the innate immune response is the inflammasome (Abulafia et al., 2009; Adamczak et al., 2014; Anrather and Iadecola, 2016). This multiprotein complex – the inflammasome – activates caspase-1 and processing of inflammatory cytokines IL-1β and IL-18 (de Rivero Vaccari et al., 2008; Lotocki et al., 2009). It has been shown that sex steroids regulate activation of the inflammasome in the brain (Slowik and Beyer, 2015).
In general, activation of inflammasomes can occur in response to either pathogen associated molecular patterns (PAMPs), derived from invading pathogens, or damage-associated molecular patterns (DAMPs), released by dying cells (Guo et al., 2015). The latter is the result of endogenous stress that activates pattern-recognition receptors (PRRs), such as nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), which form inflammasomes like the NLR family pyrin domain containing 1 (NLRP1) or NLRP3 inflammasomes. Several families of PRRs, including NLRs and the absent in melanoma 2 (AIM)-like receptors (ALRs) are important components of the inflammasome complex (Takeuchi and Akira, 2010). These PRRs (NLRs or ALRs) oligomerize to form a caspase-1–activating scaffold following sensing of stimuli (PAMPs or DAMPs). Once active, caspase-1 cleaves the pro-inflammatory cytokines IL-1 into their bioactive forms, IL-1β and IL-18 (Figure 1). In addition, this activation of caspase-1 may result in an inflammatory-mediated cell death mechanism known as pyroptosis (Lamkanfi and Dixit, 2012; Strowig et al., 2012).
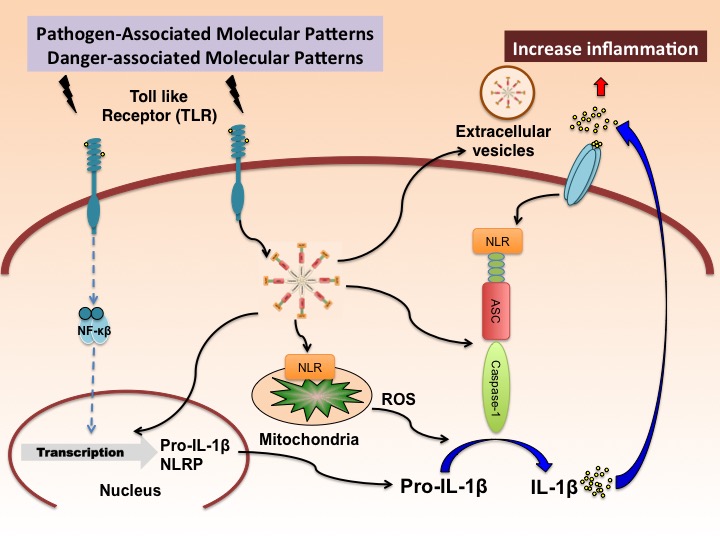
In a new window | Download PPT
Figure 1: Putative mechanism of inflammasome activation in the brain. Pathogen-associated molecular patterns (PAMPs) and Damage-associated molecular patterns (DAMPs) activate pattern recognition receptors (PRRs) such as toll like receptors (TLRs) to produce pro-IL-1β in a NF-κB-dependent manner. This process is referred to as priming. Upon activation of the inflammasome by reactive oxygen species (ROS), the inflammasome activates pro-caspase-1 into caspase-1, resulting in the processing of pro-IL-1β into IL-1β. Once active, IL-1β is secreted resulting in a spread of the inflammatory response into neighboring cells. Similarly, extracellular vesicles containing inflammasome proteins get secreted, thus also contributing to the spread of the inflammatory response.
A sub-lethal ischemic episode increased serum levels of IL-1 (α and β) between 1 and 3 days in gerbils. This sub-lethal ischemia protected CA1 neurons from an otherwise lethal ischemia induced 3 days later. Inhibition of IL-1 receptor antagonist abolished ischemic tolerance, suggesting that IL-1 is necessary for induction of IPC in the gerbil (Ohtsuki et al., 1996). In this study, the sex of animals used was not identified, thus whether similar results will be attained in female gerbils is not clear. A recently published study showed significantly higher levels of IL-1β protein levels in reproductively senescent female rats compared to their young or male counterparts (Raval et al., 2018). Such a variation may play an important role in conferring IPC or EPC-induced ischemic protection. Additionally, as mentioned before in this review, ER(s) availability in brain changes with age, and ER-β regulates inflammasome activation in the brain of reproductively senescent female rats (Figure 2). Specifically, silencing of hippocampal ER-β increases inflammasome activation, whereas periodic ER-β agonist pretreatments reduces activation of the inflammasome, consistent with decreased processing of IL-1β. Therefore, these findings advocate for a role for ER-β in the regulation of the innate immune response through the inflammasome. Elevations in inflammasome proteins have been previously reported in the hippocampus of aged rats (Mawhinney et al., 2011) as well as the cortex (Mejias et al., 2018). Consistent with our findings, a study showed increased pro-inflammatory cytokine levels in middle-aged female rats (Sarvari et al., 2014).
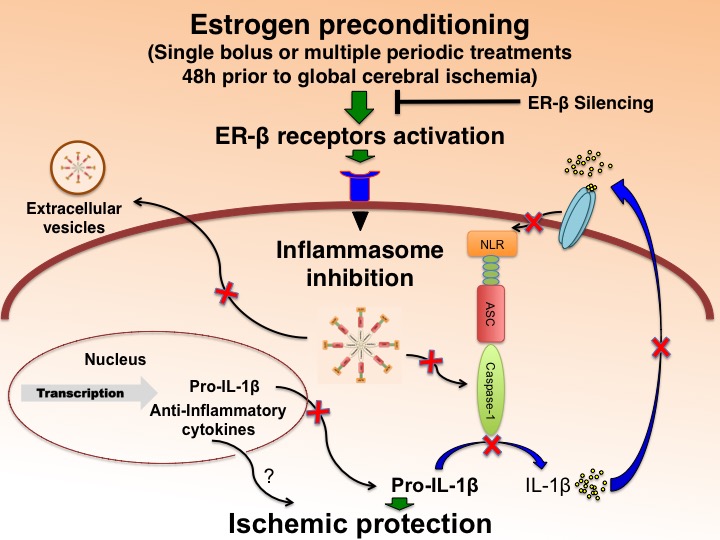
In a new window | Download PPT
Figure 2: E2/ER-β agonist preconditioning regulates inflammasome activation and protects the brain from global ischemic damage in female rats. Silencing of ER-β attenuated 17β-estradiol-mediated decrease in caspase-1, ASC and IL-1β. Silencing of ER-β was achieved by antisense delivery to the hippocampus of female rats as described previously (Raval et al., 2012) On the contrary, long-term ER-β agonist preconditioning significantly decreased inflammasome activation and increased post-ischemic neuronal survival in female rats.
ER-β activation regulates NLR inflammasome in the female brain
NLRs are cytosolic sensors and contain a carboxy-terminal leucine-rich repeat (LRR), a nucleotide binding domain called the NACHT domain, and an N-terminal pyrin domain (PYD). The PYD domain of the inflammasome binds to a PYD domain of an adaptor protein called apoptosis-associated speck-like protein (ASC) (Walsh et al., 2014). The adapter protein, ASC, has a caspase activation and recruitment domain (CARD), and it is via the CARD domain that ASC is bound to the procaspase-1 enzyme.
NLRs are encoded by a family of 22 genes, and more than twenty members of the NLR family of receptors have been identified thus far. There are four subfamilies of NLRs on the basis of their different N-terminal regions: NLRA, which consists of a class II transactivator (CIITA); NLRB, which consists of a neural apoptosis inhibitory protein (NAIP); NLRC, which consists of NOD1, NOD2, NLRC3–NLRC5, and NLRX1; and NLRP, which consists of NLRP1–NLRP14. Using an RNA–Seq transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex, a study showed the expression pattern of all the NLR family proteins in the brain (Kong et al., 2017). Only NOD1 and NLRX1 are highly expressed in all cell types in the brain, including astrocytes, neurons, oligodendrocyte precursor cells, newly formed oligodendrocytes, myelinating oligodendrocytes, microglia, and endothelial cells. NOD2 is highly expressed in microglia and endothelial cells, whereas NLRC3 is only highly expressed in endothelial cells. The most studied NLR protein in the brain is NLRP3, which is abundant in microglia and to a lesser extent in oligodendrocyte precursor cells (OPCs). Other NLR proteins in this family are absent or are expressed at very low levels (Walsh et al., 2014). In the heart, deletion of the innate immune NLRP3 receptor abolishes IPC (Zuurbier et al., 2012). The proteins of the NLR family are linked to the pathophysiology of neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), multiple sclerosis (MS), and psychological diseases (Syed et al., 2018).
The mechanisms of NLRP3 activation supported by most studies include potassium efflux from the cell, the generation of mitochondrial reactive oxygen species (ROS), the translocation of NLRP3 to the mitochondria, the release of mitochondrial DNA or cardiolipin, and the release of cathepsins into the cytosol after lysosomal destabilization (Lamkanfi and Dixit, 2014; Sutterwala et al., 2014; Guo et al., 2015; Vanaja et al., 2015). However, not all these events are induced by all NLRP3 agonists, so the precise mechanism of NLRP3 activation is still being debated (Guo et al., 2015). While the mechanism of NLRP3 activation is obscure, it is apparent that NLRP3 plays a role in ischemic pathology, as NLRP3 knockout animals have significantly reduced infarct size and neurovascular damage after focal cerebral ischemia (Yang et al., 2014). With respect to E2 regulation of NLRP3 inflammasome activation, E2 has been reported in one study to suppress NLRP3 inflammasome gene expression in the cerebral cortex after focal cerebral ischemia (Slowik and Beyer, 2015). Furthermore, a study from the Brann laboratory demonstrates that the NLRP3 inflammasome is robustly activated in microglia and astrocytes in the hippocampal CA1 region after global cerebral ischemia (Thakkar et al., 2016). On the contrary, E2 pretreatment markedly inhibited NLRP3 inflammasome pathway activation, caspase-1, and proinflammatory cytokine production, as well as P2X7 receptor expression after global cerebral ischemia at both the mRNA and protein levels (Thakkar et al., 2016). This study also demonstrated that ER coregulator protein and proline-, glutamic acid-, and leucine-rich protein 1 (PELP1) is essential for the E2-induced suppression of upstream targets of the NLRP3 inflammasome. This study provides insight into the mechanisms of ER conferred anti-inflammatory actions leading to ischemic neuroprotection. In this study, use of continuous E2 treatment for days prior to induction of global cerebral ischemia and the possible effects of ER-β agonist- induced preconditioning on the NLRP3 inflammasome and ischemic outcome remains elusive and needs further investigation.
Another NLR family member, the NLRC4 inflammasome, contributes to brain injury in a rodent model of stroke (Denes et al., 2015). Higher protein levels of the NLRC4 inflammasome have been noted in the hippocampus, serum, and ovaries of reproductively senescent compared to young normally cycling female rats. Similar changes were not observed in the brain of age-matched males (Raval et al., 2018). This study also demonstrated that ovarian release of inflammasome-containing extracellular vesicles (EVs), and these EVs via blood and cerebrospinal fluid reside in the brain of reproductively senescent female rats, which may make the brain more susceptible to subsequent stress/ischemia later in life (Raval et al., 2018). This finding is also confirmed by testing cerebrospinal fluid of peri-menopausal women. Increased expression of inflammasome complex proteins in the cerebrospinal fluid of peri-menopausal women was observed that is suggestive of altered inflammatory status during the peri-menopausal phase. In an adaptive transfer experiment in which isolated extracellular vesicles from the serum of peri-menopausal women were delivered to young rats, heightened inflammasome activation in the brain was observed, indicating that EV in menopausal women are responsible for the increased inflammatory response present in the brain of young female rats (Raval et al., 2018). Therefore, initiating ER-β agonist preconditioning therapy during peri-menopausal phase might be more beneficial in taming heightened inflammasome activation and may help prevent/reduce the incidence/severity of ischemic brain damage in reproductively senescent/post-menopausal female/women.
Conclusion
Women are more susceptible to cerebral ischemia, and we have limited understanding of the underlying mechanisms for increased ischemic severity and outcome. There are women-specific risk factors such as pregnancy, pre-eclampsia, gestational diabetes, oral contraceptives (OC), menopause, and hormone replacement therapy (HRT), which may make women more susceptible to ischemic incidence, prevalence, and mortality (Di Carlo et al., 2003; Romero et al., 2008; Aoki and Uchino, 2011; Girijala et al., 2017). What is more striking is that even among women, there are studies that clearly link more damaging effects when OC/HRT and cigarette smoking are combined, which is a gender-neutral risk factor for ischemia. Simultaneous exposure to smoking-derived nicotine and OC decreases hippocampal ER-β availability (Raval et al., 2012). Furthermore, nicotine alters ER-β-regulated inflammasome activity and exacerbates ischemic brain damage in female rats (d'Adesky et al., 2018), which may compromise the beneficial effects of estrogen/ER-β agonist-induced ischemic tolerance. Therefore, future studies investigating the efficacy of EPC/IPC in females should be designed with caution, and consideration should be given to female-specific risk factors of cerebral ischemia.
Conflicts of interest
Helen M. Bramlett and Juan Pablo de Rivero Vaccari are co-founders and managing members of InflamaCORE, LLC, a company dedicated to developing therapies and diagnostic tools focusing on the inflammasome. Helen M. Bramlett and Juan Pablo de Rivero Vaccari are Scientific Advisory Board Members of Variant Pharmaceuticals, Inc. All other authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by an Endowment from Drs. Chantal and Peritz Scheinberg (APR), Florida Department of Heath#7JK01 funds (HMB & APR), the American Heart Association Grant-in-aid # 16GRNT31300011 (APR), The Miami Project to Cure Paralysis (HMB), and National Institutes of Health/National Institute of Neurological Disorders and Stroke R01 NS34773 and NS45676 (MAPP).
References
Juan Pablo de Rivero Vaccari1
1Department of Neurological Surgery and The Miami Project to Cure Paralysis
Helen M. Bramlett1,2
1Department of Neurological Surgery and The Miami Project to Cure Paralysis
2Bruce W. Carter Department of Veterans Affairs Medical Center, Miami
Miguel A. Perez-Pinzon3
3Peritz Scheinberg Cerebral Vascular Disease Research Laboratories, Department of Neurology, Leonard M. Miller School of Medicine, University of Miami, Miami, Florida 33136, U.S.A.
Ami P. Raval3
3Peritz Scheinberg Cerebral Vascular Disease Research Laboratories, Department of Neurology, Leonard M. Miller School of Medicine, University of Miami, Miami, Florida 33136, U.S.A.
Corresponding Author:
Ami P. Raval
Email: ARaval@med.miami.edu
In a new window | Download PPT
Figure 1: Putative mechanism of inflammasome activation in the brain. Pathogen-associated molecular patterns (PAMPs) and Damage-associated molecular patterns (DAMPs) activate pattern recognition receptors (PRRs) such as toll like receptors (TLRs) to produce pro-IL-1β in a NF-κB-dependent manner. This process is referred to as priming. Upon activation of the inflammasome by reactive oxygen species (ROS), the inflammasome activates pro-caspase-1 into caspase-1, resulting in the processing of pro-IL-1β into IL-1β. Once active, IL-1β is secreted resulting in a spread of the inflammatory response into neighboring cells. Similarly, extracellular vesicles containing inflammasome proteins get secreted, thus also contributing to the spread of the inflammatory response.
In a new window | Download PPT
Figure 2: E2/ER-β agonist preconditioning regulates inflammasome activation and protects the brain from global ischemic damage in female rats. Silencing of ER-β attenuated 17β-estradiol-mediated decrease in caspase-1, ASC and IL-1β. Silencing of ER-β was achieved by antisense delivery to the hippocampus of female rats as described previously (Raval et al., 2012) On the contrary, long-term ER-β agonist preconditioning significantly decreased inflammasome activation and increased post-ischemic neuronal survival in female rats.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14055 | 30 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA