Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Induced pluripotent stem cells for modeling energetic alterations in hypertrophic cardiomyopathy
Time:2019-09-04
Number:14176
Author Affiliations

Conditioning Medicine 2019. 2(4): 142-151.
Abstract
Hypertrophic cardiomyopathy (HCM) is one of the most commonly inherited cardiac disorders that manifests with increased ventricular wall thickening, cardiomyocyte hypertrophy, disarrayed myofibers, and interstitial fibrosis. The major pathophysiological features include diastolic dysfunction, obstruction of the left ventricular outflow tract, and cardiac arrhythmias. Mutations in genes that encode mostly for sarcomeric proteins have been associated with HCM. However, despite the abundant research conducted to decipher the molecular mechanisms underlying the disease, it remains unclear as to how a primary defect in the sarcomere could lead to secondary phenotypes such as cellular hypertrophy. Mounting evidence suggests energy deficiency could be an important contributor of disease pathogenesis as well. Various animal models of HCM have been generated to gain deeper insight into disease pathogenesis, however species variation between animals and humans, as well as the limited availability of human myocardial samples, has encouraged researchers to seek alternative “humanized” models. Using induced pluripotent stem cells (iPSCs), human cardiomyocytes (CMs) have been generated from patients with HCM to investigate disease mechanisms. While these HCM-iPSC models demonstrate most of the phenotypic traits, it is important to ascertain if they recapitulate all pathophysiological features, especially that of energy deficiency. In this review we discuss current established HCM-iPSC models with emphasis on altered energetics.
Keywords: Hypertrophic cardiomyopathy (HCM), induced pluripotent stem cells (iPSCs), cardiomyocytes, disease modeling; energetics, metabolism
Abstract
Hypertrophic cardiomyopathy (HCM) is one of the most commonly inherited cardiac disorders that manifests with increased ventricular wall thickening, cardiomyocyte hypertrophy, disarrayed myofibers, and interstitial fibrosis. The major pathophysiological features include diastolic dysfunction, obstruction of the left ventricular outflow tract, and cardiac arrhythmias. Mutations in genes that encode mostly for sarcomeric proteins have been associated with HCM. However, despite the abundant research conducted to decipher the molecular mechanisms underlying the disease, it remains unclear as to how a primary defect in the sarcomere could lead to secondary phenotypes such as cellular hypertrophy. Mounting evidence suggests energy deficiency could be an important contributor of disease pathogenesis as well. Various animal models of HCM have been generated to gain deeper insight into disease pathogenesis, however species variation between animals and humans, as well as the limited availability of human myocardial samples, has encouraged researchers to seek alternative “humanized” models. Using induced pluripotent stem cells (iPSCs), human cardiomyocytes (CMs) have been generated from patients with HCM to investigate disease mechanisms. While these HCM-iPSC models demonstrate most of the phenotypic traits, it is important to ascertain if they recapitulate all pathophysiological features, especially that of energy deficiency. In this review we discuss current established HCM-iPSC models with emphasis on altered energetics.
Keywords: Hypertrophic cardiomyopathy (HCM), induced pluripotent stem cells (iPSCs), cardiomyocytes, disease modeling; energetics, metabolism
Hypertrophic cardiomyopathy
Being one of the most commonly inherited cardiac disorders, hypertrophic cardiomyopathy (HCM) affects approximately 1 in 500 individuals globally (Semsarian et al., 2015; Marian and Braunwald, 2017). Characterized by thickening of the ventricular wall (≥13 mm) (Gersh et al., 2011; Authors/Task Force et al., 2014), HCM has a similar prognosis across different ethnicities (Sheikh et al., 2016) and is more predominant in males (Olivotto et al., 2005; Kubo et al., 2010), although recent reports describe a higher incidence of HCM-related mortality in women (Kubo et al., 2010; Wang et al., 2014b; Geske et al., 2017). HCM is also the leading cause of sudden cardiac death (SCD) in adolescents, young adults, and athletes (Maron et al., 2009; Maron et al., 2016; Malhotra and Sharma, 2017). Due to the abnormal thickening of the ventricular wall and increased interstitial fibrosis, the ventricular cavity is reduced thereby impairing diastolic filling, which results in diastolic dysfunction, one of the major pathophysiological features of HCM (Shah, 2003; Finocchiaro et al., 2014). On the histological level, in addition to interstitial fibrosis, cardiomyocytes are disorganized and exhibit increased cell size (hypertrophy) with disarrayed myofibers (Hughes, 2004). The most common cause of death, however in patients with HCM, is SCD mediated primarily by ventricular tachycardia and fibrillation, which has been attributed to changes in cardiomyocyte size and organization, disruption of intercalated discs (Sepp et al., 1996), abnormal calcium handling, and increased myofilament calcium sensitivity (Baudenbacher et al., 2008). When considering patient management strategies, because most patients demonstrate minimalistic symptoms, they are not routinely given pharmacological agents such as β-blockers and L-type calcium channel blockers. For those at high risk of SCD, intervention with an implanted cardioverter defibrillator (ICD) has shown to be effective (Maron et al., 2000). Currently, there are no specific treatments available that could prevent or arrest the development of HCM. For a more comprehensive review on patient management strategies we direct the reader to the excellent review by Spirito and Autore (Spirito and Autore, 2006).
Molecular basis of hypertrophic cardiomyopathy
Over the past few decades, molecular genetics has associated over 1400 mutations, mostly in genes encoding for sarcomeric proteins, as causal factors of HCM (Maron et al., 2012; Sabater-Molina et al., 2018), which therefore demonstrates an autosomal dominant pattern of inheritance with approximately 60% of patients having a clear familial predisposition. The two most common genes that have been associated with causality is MYH7 and MYBPC3, which together account for more than 50% of patients with familial HCM (Sabater-Molina et al., 2018). These genes encode for β-myosin and cardiac myosin binding protein-C, respectively. Although uncommon, other genes encoding for sarcomeric proteins (TNNT2, TNNI3, TPM1, ACTC1, MYL2 and MYL3), have also been linked to HCM (Marian and Roberts, 2001; Marsiglia and Pereira, 2014). There are several theories as to how mutations in sarcomeric proteins could lead to disease manifestation. The first is the poison peptide theory by which missense mutations alter the conformational structure of the protein, and when subsequently incorporated into the myofibrils, these mutated proteins exert a dominant-negative effect by impeding contractile performance (Vikstrom et al., 1996; Rust et al., 1999; Fatkin et al., 2000; Frey et al., 2000). The second manner in which disease manifestation is thought to occur is through haploinsufficiency, whereby transcripts containing insertions or deletions that result in a frame-shift and consequent truncated protein, undergo nonsense-mediated decay, leading to an overall loss of total protein (Marston et al., 2009; van Dijk et al., 2009; Barefield et al., 2014). Finally, mutations in these sarcomeric proteins could have direct implications on myofibrillar mechanics, resulting in abnormal calcium sensitivity or force generation, thereby compromising heart function (Westfall et al., 2002; Tardiff, 2005; Fraysse et al., 2012)
Despite a single mutation being sufficient to cause HCM, the penetrance is variable and the phenotype that presents itself is not only a result of the causal gene, but also a result of the compounding effects mediated by other genetic influences such as modifier genes, as well as environmental factors, which makes genotype-based prognosis a considerable challenge. Furthermore, the mechanisms by which a sarcomeric mutation leads to cardiomyocyte hypertrophy and additional pathophysiological features remains unclear. To gain deeper understanding into disease pathogenesis and progression, a number of transgenic, knock-in, and knock-out animal models have been developed over the past few decades (Marian and Roberts, 2001). While these animal models demonstrate most of the phenotypic traits of HCM, there are concerns as to whether they truly recapitulate disease pathogenesis in humans. These concerns are based on the findings that a number of animal models have mutations that are known to be causal for HCM in humans, despite developing pathophysiological features, such as diastolic dysfunction, interstitial fibrosis and cardiomyocyte disorganization, fail to show significant hypertrophy, which is the key phenotypic trait of the disease (Geisterfer-Lowrance et al., 1996; Oberst et al., 1998; Yang et al., 1998; Muthuchamy et al., 1999; Miller et al., 2001). Furthermore, the major myosin isoforms are different between human and mouse, with β-myosin being predominant in the former and α-myosin (encoded by the MYH6 gene) in the latter. Hence, in order to study causal mutations in β-myosin, these mutations would need to be introduced into a MYH6 backbone in mice and though this is feasible and does lead to the development of pathophysiological features, the contractile kinetics are significantly different between the two isoforms (Malmqvist et al., 2004; Deacon et al., 2012; Lowey et al., 2013). While this could be overcome by studying animal species that have a similar myosin composition to that of humans (Marian et al., 1999; Lowey et al., 2018), they would lack the genetic background, which could determine disease severity. Hence, while these animal models help lay the foundation for identifying various mechanisms involving abnormal contractile kinetics and electrophysiological properties in HCM, caution is advised when extrapolating these findings to a human setting.
Induced pluripotent stem cells for modeling hypertrophic cardiomyopathy
Ideally, the most suitable model for studying disease manifestation in HCM would be human cardiac tissue. However, the invasiveness required to obtain primary cardiomyocytes, together with the inability to culture them for prolonged periods of time, renders it a challenge. With the discovery of induced pluripotent stem cells (iPSCs) over a decade ago (( Takahashi et al., 2007 )), researchers are able to overcome the challenges associated with primary cardiac tissue and instead are able to generate human cardiomyocytes when required, using a number of established cardiac differentiation protocols (Lian et al., 2012; Burridge et al., 2014; Mehta et al., 2014a). Using iPSC technology, cardiomyocytes have been generated from patients with channelopathies (Moretti et al., 2010; Mehta et al., 2014b), cardiomyopathies (Sun et al., 2012; Lan et al., 2013; Viswanathan et al., 2018), and other cardiac disorders (Huang et al., 2011; Kim et al., 2013; Wang et al., 2014a). These iPSC-CMs not only serve as a platform for modeling cardiac disorders but could also be used for drug screening applications with the aim of identifying novel therapeutic strategies (Mehta et al., 2018; Schwartz et al., 2019) (Figure 1). iPSCs generated from patients with HCM have allowed researchers to model this complex disorder in a dish and as they consist of the patient’s genetic background, causal mutations acting in concert with modifier genes could be studied effectively with the aim of further understanding variable penetrance and disease severity.
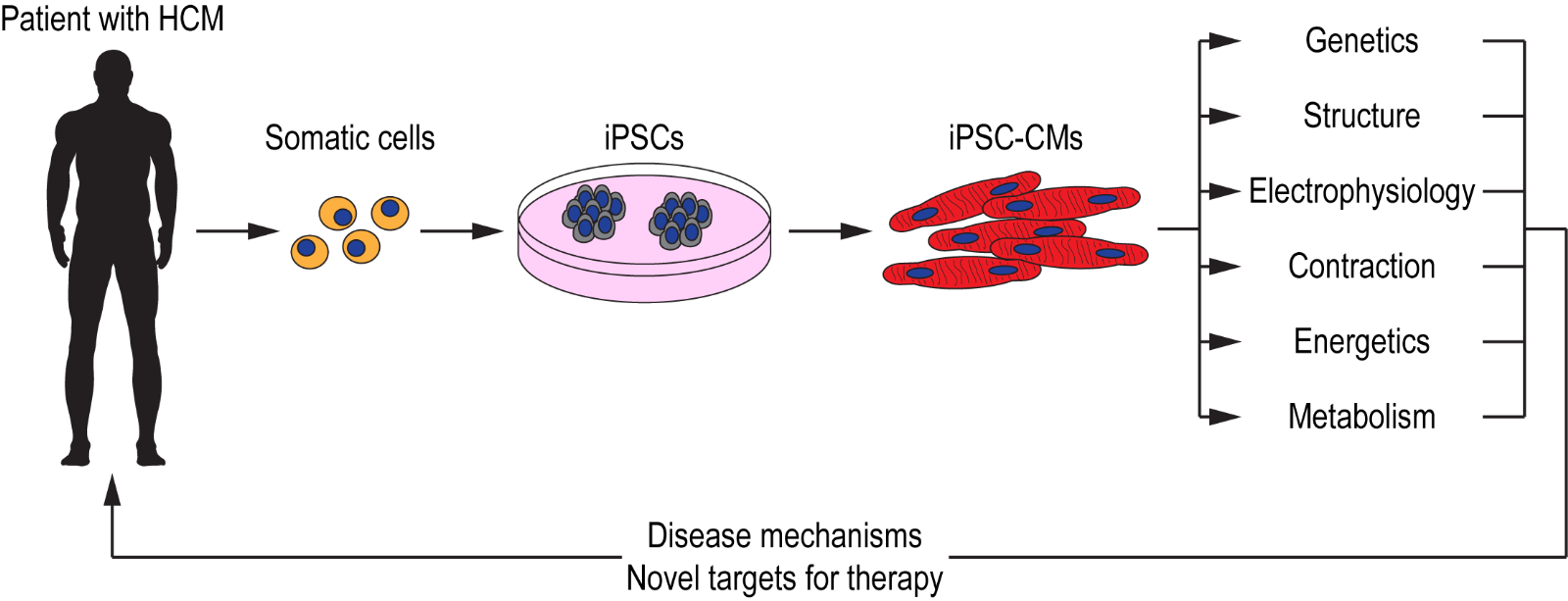
In a new window | Download PPT
Figure 1: Schematic illustration of iPSC technology for deciphering disease mechanisms and identifying novel therapeutic targets for HCM. Somatic cells (e.g. blood, dermal fibroblasts) obtained from patients with HCM are reprogrammed to generate iPSCs. These iPSCs are then differentiatied into functional cardiomyocytes (iPSC-CMs) that recapitulate the patient’s cardiac phenotype. These iPSC-CMs could serve as a platform for investigating various cellular, molecular, and functional properties with the aim of deciphering disease mechanisms, as well as for identifying novel targets for patient-specific therapies.
Altered calcium homeostasis and impaired myofilament function
The first iPSC study investigating HCM assessed the effect of a hereditary Arg663His mutation in the β-myosin protein (Lan et al., 2013). The diseased iPSC-CMs showed hallmark phenotypic traits of HCM, in that they were larger in size and demonstrated contractile arrhythmia. Importantly, the study demonstrated that dysregulated calcium cycling and elevated intracellular calcium played a key role in disease pathogenesis. Similarly, iPSC-CMs generated from a patient bearing a R58Q mutation in the myosin light chain-2 also demonstrated impaired intracellular calcium homeostasis, delayed decay time, reduced calcium current densities and arrhythmia (Zhou et al., 2019). These studies suggest abnormal calcium handling is a key phenotypic trait of HCM, but it remains to be determined if altered calcium homeostasis is a result of the causal sarcomeric mutation or due to a compound effect mediated by other genetic and environmental factors. This could be addressed by using gene editing technologies, whereby a single causal mutation is introduced into iPSCs with a known genetic background. This is very much similar to the generation of animal models of HCM but in a humanized setting. The advantage of this technique is that researchers are able to decipher the genotype-phenotype relationship more accurately, as it eliminates compounding factors such as modifier genes and environmental influences. Using CRISPR/Cas9 technology, an I79N mutation was introduced into cardiac troponin T, and the iPSC-CMs harboring this mutation when compared to their isogenic controls exhibited disorganized sarcomeres, enhanced contractility, and impaired relaxation (Wang et al., 2018). The impeded contractile performance was attributed to an increased sensitivity of the myofilaments towards calcium. In contrast, iPSC-CMs harboring mutations introduced into β-myosin (R403Q and V606M) and cardiac myosin binding protein-C (Trp792ValfsX41 and R502W) exhibited hypercontractility but with no changes in calcium handling (Cohn et al., 2019). More recently, iPSCs were generated from patients having an E848G variant in β-myosin. Although, the diseased iPSC-CMs demonstrated decreased contractile function, which was attributed to reduced interactions between β-myosin and cardiac myosin binding protein-C, calcium handling abnormalities were also absent in this model (Yang et al., 2018). Studies have reported altered calcium homeostasis to be the down-stream effect of the sarcomeric mutation found in HCM, due to a ‘calcium trapping’ phenomenon of the sarcomeres (Semsarian et al., 2002; Ashrafian et al., 2011). Therefore, these contrasting findings in iPSC-CMs are of considerable importance as it would seem that altered calcium homeostasis may not be a universal phenomenon and could perhaps be mutation-dependent or even be a result of compounding factors acting in concert with the causal gene. This hypothesis is supported by the study by Ojala and colleagues (Ojala et al., 2016) who generated iPSC-CMs from patients bearing Finnish founder mutations in either cardiac myosin binding protein-C (Gln1061X) or α-tropomyosin (Asp175Asn). In this study, although both diseased iPSC-CM lines exhibited hypertrophic features when compared to controls, differences in cell size, calcium handling, electrophysiological properties, and gene expression profiles were observed between the two diseased lines as well.
Environmental factors and key signaling cascades
Apart from causal and modifier genes, environmental factors such as life-style and diet (Stauffer et al., 2006) are also known to influence disease pathogenesis. In an attempt to decipher interactions between a patient’s genetic background and environmental factors, Tanaka and colleagues generated iPSCs from two patients bearing no mutations in the major sarcomeric genes known to be associated with HCM and from a single patient bearing a Gly999-Gln1004del mutation in cardiac myosin binding protein-C (Tanaka et al., 2014). Interestingly, the diseased iPSC-CMs demonstrated a mild phenotype under basal conditions, but upon treatment with an exogenous hypertrophy promoting stimulant such as endothelin-1, morphological and high-speed video assessment revealed increased cardiomyocyte hypertrophy, myofibrillar disarray, and variability in the direction of contraction. These findings suggest that under certain circumstances the hypertrophic phenotype could remain masked until exposed by a particular trigger. Similarly, when Prajapati and colleagues treated two diseased iPSC-CM lines harboring mutations in either cardiac myosin binding protein-C or α-tropomyosin with adrenaline (Prajapati et al., 2018), while arrhythmias were observed in a dose-dependent manner, the two iPSC-CM lines exhibited differential arrhythmia patterns, with the line harboring the cardiac myosin binding protein-C mutation exhibiting more delayed afterdepolarizations, while the line harboring the α-tropomyosin mutation was more prone to ventricular tachycardia. This study is particularly relevant to athletes who exhibit HCM, as unfortunately in 80% of these cases, SCD is the first cardiac event that occurs during or immediately after intense exercise (Pelliccia et al., 1991; Maron et al., 2009).
With technological advancement of various ‘omics’ platforms, studies that investigate changes in gene regulatory networks could prove to be beneficial in identifying critical signaling cascades involved in HCM pathogenesis. MicroRNA transcriptome profiling (Kuster et al., 2013) and RNA-seq profiling (Ren et al., 2016) performed on cardiac tissue of patients with HCM revealed remarkable differences in gene signatures when compared to healthy donors. However, it must be noted that cardiac tissue is comprised of a mixture of myocytes and non-myocytes and hence, identifying gene regulatory changes purely associated with sarcomeric mutations in cardiomyocytes is challenging. This is another avenue where iPSC-CMs could prove to be advantageous, as a relatively homogeneous population could be generated with current protocols. In lieu of this, Han and colleagues (Han et al., 2014) performed whole transcriptome sequencing followed by pathway enrichment analysis in a model bearing an Arg442Gly mutation in β-myosin. When compared against controls, the diseased iPSC-CMs showed an increase in genes responsible for cell proliferation which was mainly governed by WNT1. Genes involved in key development pathways such as Notch and fibroblast growth factor were also increased, suggesting cross-talk between multiple signaling pathways in the development of HCM. In a more recent study (Cohn et al., 2019), β-myosin and cardiac myosin binding protein-C mutations that resulted in hypercontractility were also shown to induce p53-mediated oxidative stress, which resulted in reduced cardiomyocyte viability under conditions of metabolic stress. Such studies could pave the way for gaining insight into the molecular mechanisms of the disease with regard to changes in global gene signatures.
Energetic alterations in hypertrophic cardiomyopathy
Mitochondria are the powerhouse of the cell and with the heart being the most energy consuming organ in the human body, it is not surprising that these organelles occupy about a third of cardiomyocyte volume (Piquereau et al., 2013). The ATP generated from the mitochondria is utilized for essential cellular functions including cardiomyocyte growth, contraction, ionic homeostasis, and survival. Mitochondria themselves are dynamic organelles able to change their shape and distribution via fusion and fission-mediated processes (Ong and Hausenloy, 2010). Such dynamisms are critical for normal mitochondria function, as imbalances in the fusion, fission, and mitophagy pathways could lead to the onset of various cardiomyopathies (Dorn, 2016). In order to sustain the high energy requirements associated with cardiac function, adult cardiomyocytes rely heavily on oxidative phosphorylation (OXPHOS), which is mainly fueled by fatty acid β-oxidation and to a lesser extent glucose oxidation and glycolysis (Lopaschuk and Jaswal, 2010), the latter being the major energy derivative pathway in fetal cardiomyocytes due to reduced OXPHOS and poorly developed mitochondrial networks (Porter et al., 2011). Multiple studies have reported decreased fatty acid oxidation with concurrent decline in mitochondrial energetics during heart failure (Pereira et al., 2014; Fillmore et al., 2018). Interestingly, in rats that underwent thoracic aortic constriction, mitochondrial respiratory capacity remained relatively intact during the initial stages of compensated hypertrophy with diastolic dysfunction and preserved ejection fraction. Mitochondrial dysfunction was only observed during the final stages of heart failure with systolic dysfunction and reduced ejection fraction (Doenst et al., 2010). This would suggest that despite the increase in cardiac workload, the mitochondria are not yet energy compromised. However, the initial stages of compensated hypertrophy are accompanied by a drastic decline in fatty acid oxidation, and hence the mechanisms by which respiratory capacity is preserved remain unclear.
While the aforementioned iPSC studies corroborate the feasibility to model most phenotypic traits of HCM, including cardiomyocyte hypertrophy, myofiber disarray, impaired contractility, altered myofilament calcium sensitivity, abnormal calcium handling, and arrhythmias, studies aimed at investigating energy alterations in HCM-iPSC models are still in their infancy. It is hypothesized that the sarcomeric mutation in HCM that results in increased myofilament calcium sensitivity, due to a calcium trapping phenomenon, would promote increased ATP consumption at the sarcomeres (Semsarian et al., 2002; Ashrafian et al., 2011) (Figure 2). This could have direct implications on energy homeostasis, as mitochondria energy demands would increase. The shunting of ATP molecules towards sarcomere function and away from other cellular processes, such as ion channel regulation, could lead to adverse cellular complications. Approximately 30-40% of ATP generated in the heart is used for regulating various ion pumps including SERCA (Doenst et al., 2013), and failure of SERCA to efficiently re-uptake calcium into the sarcoplasmic reticulum has been postulated to be the main reason for diastolic dysfunction and consequent incidences of arrhythmias (Miyamoto et al., 2000; Periasamy and Janssen, 2008; Yang et al., 2014a). Furthermore, mitochondria energetics are tightly regulated via the mitochondrial calcium uniporter (MCU), which facilitates the intake of calcium for the normal function of key enzymes of the Krebs cycle and for proteins of the electron transport chain (Williams et al., 2015). Calcium within the mitochondria also regulates the NADH/NAD+ redox state, which protects against reactive oxygen species (ROS) accumulation. Hence, it could be speculated that due to increased calcium sensitivity, the accumulation of calcium at the sarcomeres could also cause a calcium deficit within the mitochondria, thereby inducing an energetic collapse and increased ROS production over time. Recently, Mosqueira and colleagues attempted to validate the energy deficiency theory in HCM using PSCs-CMs harboring a R453C mutation in β-myosin (Mosqueira et al., 2018). Interestingly, though the diseased PSC-CMs exhibited a reduction in contractile force with negative clinotropic effects, an augmented mitochondrial oxygen consumption rate resulting in increased ATP production was observed in comparison to isogenic controls. Moreover, the similar levels in ROS between control and diseased PSC-CMs would suggest that the mitochondria are not overburdened, but in a state of homeostasis, which is paradoxical to the energy deficiency theory. The importance for studying energetic alterations also stems from findings that energy deficiency may be one of the earliest features of disease pathogenesis, which in turn leads to secondary clinical phenotypes such as diastolic dysfunction, heart failure, and SCD. This hypothesis is supported by the findings of low PCr (phospho-creatine)/ATP ratios in patients with HCM as well as in individuals who harbour causal genes but who had yet to develop hypertrophy (Crilley et al., 2003; Abraham et al., 2013).
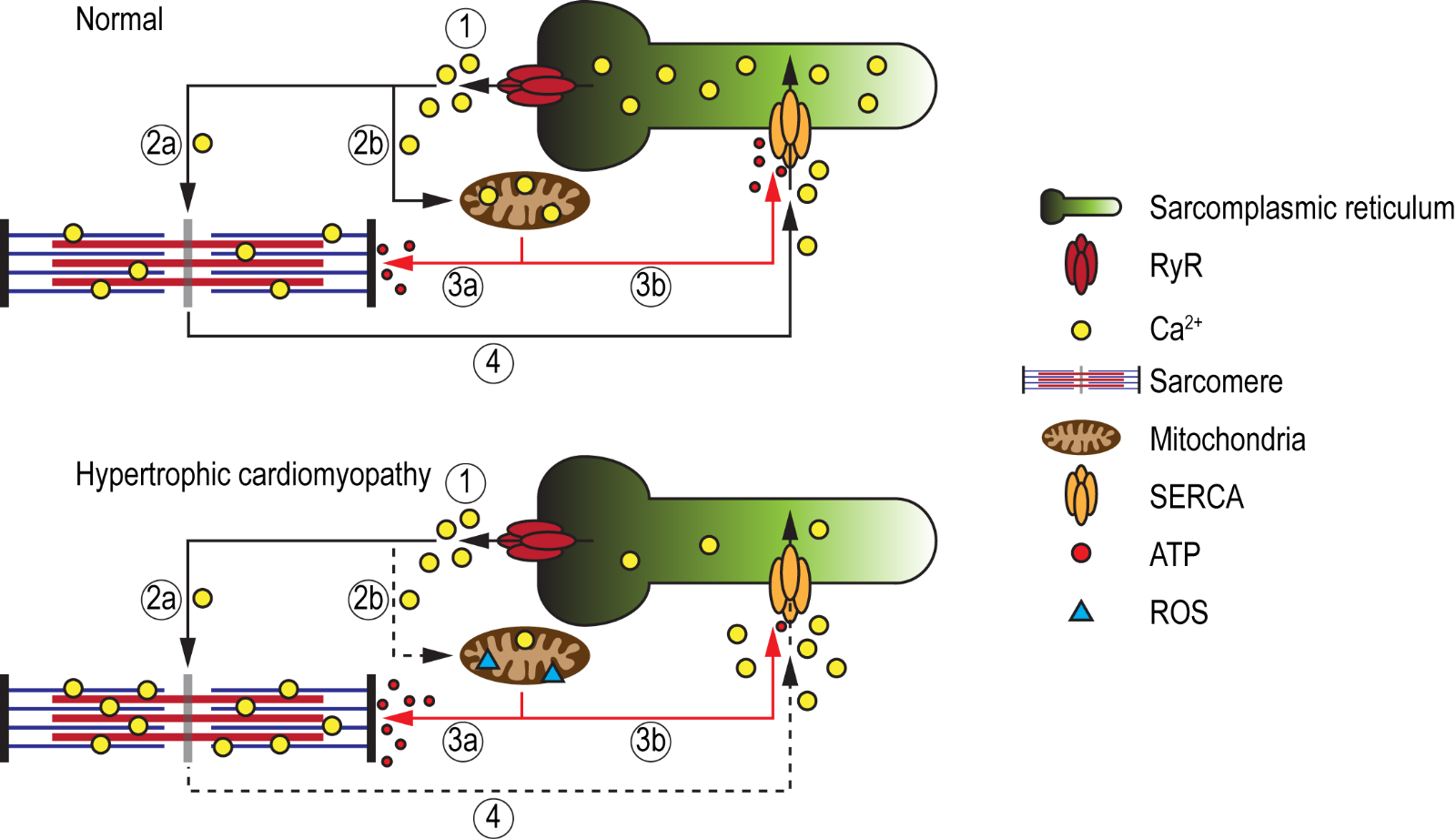
In a new window | Download PPT
Figure 2: Schematic illustration of the energy deficiency theory in HCM. Under normal conditions, calcium ions are released from the sarcoplasmic reticulum (SR), via ryanodine receptors (RyR) (1), where they bind to the myofibrils (2a) as well as enter the mitochondria (2b) to maintain energy production and redox state. The mitochondria in turn produce sufficient amounts of ATP that will be utilized by the sarcomeres for relaxation (3a) via cross-bridge detachment, as well as for the regulation of SERCA (3b), which mediates calcium re-uptake into the SR (4), thereby maintaining calcium homeostasis. In HCM, due to increased calcium sensitivity, calcium ions released from the SR are trapped within the myofibrils (2a), resulting in a calcium deficit within the mitochondria (2b). This would eventually lead to an energetic collapse and an increase in ROS production. To provide sufficient energy for relaxation (i.e. cross-bridge detachment), higher ATP consumption at the sarcomeres (3a) will shunt ATP away from SERCA (3b), impairing its ability to re-uptake calcium into the SR (4). The resultant increase in cytosolic calcium could lead to diastolic dysfunction and cardiac arrhythmia.
Another reason for modeling energetic alteration in HCM is that patients with inherited syndromes, where energy production is impaired (e.g. mitochondria disorders), develop cardiac hypertrophy (Zeviani et al., 1995; Bates et al., 2012). iPSC-CMs generated from patients harboring mutations in the SCO2 gene that leads to COX deficiency (Papadopoulou et al., 1999), exhibit ultrastructural abnormalities and abnormal calcium handling due to reduced SERCA activity, which was attributed to a shortage of ATP (Hallas et al., 2018). Similarly, iPSC-CMs generated from patients with HCM bearing the mitochondrial mutation 2336T>C in the MT-RNR2 gene exhibited defects in mitochondria ultrastructure, reduction in ATP/ADP ratio, and diminished mitochondrial membrane potential, which resulted in altered calcium homeostasis and abnormal electrophysiological properties (Li et al., 2018). With regard to metabolism, despite glucose being the more energy efficient substrate, the healthy human heart generates approximately 70-90% of its ATP by oxidation of fatty acids (Doenst et al., 2013). When ATP consumption increases due to increase in workload (which is thought to occur in HCM), a metabolic shift from fatty acid to glucose oxidation is thought to take place as a compensatory mechanism to provide more energy to the already energy deficient failing heart (Lionetti et al., 2011). This condition is exacerbated under hypoxic conditions and increased work load as a large part of glucose is then converted to the less energy efficient lactate (Neglia et al., 2007). Similar to mitochondria-related disorders, deficient fatty acid oxidation and uptake could also result in an HCM-like phenotype (Bautista et al., 1990; Aoyama et al., 1995). For these reasons, modeling energy alterations in HCM-iPSC models is of paramount importance to understanding mechanisms involved in early disease pathogenesis.
Challenges in modeling energetic alterations
To model energetic alterations, there are two cellular and biochemical components that need to be considered: the state of the mitochondria and its metabolic substrate selectivity. It is widely recognized that iPSC-CMs exhibit a fetal-like phenotype (Tan and Ye, 2018) and hence, (i) do not possess sufficient numbers of mitochondria, (ii) the mitochondrial ultrastructure is poorly developed, (iii) the mitochondria are mostly round in shape, and (iv) are localized mainly in the perinuclear region. This is in stark contrast to their adult counterparts which contain densely packed, highly developed, elongated mitochondrial networks that show inter-myofibril, peri-nuclear, and sub-sarcolemma distribution patterns. Furthermore, being fetal-like in nature, these iPSC-CMs consist of a glucose-based metabolism as opposed to fatty acid oxidation. Taking this into consideration, it could be challenging to model energetic alterations in HCM-iPSC models, as energy deficiency could either be due to disease pathogenesis or an immature cellular phenotype.
Having said this, researchers are constantly developing protocols which could promote the maturity of iPSC-CMs. Biochemical approaches such as the treatment with small molecules (Yang et al., 2014b) and environmental manipulation, such as the incorporation of electrical/mechanical stimulation (Ruan et al., 2016; Ulmer et al., 2018), growth on various matrices (Parikh et al., 2017), or even the substitution of metabolic substrates with fatty acids (Correia et al., 2017; Ramachandra et al., 2018) in culture media has shown to significantly enhance structural, electrophysiological, and bioenergetic properties in iPSC-CMs. More recently, by incorporating iPSC-CMs into a 3D structure such as engineered heart tissue (EHT) (Breckwoldt et al., 2017; Tiburcy et al., 2017) or a cardiac organoid (Mills et al., 2017), a model more relevant to that of adult physiology could be generated. Exposure to an adult-like metabolic environment by supplementing fatty acids into culture media may aggravate the disease phenotype of HCM-iPSC models. Besides the induction of maturation, substitution of metabolic substrates to fatty acids could help unravel the mechanisms that preserve mitochondrial respiratory capacity during the early onset of cardiac hypertrophy, where workload is increased, despite the decline in fatty acid oxidation (Doenst et al., 2010). This in turn could lead to the identification of novel targets that could help prevent mitochondrial dysfunction during the later stages of disease progression.
Concluding remarks
Being one of the most commonly inherited cardiac disorders and the leading cause of SCD in adolescents, young adults, and athletes, considerable efforts should be made to decipher the underlying mechanisms of HCM, especially those which contribute to early pathogenesis such as energetic alterations. Current pharmacological agents used to treat symptoms are generic and are not tailored for patient specificity, hence gaining deeper mechanistic insight into energy deficiency by using HCM-iPSC models could lay the foundation for identifying new targets for stratified therapies. Having said this, further research on iPSC-CM maturation is required until a common consensus is agreed upon that they are mature enough to model various structural, electrophysiological, energetic, and metabolic features of adult cardiomyocytes. Despite this, as the most relevant humanized model that closely resembles human cardiac physiology, iPSC-CMs will continue to play a defining role in identifying novel molecular mechanisms and drug targets for HCM in the near future.
Acknowledgments
Chrishan Ramachandra was supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (OF-YIRG) – [NMRC/OFYIRG/0073/2018] and through the National Health Innovation Centre Singapore under its Innovation to Develop Grant (NHIC-I2S-1811007). William A. Boisvert was supported by National Institutes of Health grant HL081863. Derek Hausenloy was supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
References
Chrishan J.A. Ramachandra1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore; 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Myu Mai Ja KP1
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
Ying-Hsi Lin1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore; 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Winston Shim3
3Health and Social Sciences Cluster, Singapore Institute of Technology, Singapore.
William A. Boisvert4
4Center for Cardiovascular Research, John A. Burns School of Medicine, University of Hawaii, USA.
Derek J. Hausenloy1,2,5,6,7,8
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore; 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore; 5Yong Loo Lin School of Medicine, National University Singapore, Singapore; 6The Hatter Cardiovascular Institute, University College London, London, UK; 7The National Institute of Health Research University College London Hospitals Biomedical Research Centre, Research & Development, London, UK; 8Tecnologico de Monterrey, Centro de Biotecnologia-FEMSA, Nuevo Leon, Mexico.
Corresponding author:
Chrishan J.A. Ramachandra
Email: chrishan.ramachandra@nhcs.com
In a new window | Download PPT
Figure 1: Schematic illustration of iPSC technology for deciphering disease mechanisms and identifying novel therapeutic targets for HCM. Somatic cells (e.g. blood, dermal fibroblasts) obtained from patients with HCM are reprogrammed to generate iPSCs. These iPSCs are then differentiatied into functional cardiomyocytes (iPSC-CMs) that recapitulate the patient’s cardiac phenotype. These iPSC-CMs could serve as a platform for investigating various cellular, molecular, and functional properties with the aim of deciphering disease mechanisms, as well as for identifying novel targets for patient-specific therapies.
In a new window | Download PPT
Figure 2: Schematic illustration of the energy deficiency theory in HCM. Under normal conditions, calcium ions are released from the sarcoplasmic reticulum (SR), via ryanodine receptors (RyR) (1), where they bind to the myofibrils (2a) as well as enter the mitochondria (2b) to maintain energy production and redox state. The mitochondria in turn produce sufficient amounts of ATP that will be utilized by the sarcomeres for relaxation (3a) via cross-bridge detachment, as well as for the regulation of SERCA (3b), which mediates calcium re-uptake into the SR (4), thereby maintaining calcium homeostasis. In HCM, due to increased calcium sensitivity, calcium ions released from the SR are trapped within the myofibrils (2a), resulting in a calcium deficit within the mitochondria (2b). This would eventually lead to an energetic collapse and an increase in ROS production. To provide sufficient energy for relaxation (i.e. cross-bridge detachment), higher ATP consumption at the sarcomeres (3a) will shunt ATP away from SERCA (3b), impairing its ability to re-uptake calcium into the SR (4). The resultant increase in cytosolic calcium could lead to diastolic dysfunction and cardiac arrhythmia.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14176 | 30 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA