Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
New insights provided by myofibril mechanics in inherited cardiomyopathies
Time:2019-11-02
Number:13154
Author Affiliations

Conditioning Medicine 2019. 2(5):213-224.
Abstract
Cardiomyopathies represent a heterogeneous group of cardiac disorders that perturb cardiac contraction and/or relaxation, and can result in arrhythmias, heart failure, and sudden cardiac death. Based on morphological and functional differences, cardiomyopathies have been classified into hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and restrictive cardiomyopathy (RCM). It has been well documented that mutations in genes encoding sarcomeric proteins are associated with the onset of inherited cardiomyopathies. However, correlating patient genotype to the clinical phenotype has been challenging because of the complex genetic backgrounds, environmental influences, and lifestyles of individuals. Thus, “scaling down” the focus to the basic contractile unit of heart muscle using isolated single myofibril function techniques is of great importance and may be used to understand the molecular basis of disease-causing sarcomeric mutations. Single myofibril bundles harvested from diseased human or experimental animal hearts, as well as cultured adult cardiomyocytes or human cardiomyocytes derived from induced pluripotent stem cells, can be used, thereby providing an ideal multi-level, cross-species platform to dissect sarcomeric function in cardiomyopathies. Here, we will review the myofibril function technique, and discuss alterations in myofibril mechanics, which are known to occur in sarcomeric genetic mutations linked to inherited HCM, DCM, and RCM, and describe the therapeutic potential for future target identification.
Keywords: Hypertrophic cardiomyopathy; dilated cardiomyopathy; restrictive cardiomyopathy; myofibrils mechanics; sarcomere; heart failure
Abstract
Cardiomyopathies represent a heterogeneous group of cardiac disorders that perturb cardiac contraction and/or relaxation, and can result in arrhythmias, heart failure, and sudden cardiac death. Based on morphological and functional differences, cardiomyopathies have been classified into hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and restrictive cardiomyopathy (RCM). It has been well documented that mutations in genes encoding sarcomeric proteins are associated with the onset of inherited cardiomyopathies. However, correlating patient genotype to the clinical phenotype has been challenging because of the complex genetic backgrounds, environmental influences, and lifestyles of individuals. Thus, “scaling down” the focus to the basic contractile unit of heart muscle using isolated single myofibril function techniques is of great importance and may be used to understand the molecular basis of disease-causing sarcomeric mutations. Single myofibril bundles harvested from diseased human or experimental animal hearts, as well as cultured adult cardiomyocytes or human cardiomyocytes derived from induced pluripotent stem cells, can be used, thereby providing an ideal multi-level, cross-species platform to dissect sarcomeric function in cardiomyopathies. Here, we will review the myofibril function technique, and discuss alterations in myofibril mechanics, which are known to occur in sarcomeric genetic mutations linked to inherited HCM, DCM, and RCM, and describe the therapeutic potential for future target identification.
Keywords: Hypertrophic cardiomyopathy; dilated cardiomyopathy; restrictive cardiomyopathy; myofibrils mechanics; sarcomere; heart failure
Overview
Heart failure (HF) is one of the leading causes of death and disability worldwide (Mozaffarian et al., 2016), with over 37.7 million individuals suffering from HF globally, and 50% mortality at 5 years (Levy et al. 2002; Bleumink et al. 2004; Vos et al., 2012; Ziaeian et al., 2016; Shah et al. 2017). The estimated cost for HF reached $108 billion in 2012 globally (Cook et al, 2014), and is projected to double by 2030 (Heidenreich et al., 2013). In most cases, the cardiomyopathies responsible for causing HF are chronic and progressive conditions with diverse etiologies. They have been classified as follows (Elliott et al., 2008): (1) hypertrophic cardiomyopathy (HCM), characterized by unexplained left ventricular (LV) hypertrophy, diastolic dysfunction, and normal or increased ejection fraction; (2) dilated cardiomyopathy (DCM), characterized by LV dilatation and reduced LV systolic function; (3) restrictive cardiomyopathy (RCM), characterized by impaired LV filling due to increased myocardial stiffness, and diastolic dysfunction; (4) arrhythmogenic right ventricular dysplasia (ARVD), characterized by progressive replacement of the right ventricle with fibrofatty infiltration and scar tissue; and (5) those which are unclassifiable. In this article we will focus on those inherited cardiomyopathies, HCM, DCM, and RCM, which primarily affect left ventricular function.
Cardiomyopathies can be inherited (familial) or acquired (non-familial). The first familial β-myosin heavy chain mutation (R403Q) associated with HCM was described in the early 1990s by the Seidman group (Geisterfer-Lowrance et al., 1990), and since then more than 1000 sarcomeric gene mutations have been identified to be pathologic variants of inherited cardiomyopathies (Yotti et al., 2019). However, despite nearly three decades of research, it has been challenging to correlate patient genotype with clinical phenotype and guide management of the cardiomyopathy. This can be explained by the complexity of the pathophysiology of inherited cardiomyopathies, which for the most part, is incompletely understood. The majority of carriers of disease-causing sarcomeric mutations do not have symptoms (genotype+/ phenotype-) in their younger ages, and the abnormal sarcomeric function results in decades of cardiac remodeling before clinical manifestations later in age (genotype+/ phenotype+) (Deranek et al., 2019). The phenotype can be affected by genetic variation, ethnic background, environmental/cultural exposure, and even the lifestyle of each individual (Garfinkel et al., 2018). Therefore, in order to better identify and prevent the onset and progression of cardiomyopathies, it is necessary to elucidate the molecular basis of these sarcomeric pathologic variants. In this review article, we discuss how research focused on investigating the basic contractile units of the heart muscle, the myofibrils, can shed light on the pathophysiology of inherited cardiomyopathies, and therefore aid the discovery of novel therapeutic targets for these conditions.
Sarcomere structure
The sarcomere is the basic functional unit of the cardiomyocyte myofibril. The word “sarcomere” is composed of two Greek words “sarc (flesh)” and “meros (part),” meaning “the basic unit of meat (muscle).” The earliest observation of the sarcomere came from the striations observed under light microscopy in which there is a repeating structure limited by two dark Z-lines, between which are the I-band, A-band, and H-zone (Hanson et al., 1954; Eisenberg et al., 1983). The distances between the two Z-lines along with the width of the I-Band and H-zone oscillate throughout the cardiac cycle of muscle contraction and relaxation. It was later shown by electronic microscopy that the A-band corresponds to the full length of thick filaments, and the I-band corresponds to the thin filaments interdigitating with the thick filaments. In 1957, Sir Andrew F. Huxley established the sliding filament model, noting that muscle contraction and relaxation are achieved by alternating sliding action between the thin and thick filaments toward or away from the center of the sarcomere (Huxley et al., 1957) (Fig. 1A).

In a new window | Download PPT
Figure 1: Sliding filament model of muscle contraction and the cross-bridge cycle. (A) During muscle contraction, the thick (myosin) filaments pull the thin filaments toward the center of the sarcomere. When the thin filaments slide over the thick filaments, the I-bands and H-zones become shorter and eventually disappear. During muscle relaxation, the thick filaments release the interaction with thin filaments, and slide back to their relaxed positions. I-bands and H-zones are widened again. (B) Cross-bridge cycle. 1: Previous cross-bridge cycle ends with the binding of ATP to the myosin head. 2. Weakening actin-myosin interaction leads to releasing of the cross-bridge; 3: ATP is hydrolyzed to ADP and Pi, leading to the “cocking” of the myosin head and ready to bind to actin. The accessibility of AM.ADP.Pi is regulated by troponin complex. 4: Pi is released. The myosin head twists and bends, generating force and pulling the attached actin myofilament ahead. 5. ADP is released. Ready for ATP binding and next cycle.
The thin and thick filaments contain a specific set of proteins, which make up the contractile apparatus of the sarcomere. The thin filament is mainly composed of the filament form of sarcomeric α-actin with regulatory proteins such as troponin complex (Tn) and tropomyosin. The thick filament consists of myosin heavy chains, regulatory myosin light chains, and myosin binding protein C. The protruding globular region of myosin heavy chains, namely myosin heads or cross-bridges, can interact with actin using adenosine triphosphate (ATP) to provide the force to pull the thin filament towards the center of the sarcomere (Weber et al., 1973) (Fig. 1B). Cross-bridge cycling is highly regulated by calcium (Ca2+) levels. Indeed, the terms “contraction” and “relaxation” refer to the “on” and “off” of force-generation mediated by the cytosolic calcium concentration ([Ca2+]). During muscle contraction, elevated [Ca2+] stimulated by the action potential propels Ca2+ to bind to troponin C (TnC), resulting in conformational changes in the troponin complex. This releases the allosteric inhibitory action of tropomyosin, allowing the cross-bridges to interact with actin and enter cross-bridge cycling for force generation and contraction (Ebashi et al., 1968; Gordon et al., 2000; Solaro et al., 2013) (Fig. 2). During relaxation, Ca2+ dissociates from TnI, arresting the interaction of cross-bridges to actin, thereby allowing the sarcomere to slide back into the relaxed position.

In a new window | Download PPT
Figure 2: Scheme illustration of troponin complex structural changes in the transition from a relaxation to an active state. Left: In relaxation, troponin complex is in the “switch-off state” when tropomyosin (Tm) blocks the accessibility of the myosin head to the binding regions on the actin filament. Middle: During myofibril activation, Ca2+ binds to TnC N-terminal lobe (NT lobe), promoting the interaction between this region and the switch peptide (SwP) of TnI. Right: Movement of the SwP triggers a series of conformational changes including movement of the actin-binding peptides of TnI, intertwined regions of TnT and TnI, along with the C-terminal lobe of TnC. Troponin complex is “switched on”, which releases Tm and exposes binding sites for myosin cross-bridges on actin.
The myofibril function rig: an experimental model to dissect the molecular basis of sarcomere function
The inherited cardiomyopathies, HCM, DCM, and RCM display distinct spectrums of pathologic variants in the sarcomere (Garfinkel et al., 2018; Yotti et al., 2019). Studying alterations in sarcomeric function of these variants from heart tissue collected from cardiomyopathy patients or currently available transgenic animal models is challenging. Muscle function is regulated on many different levels: from the action potential stimulated by the neurohormonal system or pacemakers, to intracellular signaling with Ca2+ influx/uptake via the sarcoplasmic reticulum (SR), and through cross-bridge cycling between thin and thick filaments.
The traditional experimental setup to investigate “pure” muscle function is the skinned muscle fiber preparation, in which cell membrane and SR are replaced by activating solutions containing Ca2+ to exclude the effects of intracellular regulation and the calcium handling system on sarcomere activation (Wankerl et al., 1990; James et al., 2000; Montgomery et al., 2001). The limitation of this technique is that the amount of time that it takes for Ca2+ to diffuse into thick muscle fibers is too long to detect the fast kinetics of sarcomere contraction and relaxation. This issue can be circumvented by applying photosensitive caged compounds such as Nitr-7 to rapidly release Ca2+ by strong light (Ashley et al., 1991; Araujo et al., 1994). However, this approach also has its drawbacks, including the inability to assess detailed relaxation kinetics (Poggesi et al., 2005; Stehle et al., 2009).
The isolated myofibril and fast solution switching technique, or the “isolated myofibril function rig”, is a mechanical system that has the capability to accurately measure detailed parameters of contraction and relaxation of the most basic subcellular unit of the cardiomyocyte, the myofibril (Colomo et al., 1998). Isolated myofibrils are mounted on a force transducer and rapidly shifted between a solution with a high Ca2+ concentration and a solution free of Ca2+, and activation/relaxation kinetics are then quantified (Fig. 3A). The unique aspects of this system, such as the rapid solution switch technique combined with the physical characteristics of the small myofibril bundles (diameter < 2μm), enables attainment of equilibrium from the bathing solution within one millisecond. Therefore, the measurements for force generation following rapid Ca2+ delivery and relaxation following sudden Ca2+ reduction accurately reflects the kinetics of interaction between contractile proteins.

In a new window | Download PPT
Figure 3: Myofibril mechanics rig and representative activation-relaxation cycle. (A) Left: Myofibril mechanical rig set-up. Right: Scheme illustration of double barrel perfusion pipette delivering different concentrations of calcium (pCa 4.5 and pCa 9.0). Myofibrils are mounted between a force probe (calibrated to detect force in μN/μm) and a supporting stretcher. (B) Recording chart of one myofibril activation. Resting Tension (RT), force generation (FMAX), and activation kinetics (kACT and kTR) were measured. Relaxation is highlighted with the white box. (C) Representative trace of myofibril relaxation showing linear and exponential phases of relaxation.
While the myofibril rig system is capable of measuring fundamental sarcomere function such as force capacity (or maximal force, Fmax), Ca2+ sensitivity (EC50), and resting tension (RT, relevant to stiffness) of myofibrils in a similar manner to the traditional skinned muscle fiber technique, the major advantage of the myofibril rig system is the accurate measurement of activation and relaxation kinetics of single myofibrils with Ca2+ delivery/removal, allowing the interpretation of detailed cross-bridge kinetics.
Activation Kinetics
When Ca2+ is delivered to myofibrils, there is an extremely fast process of Ca2+ binding to TnC and subsequent thin filament activation with conformation changes of Tn to the “switch-on” state. Switch-on of Tn greatly increases the probability of actin-myosin interaction, propelling cross-bridge cycling toward force development until the myofibril reaches its maximal isometric tension (Fmax). This time course of force development following activation is roughly mono-exponential with a rate constant of kACT (Fig 3B). This process is almost superimposed with the force re-development after a fast re-stretch-release (kTR), suggesting that kACT is independent of Ca2+ binding and thin filament activation, and essentially represents the action of cross-bridge cycling (Colomo et al., 1998). Force generation is achieved by the combination of two cross-bridge states: (1) force-generating cross-bridge state, with the apparent rate constant of fapp defined by Brenner et al. (1988); and (2) non-force-generating state, with the apparent rate constant of gapp (Brenner et al., 1988). The activation constants kACT and kTR observed during contraction is the composition of these two states:
fapp + gapp = kACT = kTR
The value of fapp is much higher than gapp during contraction with the activated thin filament, leading to net force generation shown in Fig. 3B. For a more detailed and comprehensive review on myofibril cross-bridge kinetics we would like to direct the reader to two excellent review articles (Poggesi et al., 2005; Stehle et al., 2009).
kACT and kTR are mediated by : (1) The nature of the cross-bridges, such as myosin isoform composition - Myofibrils isolated from human ventricles have much lower kACT than that from rodents, correlating with the predominant β-myosin heavy chain (slow form) expression in human heart (Stehle et al., 2002; Krüger et al., 2003); (2) the equilibrium of cross-bridge cycling - Force development rate can be modulated by the substrates of cross-bridge cycles seen in Fig. 1B. For example, addition of inorganic phosphate (Pi) can return the cross-bridge cycle back to the non-force generation state, leading to decreased activation kinetics (Tesi et al., 2000); (3) steric hindrance of regulatory proteins - Despite the fact that kACT has been shown to be unaffected by thin filament regulation (de Tombe et al., 2007), mutations of the inhibitory peptides on cTnI (R145G) can lead to weakened Ca2+-dependent cTnI–cTnC interaction, slowing down the release of allosteric inhibition, which allows force generation (Krüger et al., 2003, 2005).
Relaxation Kinetics
Compared to contraction, muscle relaxation is less understood. During relaxation, Ca2+ is taken up from the cytosol by the sarcoplasmic reticulum (SR) through the pumping action of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). With the fall of cytosolic [Ca2+], Ca2+ dissociates from TnC, leading to the prompt switch-off of Tn back to its inactive form. Thin filament inactivation then stops force generation of the cross-bridges and initiates myofibril relaxation. These processes, however, are not as straightforward as Ca2+-activated force generation. First, unlike the rapid rise of Ca2+ transiently stimulated by action potential during activation, the fall of cytosolic [Ca2+] during relaxation is a much more gradual process (Blinks et al., 1978; Backx et al., 1995) where dissociation of Ca2+ from TnC and thin filament inactivation happen concurrently. Second, relaxation kinetics of myofibrils after Ca2+ removal is more complex with a unique “bi-phasic” pattern, which was not possible to resolve with the gradual [Ca2+] changes. With the fast switch perfusion system, another key feature of the isolated myofibril function technique is sufficient sensitivity to resolve the slow (linear) and the fast (exponential) relaxation phases (Fig. 3C) (Stehle et al., 2002; Tesi et al., 2002).
This force decay process after Ca2+ removal highlights the fundamental differences of cross-bridge cycling between contraction and relaxation. During relaxation, after Ca2+ dissociation from TnC, inactivation of the thin filament stops cross-bridges from entering the force generating state (fapp ≈ zero). However, the non-force-generating state still continues. The rate constant kREL = fapp + gapp is therefore close to gapp since fapp is negligible. This force decay based on gapp is a very slow, almost isometric process, which can be fitted with a linear function with the slope defined as the rate constant “slow kREL“ and the duration “slow tREL“ by Tesi et al. (2002). Although the cross-bridge cycling rate gapp is the rate-limiting factor of the slow relaxation phase (Stehle et al., 2002), its duration is “thin filament inactivation-related”. Slow tLIN was reported to vary with the amount of time TnC needs to be switched off (Stehle et al., 2006) and TnI isoform exchange (Kreutziger et al., 2008).
Around 50-150 ms after Ca2+ removal, slow phase relaxation is followed by a sudden and very fast exponential force decay. The rate constant (fast kREL) of this “fast relaxation phase” is roughly 10-20 times faster than the slow kREL. Interestingly, fast kREL is usually even faster than kACT and kTR, (Poggesi et al., 2005), suggesting the original cross-bridge cycling equation kREL = fapp + gapp based on chemical reaction is not enough to explain this rate of force decay. Stehle et al. (2002) found that the fast relaxation phase starts from a sudden lengthening of the “structurally weakest” sarcomere, and propagates to the adjacent sarcomeres. This phenomenon can be explained by a “two detachment model” (Stehle et al., 2009). During contraction, the power-strokes generated by cross-bridges cause the distortion of the elastic elements (e.g. titin) of the myofibrils. After Ca2+ is removed, cross-bridges cycling is blocked (step 3 of Fig. 1B), leading to slow force decay without recruiting new force-generating cross-bridges. This can reach a point where the load released from detached cross-bridges exerts exceeding strain on other cross-bridges and forces their detachment in a “backward” path along with Pi rebinding (the reverse of the “step 4” in Fig. 1B) - and therefore the half-sarcomere is lengthened. Meanwhile, the adjacent half-sarcomere is shortened, facilitating the “forward detachment” by transferring mechanical energy into adenosine diphosphate (ADP) release and ADP.Pi (ATP) binding (“step 5” of Fig. 1B). This model of rapid relaxation propagation with mechanical-chemical energy coupling fits well with the experimental observation where the addition of ADP significantly slows down sarcomere relaxation in myofibrils and skinned muscle fibers (Lipscomb et al., 1999; Tesi et al., 2002). The fast phase of relaxation occurs independent of thin filament inactivation. Instead, it is more relevant to cross-bridge cycling, the affinity between myosin heads and actin, and the physical properties of the myofibrils such as the elasticity provided by titin. Thus, observing the alterations in fast and slow relaxation phases may provide insights into the molecular basis of a certain disease state, and provide potential target proteins for investigation.
Advantages
Working on the isolated myofibril function rig has several advantages for the study of cardiomyopathies. First, the mechanical parameters measured from myofibrils of diseased heart tissue provide a complementary approach to validate cross-bridge kinetics previously only obtainable from ex vitro preparations, such as reconstituted troponin complex (Dong et al., 1997), reconstituted thin filaments (Tn·Tm·actin, Kobayashi et al., 2005), and actin-myosin interactions (Rosenfeld et al., 1987). Second, resolution of the slow and fast phases of relaxation is crucial to dissect the molecular basis of relaxation impairment in diastolic heart failure (Bhatia et al., 2006; Massie et al., 2008; Shah et al., 2008). Furthermore, the readout from this technique allows investigators to focus on pure sarcomere function independent of intracellular regulation distinguishing it from whole cell/tissue systems such as Ion-Optix or engineered heart tissue. Experimentally, the myofibril rig can be employed on both fresh and frozen tissues with a minimal quantity of tissue (down to ~ 5μg), such as small pieces of bioptomes. Also, the fast equilibration with the bathing solution allows the analysis of the functional response of sarcomeric proteins to defined concentrations of cross-bridge substrates, as well as specific enzymes such as kinases and deacetylases (Cheng et al., 2016; Dvornikov et al., 2016; Jeong et al., 2018). More importantly, recent reports have shown that the myofibril rig can analyze myofibrils from cultured adult myocytes (Woulfe et al., 2019) and cardiomyocytes derived from human induced pluripotential stems cells (hiPSCs) (Pioner et al., 2016; Iorga et al., 2018; Pioner et al., 2019). Thus, this multi-level, cross-species platform makes it stand out from current techniques in the study of sarcomere function.
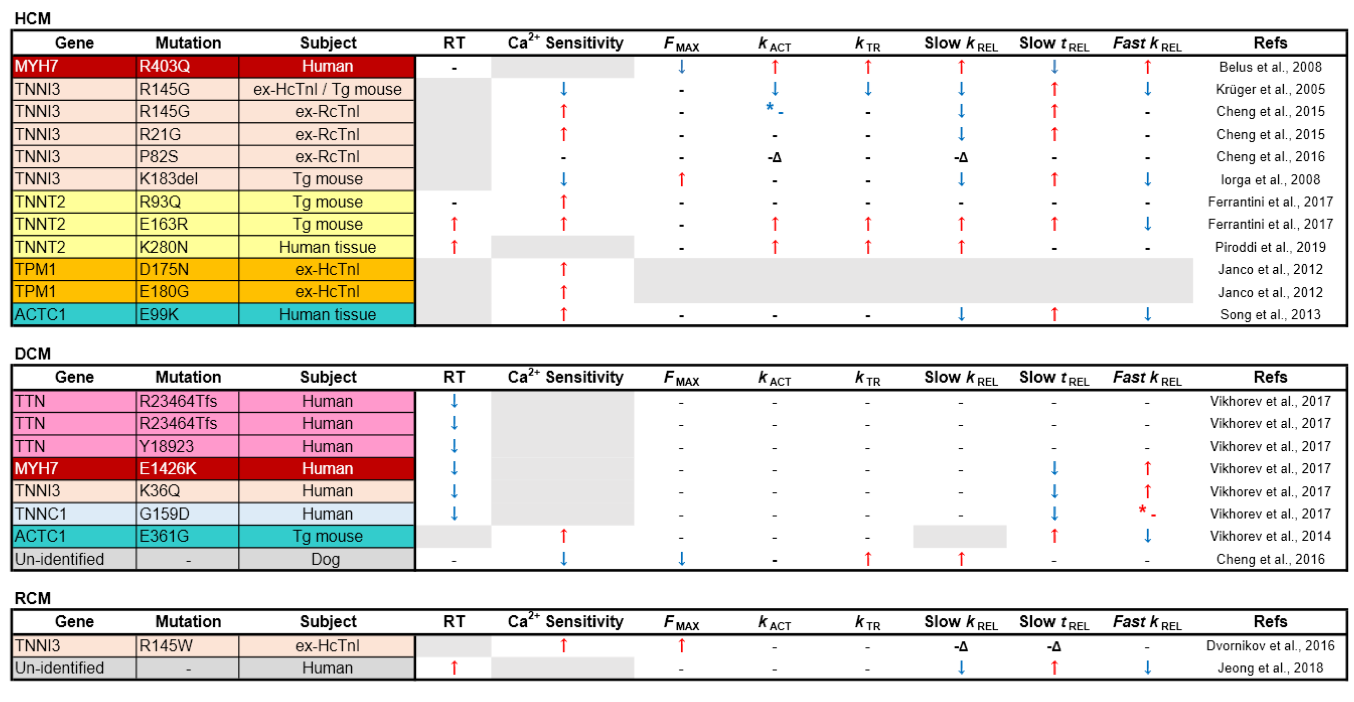
Given that many different and complex pathways regulate whole heart function, the myofibril function rig provides crucial insights about whether specific mutations cause inherited cardiomyopathies directly through impacting pure sarcomere function. In the cases of inherited HCM, many HCM-linked sarcomeric mutations lead to relaxation impairment (prolonged slow tREL, lower slow tREL and fast kREL) of the myofibrils (Table 1), corresponding to the whole heart function of this disease, details of which will be elucidated in the next section. Furthermore, the majority of myofibrils of HCM hearts have elevated Ca2+ sensitivity, which might explain their hyper-contractile features. In contrast, the alterations of myofibril mechanics in some mutations are not necessarily seen at the level of whole hearts, suggesting the phenotypes might have resulted from compensatory effects or other mechanisms. In the following sections, we discuss the mechanical changes and the possible mechanisms underlying sarcomeric genetic mutations associated with the inherited cardiomyopathies at the myofibril level in HCM, DCM, and RCM.
Table 1. Comparison of mechanical parameters from myofibrils isolated from inherited cardiomyopathy models. RT: resting tension; ex-HcTnI: ex vivo myofibrils exchanged with human cardiac troponin complex; ex-RcTnI: ex vivo myofibrils exchanged with exogenous rat cardiac troponin complex; Δ: uncoupled from PKA regulation; * - (red): non-significant increase; * - (blue): non-significantly increase.
Hypertrophic cardiomyopathy
HCM is a common inherited cardiomyopathy affecting 1 in 500 people (Maron et al., 1995), and is distributed fairly equally in different countries and ethnicities (Hada et al., 1987; Zou et al., 2004; Maron et al., 2004). Clinically, adults with non-dilated left ventricle chamber size, with ≥ 13 mm end-diastolic wall thickness are recognized as having HCM (Maron et al., 1999; Marian et al., 2017). Thirty to sixty percent of HCM probands are found to carry disease-causing mutations (Burke et al., 2016). Patients rarely have symptoms at very young ages, but manifestations start to occur after adolescence (Maron et al., 2003; Masarone et al., 2018). HCM is known to be the leading cause of sudden cardiac death (SCD) of young adults and athletes (Elliott et al., 2008). Thus, it is crucial to understand the molecular basis of how these mutations cause their clinical phenotype for better prediction and prognosis of HCM. Echocardiographic analyses of individuals carrying HCM pathological variants without overt clinical manifestations have shown that the majority have hyper-contractility with diastolic dysfunction before developing heart failure (Ho et al., 2002).
Myofibril mechanics in HCM with thick filament mutations
The majority of HCM-linked mutations (~75%), including the earliest identified β-MHC R403Q, were found in the thick filament, either in the β-MHC or myosin binding protein C3 (MyBP-C3) (Burke et al., 2016). Interestingly, despite being the main pathologic variants for HCM, research on the myofibril mechanics on MYH7 (gene for β-MHC) mutations are very limited. One reason for this is that the transgenic murine model is a mutation in α-MHC, which does not replicate the human β-MHC mutation (Lowey et al., 2002; 2008).
It was not until 2008 that Belus et al. (2008) directly obtained heart tissues from patients with β-MHC R403Q mutation and performed the first myofibril mechanics analysis on pathologic variants of the thick filament. In contrast to the hyper-contractility with relaxation impairment observed at the whole heart level, myofibrils with β-MHC R403Q were shown to contract with less force (Fmax) with significantly faster kACT and slow kREL (Belus et al., 2008; Witjas-Paalberends et al., 2014). Garfinkel et al. (2018) proposed that thick filament mutations cause HCM based on the ratio of two groups of myosin heads during relaxation: disordered relaxation (DRX, half-activated) and super relaxation (SRX, complete relaxed), noting that HCM variants have myosin heads toward the DRX state, leading to relaxation impairment (Alamo et al., 2017; Garfinkel et al., 2018). Interestingly, the finding from Belus et al (2008) did not fit the general features of HCM described by Garfinkel as R403Q was shown to be one of the MYH7 variants to be in favor of DRX states (Anderson et al., 2018). However, the myofibrils had significantly increased energy costs despite having faster cross-bridge kinetics, which may explain the phenotype of energy depletion in some HCM cases (Ashrafian et al., 2011). Unfortunately, apart from the report of Belus et al. (2008), myofibril mechanics of most HCM-linked variants on β-MHC and MYBP-C3, especially the latter, are still unknown, and more studies are needed.
Myofibril mechanics in HCM with thin filament mutations
In contrast to pathologic variants in the thick filament, mutations in the thin filament (TPM1, TNNT2, TNNI3, and ACTC1, which encode cardiac tropomyosin alpha-1 chain, troponin T, troponin I, and sarcomeric α-actin, respectively) are rarer than thick filament variants (< 10%) but are more well-studied at the myofibril level, especially those in Tn. Reconstituted Tn with disease-causing mutations on one of its subunits can be substituted into myofibrils in the ex vivo environment, replacing endogenous Tn for myofibril function analysis (Brenner et al., 1999). In an alternative approach, Westfall et al. (1997) reported that infecting adult cardiomyocytes with adenovirus expressing troponin subunits can also achieve up to 70-80% of exchange rate at the cellular level, comparable to the ex vivo exchange protocol.
Troponin I
Cardiac troponin I (cTnI) is the inhibitory subunit of Tn. The major difference between cTnI and skeletal muscle TnI is that the former has additional phosphorylation sites (Solaro et al., 2013). The most well-known is the phosphorylation of S22/23 (in human) and S23/24 (in rodents) by protein kinase A (PKA), which can reduce Ca2+ sensitivity of the myofibrils, thereby accelerating force decay, the result of which is beneficial for relaxation (Zhang et al., 1995). This highly modifiable feature makes cTnI a central regulator in myocardial contraction and relaxation. Using a reconstituted Tn exchange technique and a transgenic mouse model, the R145G (or R146 in mouse), cTnI mutation was the first HCM variant to be tested for myofibril mechanics (Kruger et al., 2005). In this study, Fmax, kACT, and slow tREL were all reduced in wild type mouse myofibrils exchanged with human cTnI R145G. In transgenic mice, there was a similar trend but the changes were less pronounced probably because of the low replacement rate (40%) of the mutated cTnI (James et al., 2000). These findings were recapitulated by Cheng et al. (2015) with rat cardiac myofibrils exchanged with rat cTnI R146G except there was also increased Ca2+ sensitivity, a finding that has been supported by multiple reports using skinned muscle fibers (James et al., 2000; Takahashi-Yanaga et al., 2001; Lang et al., 2002). In the same report, cTnI R146G and another HCM-linked cTnI (R21C) mutation were found to antagonize the effect of PKA phosphorylation on Ca2+ sensitivity and relaxation. By using computational simulation, Cheng et al. (2015) reported that these two mutations caused reduced intramolecular interaction between the N-terminus and the inhibitory peptides of cTnI, which is the mechanism through which PKA regulates TnI function based on their earlier report (Lindert et al., 2015; Rao et al., 2014). Other HCM-linked variants on cTnI that have been analyzed at the myofibril level include K183 deletion and P82S (Iorga et al., 2008; Cheng et al., 2016). Interestingly, like the other cTnI variants, mutations of these two sites resulted in blunted relaxation function or insensitivity to PKA signaling. Thus, HCM-linked variants on cTnI seem to share a similar feature with impaired relaxation of the myofibrils possibly by impeding cTn’s capacity to be switched off, which reflects the diastolic dysfunction observed in HCM.
Troponin T
Troponin T (TnT) is the longest and largest subunit of Tn, and is known to be the “molecular linker” of the whole regulatory machinery (Tobacman et al., 1996; Takeda et al., 2003). Because of its two large binding areas to tropomyosin (Jin et al., 2010), TnT is the key subunit of Tn for regulating the position of tropomyosin during muscle contraction (Kobayashi et al., 2005). Although HCM-linked mutations of cardiac TnT (cTnT) is the largest class among the 3 subunits of Tn (5-10% of all familial HCM), studies on myofibril mechanics of HCM-causing cTnT mutations are less advanced than those on cTnI. In 2017, Ferrantini et al. (2017) first analyzed the detailed kinetic parameters of myofibrils from two mouse models with HCM-causing mutations cTnT R93Q and E163R. Interestingly, despite the fact that both mouse models developed similar HCM-like heart function and contractile properties of the intact muscle fibers, their myofibril mechanics were substantially different. The myofibrils from cTnT E163R transgenic mice had faster kACT and slow kREL, but significantly prolonged slow tREL and increased passive stiffness. In contrast, in cTnT R93Q transgenic mice the kinetic properties of myofibrils were not much different except for a drastic increase in Ca2+ sensitivity. The authors concluded that similar HCM phenotypes can be generated through different pathways: (1) impaired myofibril relaxation and tension cost (E163R), or (2) delayed intact muscle fiber relaxation with higher Ca2+ sensitivity because of Ca2+ mishandling (Coppini et al., 2017). Recently, the same group analyzed the myofibrils from HCM human patients with homozygous cTnT K280N, another HCM-causing mutation (Piroddi et al., 2019). They found this mutation at the C-terminus of cTnT led to yet another myofibril phenotype: impaired efficient energy usage, increased kACT and slow kREL but unchanged slow tREL. All of these reports emphasize the need for a precision medicine approach to treatment of HCM, given the different observed phenotypes with mutations in the same protein.
Tropomyosin and actin
Tropomyosin is an α-helical coiled dimer, which is positioned along the length of actin filament and plays a crucial role in providing steric hindrance of the cross-bridge actin-myosin interaction regulated by troponin complex (Tobacman et al., 1996; Kobayashi et al., 2005) (Fig. 2). Mutations in TPM1, the gene encoding α-tropomyosin, are another major group of HCM-causing variants. Not much is known regarding myofibril mechanics in TPM1 mutations. However, it is well documented that HCM-causing TPM1 variants mainly cause increased Ca2+ sensitivity (Robinson et al., 2018), a feature that is also observed at the myofibril level in two HCM-linked variants, D175N and E180G (Janco et al., 2012). Furthermore, S283 pseudo-phosphorylation of α-tropomyosin has been shown to impair myofibril relaxation (Nixon et al., 2013). It has been shown that the S283 phosphorylation level is elevated in E180G transgenic HCM mice, and the impaired cardiac function is rescued by phosphorylation-null mutation S283A (Schutz et al., 2013). Whether enhanced S283 phosphorylation levels in HCM hearts could causes the relaxation impairment of the myofibrils is still unknown. Another HCM-linked mutation being studied at the myofibril level is ACTC1, the gene encoding sarcomeric α-actin. A mouse model for HCM-linked ACTC1 E99K mutation has demonstrated that myofibrils from these animals had increased Ca2+ sensitivity with impairment of all relaxation kinetics, slow tREL, slow kREL, and fast kREL, which reflects the phenotypes of HCM (Song et al., 2013). Further investigation is required to understand whether this finding can be applied to other mutations in ACTC1. For a more comprehensive comparison of myofibril mechanics alterations among all HCM variants please see Table 1.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is defined by the presence of left ventricular or biventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions (hypertension, valve disease) or coronary artery disease sufficient to cause global systolic impairment. The causes of DCM can be classified as genetic or acquired (please see review for more information, Pinto et al., 2016). DCM was originally estimated to be present 1 in 2,500 individuals (Codd et al., 1989), but recent reports have suggested a much higher prevalence (≥ 1 in 250 individuals) (Hershberger et al., 2013; McNally et al., 2017). Clinical manifestations of DCM usually begin at 30 to 40 years old, although it can present at all ages (Bozhurt et al., 2016), and it is the predominant cardiomyopathy in children (Towbin et al., 2006). It often leads to progressive heart failure, and is the leading cause of heart transplantation among all the cardiomyopathies (Kirt et al., 2009). About 20-50% of DCM patients are found to be familial, and ~40% of familial DCM have identifiable genetic mutations (Ganesh et al., 2013; Sweet et al., 2015). Genetic carriers without phenotypic clinical manifestation were found to have reduced longitudinal left ventricular strain, an indicator of abnormal systolic function (Lakdawala et al., 2012; Japp et al. 2016).
Myofibril mechanics in DCM with titin-truncating variants
Titin is the largest human protein, being 35,000 amino acids in length, with one single titin molecule spanning from the Z disc to the central M line. Titin not only provides structural support and elasticity of the myofibrils, but it also plays a crucial role in sarcomere assembly (Ehler and Gautel, 2008). Titin-truncating variants (TTNtvs) are the most common sarcomeric mutations causing DCM, accounting for up to 25% of familial DCM (Herman et al., 2012). Since titin is such a large protein with a large number of variants, TTNtvs are also present in 2% of normal individuals (a prevalence which is much higher than that of DCM), making it challenging to correlate genotype with clinical manifestations. In 2015, Hinson et al. (2015) first generated hiPSCs-derived cardiomyocytes from patients carrying TTNtvs, and found that heterozygous TTNtv cells had titin “haplo-insufficiency”, substantially reduced sarcomere content and impaired force generation, indicating the importance of titin in sarcomerogenesis. In an animal model of DCM-causing TTNtvs, truncated titin isoforms could not be identified, suggesting a rapid turnover of the “malfunctioned titin” (Schafer et al., 2016). Vikhorev et al (2017) have studied myofibril mechanics in 3 DCM patients with TTNtvs. They found that mechanical kinetics including Fmax, activation and relaxation were mostly unchanged. Interestingly, the stiffness of TTNtv myofibrils was reduced by about 40% among all three samples tested. Therefore, the compliant myofibril might be a feature of DCM TTNtvs, and indirectly cause impaired contractility of the heart by over-stretching the myocardium and decreasing systolic capacity (Vikhorev et al., 2018; Garfinkel et al., 2018). The underlying mechanism of the stiffness alteration needs to be investigated in future studies. Also, this feature may not be present in all DCM patients with TTNtvs, as the latter may involve different functional domains and result in different myofibril mechanics.
Myofibril mechanics in DCM with mutations on contractile proteins
DCM due to mutations in sarcomeric proteins is relatively rare but include MYH7 (β-cardiac myosin heavy chain), TPM1 (tropomyosin alpha-1 chain), and cardiac TNNT2 (troponin T), TNNI3 (troponin I), and TNNC1 (troponin C) (Burke et al., 2016). Despite overlapping with sarcomeric proteins responsible for HCM (Marian et al., 2016), the disease phenotype is very different. Vikhorev et al (2017) investigated DCM-causing sarcomeric mutations in thin filament proteins (cTnI K36Q and cTnC G159D), and the thick filament (β-MHC E1426K), and found that Fmax was unchanged, but myofibril relaxation kinetics (tREL and slow kREL) were significantly shortened, suggesting decreased Ca2+ sensitivity. These might be relevant to the reduced systolic function observed with DCM. Similar results were found in an inbred canine model of DCM (Cheng et al., 2016). In contrast, an earlier study in mice with DCM due to ACTC1 E361G mutation found that myofibrils had unchanged activation kinetics (kACT), but slower relaxation kinetics (slow tREL and slow kREL) with higher Ca2+ sensitivity (Vikhorev et al., 2014), a phenotype more suggestive of HCM. Interestingly, a recent longitudinal clinical follow-up of DCM genotyped patients found that in some patients who developed end-stage DCM, they had originally presented as having transitory HCM (Ho et al., 2017), suggesting perhaps an overlap between HCM and DCM in some patients.
In summary, current knowledge of myofibril mechanics alterations in DCM is much less advanced than that in HCM, and has only been investigated by a few groups. Given its comparable prevalence with HCM, further research effort is required to understand a more generalized nature of the myofibril mechanics in inherited DCM hearts. For a more comprehensive comparison of myofibril mechanics alterations among all DCM variants please see Table 1.
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCM) is the least common cause of cardiomyopathy, accounting for less than 5% of all cardiomyopathies (Muchtar et al., 2017). RCM is characterized by impaired diastolic function due to increased myocardial stiffness in the presence of normal wall thickness and systolic function (Elliott et al., 2008). It is associated with a variety of pathologic conditions, and is therefore very heterogeneous, and can be difficult to diagnose (Kushwaha et al., 1997; Muchtar et al., 2017). RCM was mostly regarded as an acquired (non-familial) cardiomyopathy until a mutation in cardiac troponin I (TNNI3 D190H) was identified as causing RCM (Morgensen et al., 2013). Since then, RCM-causing variants have also been found in most major sarcomeric genes such as TNNT2, MYH7, TPM1, MYL3, MYL2, TTN and DES (Peddy et al., 2006; Arbustini et al., 2006; Kubo et al. 2007; Monserrat et al, 2007; Caleshu et al., 2011; Peled et al., 2014), but it has been difficult to correlate them with specific RCM phenotypes (Gigli et al., 2016).
Myofibril mechanics in RCM
The only published myofibril study in inherited RCM was performed by Dvornikov et al (2016), who reconstituted cardiac troponin complex containing RCM-linked cTnI R145W mutation into myofibrils from normal human heart tissue. Similar to previous work on HCM-linked cTnI R146G mutation in rat cardiac myofibrils (Cheng et al., 2015), R145W mutation in cTnI resulted in increased Ca2+ sensitivity and uncoupled myofibrils from PKA regulation, and relaxation was non-significantly prolonged. Recently, Jeong et al (2018) investigated myofibril mechanics of explanted LV tissue from two patients diagnosed as idiopathic RCM who underwent cardiac transplantation. They found slow tREL and slow kREL of the RCM patients suggesting delayed relaxation when compared to control donors, and went on to to show that the diastolic dysfunction was corrected using pharmacological HDAC inhibition in two different small animal models of diastolic dysfunction and preserved left ventricular ejection fraction. Whether human myofibrils with relaxation impairment can be corrected by HDAC inhibition remains to be determined.
Conclusions
New treatments are needed to treat inherited cardiomyopathies such as HCM, DCM, and RCM, in order to correct the contractile/relaxation impairment and prevent the onset of heart failure. Investigations of myofibril mechanics have provided important insight into the pathophysiology underlying these different cardiomyopathies that appear to correlate with the clinical phenotype. These studies should lead to the identification of novel therapeutic targets for improving systolic function for DCM, and improving diastolic function in HCM and RCM. The myofibril function rig allows the detailed assessment of both contractile and relaxation of myofibrils form a variety of sources including: cardiac cell lines, primary atrial and ventricular cardiomyocytes, cardiomyocytes derived from hiPSCs, animal heart tissue (both fresh and frozen), and human heart tissue (both fresh and frozen), and can therefore be used to discover novel targets for treating cardiomyopathies.
Funding
Jonathan Yap is supported by National Institutes of Health grant F31 HL139082. Derek Hausenloy was supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology). Chrishan Ramachandra is supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (OF-YIRG) – [NMRC/OFYIRG/0073/2018] and through the National Health Innovation Centre Singapore under its Innovation to Develop Grant (NHIC-I2S-1811007).
Confilct of interests:
None
References
Ying-Hsi Lin1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Jonathan Yap3
3Center for Cardiovascular Research, John A. Burns School of Medicine, University of Hawaii, USA.
Chrishan J.A. Ramachandra1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Derek J. Hausenloy1,2,4-8
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore. 4Yong Loo Lin School of Medicine, National University Singapore, Singapore. 5The Hatter Cardiovascular Institute, University College London, London, UK. 6The National Institute of Health Research University College London Hospitals. 7Biomedical Research Centre, Research & Development, London, UK. 8Tecnologico de Monterrey, Centro de Biotecnologia-FEMSA, Nuevo Leon, Mexico.
Corresponding author:
Dr. Ying-Hsi Lin
Email: ying-hsi.lin@duke-nus.edu.sg
In a new window | Download PPT
Figure 1: Sliding filament model of muscle contraction and the cross-bridge cycle. (A) During muscle contraction, the thick (myosin) filaments pull the thin filaments toward the center of the sarcomere. When the thin filaments slide over the thick filaments, the I-bands and H-zones become shorter and eventually disappear. During muscle relaxation, the thick filaments release the interaction with thin filaments, and slide back to their relaxed positions. I-bands and H-zones are widened again. (B) Cross-bridge cycle. 1: Previous cross-bridge cycle ends with the binding of ATP to the myosin head. 2. Weakening actin-myosin interaction leads to releasing of the cross-bridge; 3: ATP is hydrolyzed to ADP and Pi, leading to the “cocking” of the myosin head and ready to bind to actin. The accessibility of AM.ADP.Pi is regulated by troponin complex. 4: Pi is released. The myosin head twists and bends, generating force and pulling the attached actin myofilament ahead. 5. ADP is released. Ready for ATP binding and next cycle.
In a new window | Download PPT
Figure 2: Scheme illustration of troponin complex structural changes in the transition from a relaxation to an active state. Left: In relaxation, troponin complex is in the “switch-off state” when tropomyosin (Tm) blocks the accessibility of the myosin head to the binding regions on the actin filament. Middle: During myofibril activation, Ca2+ binds to TnC N-terminal lobe (NT lobe), promoting the interaction between this region and the switch peptide (SwP) of TnI. Right: Movement of the SwP triggers a series of conformational changes including movement of the actin-binding peptides of TnI, intertwined regions of TnT and TnI, along with the C-terminal lobe of TnC. Troponin complex is “switched on”, which releases Tm and exposes binding sites for myosin cross-bridges on actin.
In a new window | Download PPT
Figure 3: Myofibril mechanics rig and representative activation-relaxation cycle. (A) Left: Myofibril mechanical rig set-up. Right: Scheme illustration of double barrel perfusion pipette delivering different concentrations of calcium (pCa 4.5 and pCa 9.0). Myofibrils are mounted between a force probe (calibrated to detect force in μN/μm) and a supporting stretcher. (B) Recording chart of one myofibril activation. Resting Tension (RT), force generation (FMAX), and activation kinetics (kACT and kTR) were measured. Relaxation is highlighted with the white box. (C) Representative trace of myofibril relaxation showing linear and exponential phases of relaxation.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13154 | 34 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA