Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Is there a role for remote ischemic conditioning in preventing 5-fluorouracil-induced coronary vasospasm?
Time:2019-11-02
Number:11892
Author Affiliations

Conditioning Medicine 2019. 2(5):204-212.
Abstract
Cardiac ischemia associated with chemotherapy has been linked to several anti-neoplastic agents and is multifactorial in etiology. Coronary artery vasospasm is one of the most commonly reported effects of cancer therapy that can lead to myocardial ischemia or infarction. The chemotherapy agent 5-fluorouracil (5-FU) or its oral pro-drug capecitabine can result in coronary vascular endothelial dysfunction causing coronary artery spasm, and possibly coronary thrombosis. These drugs have also been shown to be associated with myocardial infarction, malignant ventricular arrhythmias, heart failure, cardiogenic shock, and sudden death. The proposed mechanisms underlying cardiotoxicity induced by 5-FU are vascular endothelial damage followed by thrombus formation, ischemia secondary to coronary artery vasospasm, direct toxicity on myocardium, and thrombogenicity. There remains a pressing need to discover novel and effective therapies that can prevent or ameliorate 5-FU associated cardiotoxicity. To this point, promising overlap has been observed between proposed remote ischemic conditioning (RIC) cardioprotective mechanisms and 5FU-associated cardiotoxic cellular pathways. RIC, in which transient episodes of limb ischemia and reperfusion (induced by inflations and deflations of a pneumatic cuff placed on the upper arm or thigh), confer both cardioprotective and vasculoprotective effects, and may therefore prevent 5-FU coronary artery spasm/cardiotoxicity. In this review, we will be discussing the following potentially therapeutic aspects of RIC in ameliorating 5-FU associated cardiotoxicity: sequential phases of 5-FU cardiotoxicity as possible targets for dual windows of cardioprotection characteristic of RIC; protective effects of RIC on endothelial function and microvasculature in relation to 5-FU induced endothelial dysfunction/microvascular dysfunction; reduction in platelet activation by RIC in the context of 5-FU induced thrombogenicity; and the utility of improvement in mitochondrial function conferred by RIC in 5-FU induced cellular toxicity secondary to mitochondrial dysfunction.
Abstract
Cardiac ischemia associated with chemotherapy has been linked to several anti-neoplastic agents and is multifactorial in etiology. Coronary artery vasospasm is one of the most commonly reported effects of cancer therapy that can lead to myocardial ischemia or infarction. The chemotherapy agent 5-fluorouracil (5-FU) or its oral pro-drug capecitabine can result in coronary vascular endothelial dysfunction causing coronary artery spasm, and possibly coronary thrombosis. These drugs have also been shown to be associated with myocardial infarction, malignant ventricular arrhythmias, heart failure, cardiogenic shock, and sudden death. The proposed mechanisms underlying cardiotoxicity induced by 5-FU are vascular endothelial damage followed by thrombus formation, ischemia secondary to coronary artery vasospasm, direct toxicity on myocardium, and thrombogenicity. There remains a pressing need to discover novel and effective therapies that can prevent or ameliorate 5-FU associated cardiotoxicity. To this point, promising overlap has been observed between proposed remote ischemic conditioning (RIC) cardioprotective mechanisms and 5FU-associated cardiotoxic cellular pathways. RIC, in which transient episodes of limb ischemia and reperfusion (induced by inflations and deflations of a pneumatic cuff placed on the upper arm or thigh), confer both cardioprotective and vasculoprotective effects, and may therefore prevent 5-FU coronary artery spasm/cardiotoxicity. In this review, we will be discussing the following potentially therapeutic aspects of RIC in ameliorating 5-FU associated cardiotoxicity: sequential phases of 5-FU cardiotoxicity as possible targets for dual windows of cardioprotection characteristic of RIC; protective effects of RIC on endothelial function and microvasculature in relation to 5-FU induced endothelial dysfunction/microvascular dysfunction; reduction in platelet activation by RIC in the context of 5-FU induced thrombogenicity; and the utility of improvement in mitochondrial function conferred by RIC in 5-FU induced cellular toxicity secondary to mitochondrial dysfunction.
Introduction
Cardiovascular disease is the leading cause of morbidity and mortality worldwide (Benjamin et al., 2019). The World Health Organization (WHO) estimates that 17 million people die each year of cardiovascular disease, accounting for 30% of all deaths (Balukumar et al., 2016). Cancer is the second leading cause of death globally and is associated with 9 million deaths each year (Leal et al., 2016). In fact, cancer has now overtaken cardiovascular disease as the leading cause of mortality in high-income countries (Dagenais et al., 2019). According to the WHO, the incidence of cancer is expected to rise by about 70% over the next 20 years (WHO, 2018). Half of those diagnosed with cancer will survive for at least a decade, but this survival rate is expected to increase significantly in the future, leading to worsening burden of cancer-related complications experienced by the global population (Lucas et al., 2017, Cancer Research UK, 2019). Significant advances in cancer therapy have greatly reduced the mortality of cancer patients, with non-malignant comorbid conditions becoming important determinants of their quality of life and overall survival (Siegel et al., 2012). Among this heterogeneous group of comorbid conditions, cardiovascular diseases are a major contributor to overall morbidity and mortality in cancer survivors and patients with active cancer (Barac et al., 2015).
Heart disease and cancer share common risk factors in an ageing population and are further linked through cardiotoxic effects of contemporary cancer treatment (Moser et al., 2006; Weaver et al., 2013; Ghosh et al., 2017). Many cancer patients have subclinical cardiovascular disease, which can be worsened by the pro-inflammatory and hypercoagulable states associated with cancer (Blann 2011; Demers et al., 2012; Ghosh et al., 2018).
Cardiotoxicity secondary to 5-fluorouracil/capecitabine
Cardiac ischemia associated with chemotherapy has been linked to several anti-neoplastic agents and is multifactorial in etiology (Iliescu et al., 2016). Coronary artery vasospasm is one of the most commonly reported effects of cancer therapy that can lead to myocardial ischemia or infarction (Stewart and Pavlakis 2010; Naib and Steingart 2011). The chemotherapy agent 5-fluorouracil (5-FU) or its oral pro-drug capecitabine can result in coronary vascular endothelial dysfunction causing coronary artery spasm, and possibly coronary thrombosis, with a wide range of reported incidence between 1% and 68% (Pai 2000; Van Cutsem et al., 2002). These agents are used to treat solid cancers, including gastrointestinal, breast, head, neck, and pancreatic cancers (Polk et al., 2014). These drugs have also been shown to be associated with myocardial infarction or malignant ventricular arrhythmias (Kosmas et al., 2008). Capecitabine is converted to 5-FU in a three-step process involving several enzymes (Malet-Martino and Jolimaitre 2002). The last step is catalyzed by thymidine phosphorylase (Malet-Martino and Jolimaitre 2002). Many body tissues express thymidine phosphorylase, but this enzyme is expressed in higher concentrations in some carcinomas than in the surrounding normal tissues (Malet-Martino and Jolimaitre 2002). Based on this theory, the concentration of 5-FU at the tumor site should be increased compared to the concentration of 5-FU in healthy tissues, resulting in fewer side-effects involving healthy tissue (Malet-Martino and Jolimaitre 2002). The incidence of capecitabine-associated cardiac side-effects is 3-35%, gathered from the few studies examining capecitabine cardiotoxicity (Van Cutsem et al., 2002; Ng and Cunningham 2005; Jensen 2006; Kosmas et al., 2008; Koca et al., 2011). Case reports of cardiotoxicity after administration of capecitabine are similar to intravenous 5-FU treatment, with the predominant symptom being chest pain (Frickhofen et al., 2002; Cardinale and Colombo 2006; Coughlin et al., 2008). Other less frequent adverse effects are cardiac arrhythmias, myocardial infarction, heart failure, cardiogenic shock, and sudden death (Saif and Shah 2009; Kelly et al., 2013; Polk et al., 2013). Chest pain onset is often abrupt during infusion of 5-FU, but can also be delayed, presenting within the first 72 hours after 5-FU administration (Wacker et al., 2003; Saif and Shah 2009). Often, angina is accompanied by electrocardiogram (ECG) changes including ST-segment depression and prolonged repolarization abnormalities (Saif and Shah 2009).
Cardiac enzymes are infrequently elevated in patients experiencing angina following 5-FU (around 14% of cases) (Holubec et al., 2007; Saif and Shah 2009), and echocardiography has shown regional or global hypokinesis that usually returns to baseline within 48 hours of 5-FU cessation (Saif and Shah 2009). In these cases, significant coronary artery disease and acute plaque rupture is usually ruled out on coronary angiography, which leads to the consideration of coronary artery vasospasm (Cardinale and Colombo 2006; Lu et al., 2006). In a review of 377 patients with 5-FU-induced cardiotoxicity, cardiovascular risk factors such as smoking, diabetes, hypercholesterolemia, and family history of heart disease were found in 37% of the patients. Smoking was the most common risk factor among these groups of patients (Saif and Shah 2009). Previous or concomitant radiation therapy may play a role in 5-FU-induced cardiac toxicity as radiation can cause small-vessel thrombosis. 5-FU is a radio-sensitizer and may enhance radiation-induced thrombosis (Fajardo 1973; May et al., 1990). There is a higher incidence of angina with administration through continuous infusion compared to bolus infusion (Sudhoff et al., 2004; Saif and Shah 2009). It is unclear if this effect is dose-dependent, and although cessation of 5-FU results in resolution of angina, symptoms have been reported to last up to 12 hours (Tsavaris et al. 2002). Re-initiation of 5-FU has been associated with increased incidence of angina with serious complications including acute coronary syndrome, hypotension, cardiac failure, and even death (Sudhoff et al., 2004; Saif and Shah 2009).
While the causative relationship is unclear, endothelin-1 levels have been noted to be elevated in angina patients with 5-FU infusion (Sudhoff et al., 2004). Patients with known pre-existing history of coronary artery disease also have a higher incidence of angina, and are considered to have an increased risk of developing cardiac ischemia (Labianca et al., 1982; Giza et al., 2017).
In addition to high doses of 5-FU, prior mantle radiation, or simultaneous administration of another cardiotoxic chemotherapeutic agent are factors that can contribute to development of cardiac ischemia in patients treated with anti-metabolite drugs (de Forni et al., 1992, Anand 1994). In one large study, myocardial ischemia was reported in 4% of patients receiving high-dose, continuous infusion of 5-FU (Tsavaris et al. 2002). However, the failure of ergonovine and 5-FU to produce direct coronary artery vasospasm during cardiac catheterization has questioned the hypothesis of abnormal vasoreactivity being the predominant mechanism causing 5-FU associated myocardial ischemia (Freeman and Constanza 1988; Stewart and Pavlakis 2010). Age of the patient did not appear to influence the occurrence of cardiotoxicity (Labianca et al., 1982).
Need for novel therapies to prevent 5-FU associated cardiotoxicity
The proposed mechanisms underlying cardiotoxicity induced by 5-FU are vascular endothelial damage followed by thrombus formation, ischemia secondary to coronary artery vasospasm, direct toxicity on myocardium, and thrombogenicity. Patients developing ischemic events usually have recurrences if the drug is subsequently administered, so consideration must be given to withholding future 5-FU therapy if a patient develops ischemic events while on the drug (Anand 1994; Akpek and Hartshorn 1999). Although treatment with vasodilators have been proposed as prophylaxis against coronary artery vasospasm, limited effectiveness of this prophylactic therapy has been observed (Patel et al., 1987). Pharmacogenomic studies and genetic profiling may help predict the occurrence and streamline the treatment of 5-FU-induced coronary artery vasospasm. Further research is required to explore the cardioprotective effect of agents such as coenzyme complex, GLP-1 analogues, and degradation inhibitors on 5-FU-induced coronary artery vasospasm.
Postulated mechanisms of 5-FU associated cardiotoxicity – an overview
Patients with coronary artery vasospasm may have ECG findings suggestive of coronary occlusion, including ST-segment elevation as well as biochemical evidence of myocardial injury with troponin elevation even in the absence of occlusive epicardial vessel disease on coronary angiography or computed tomography (CT) imaging of the coronary vessels. In fact, patients with 5-FU-associated cardiotoxicity have consistently been shown to lack significant coronary stenosis on coronary angiography (Shoemaker et al., 2004; Alter et al., 2006; Camaro et al., 2009; Atar et al., 2010; Tajik et al., 2010).
The underlying mechanism of 5-FU associated cardiotoxicity is not well established and is likely to be multifactorial (Polk et al., 2014). The mechanism to explain 5-FU cardiac effects that is best supported by preclinical and clinical data is coronary artery vasospasm (de Forni et al., 1992; Akhtar et al., 1993; Mosseri et al., 1993; Porta et al., 1998; Sudhoff et al., 2004, Alter et al., 2006, Floyd et al., 2005; Dalzell and Samuel 2009). Preclinical models provide in vitro evidence of concentration-dependent vasoconstriction by 5-FU on vascular smooth muscle cells (Mosseri et al., 1993). Clinical data include the documentation of coronary artery spasm angiographically following intravenous (IV) 5-FU, and some cases of successful prophylaxis against coronary artery vasospasm with calcium channel antagonists (Kleiman et al., 1987; Luwaert et al., 1991; Shoemaker et al., 2004; Sudhoff et al., 2004).
However, some characteristics of 5-FU cardiotoxicity are inconsistent with this hypothesis. Coronary artery vasospasm has not been consistently shown angiographically during symptomatic attacks, and reintroduction of 5-FU in patients with a previous adverse cardiac event has not resulted in coronary spasm as evidenced by coronary angiography (Burger and Mannino 1987; Mizuno et al., 1995). In some patients with suspected 5-FU-related cardiotoxicity, ergonovine provocation has failed to induce coronary artery vasospasm (Freeman and Constanza 1988). Echocardiography has demonstrated a reduced ejection fraction and global akinesia of the left ventricular myocardium during attacks, which did not correspond to the segmental distribution of the major coronary arteries (de Forni et al., 1992). Vasodilator drugs are also not consistently protective (Patel et al., 1987; Oleksowicz and Bruckner 1988; Eskilsson and Albertsson 1990; Cwikiel et al., 1996; Akpek and Hartshorn 1999; Saif and Shah 2009).
Therefore, other pathophysiologic mechanisms probably contribute, including myocarditis (Tsibiribi et al., 2006), a direct myocardial toxic effect secondary to the antimetabolite effects of the drug causing a cardiomyopathic picture (Patel et al., 1987; Jensen et al., 2010), or a thrombogenic effect due to endothelial injury (Sasson et al., 1994; Cwikiel et al., 1996; Kuropkat et al., 1999; Jensen et al., 2012). As 5-FU is rapidly cleared from the bloodstream after bolus administration with a half-life of 15 to 20 minutes, a direct effect of the drug seems unlikely to be the cause of cardiotoxicity. There is also a higher incidence of angina with administration through continuous infusion compared to bolus infusion (Sudhoff et al., 2004; Saif and Shah 2009).
Of note, the metabolite of 5-FU, alpha-fluoro-beta-alanine (FBAL), is further catabolized into fluoroacetate, which is known to be highly cardiotoxic (Arellano et al., 1998; Muneoka et al., 2005). The lack of reported cardiac toxicity from fluoropyrimidines administered with the dihydropyrimidine dehydrogenase (DPD) enzyme inhibitors eniluracil and gimeracil lends further support to the theory that metabolic pathways leading to FBAL generation may be a significant pathophysiologic component of cardiotoxicity (Marsh et al., 2002; Guo et al, 2003; Yip et al., 2003).
5-FU administration can evoke a Takotsubo type of cardiomyopathy, a transient regional myocardial dysfunction that is precipitated by physical or emotional stress and thought to be related to exaggerated sympathetic stimulation (Stewart and Pavlakis 2010; Basselin et al., 2011; Dechant et al., 2012; Grunwald et al., 2012). The ECGs of patients with presumed Takotsubo cardiomyopathy often reveal ST-segment elevation, and cardiac enzymes are frequently mildly elevated, with a characteristic pattern of left ventricular dysfunction of non-segmental distribution. Finally, individual sensitivity to cardiotoxicity might result from inherited variations in the catabolic enzyme pathways responsible for the metabolism of 5-FU, leading to variable levels of cardiotoxic degradation products.
There remains a pressing need to discover novel and effective therapies that can prevent or ameliorate 5-FU associated cardiotoxicity. In this regard, remote ischemic conditioning (RIC), in which transient episodes of limb ischemia and reperfusion (induced by inflations and deflations of a pneumatic cuff placed on the upper arm or thigh), confer both cardioprotective and vasculoprotective effects, and may therefore prevent 5-FU coronary artery spasm/cardiotoxicity (see Figure 1) (Przyklenk et al., 1993; Chong et al., 2017; Chong et al., 2019).
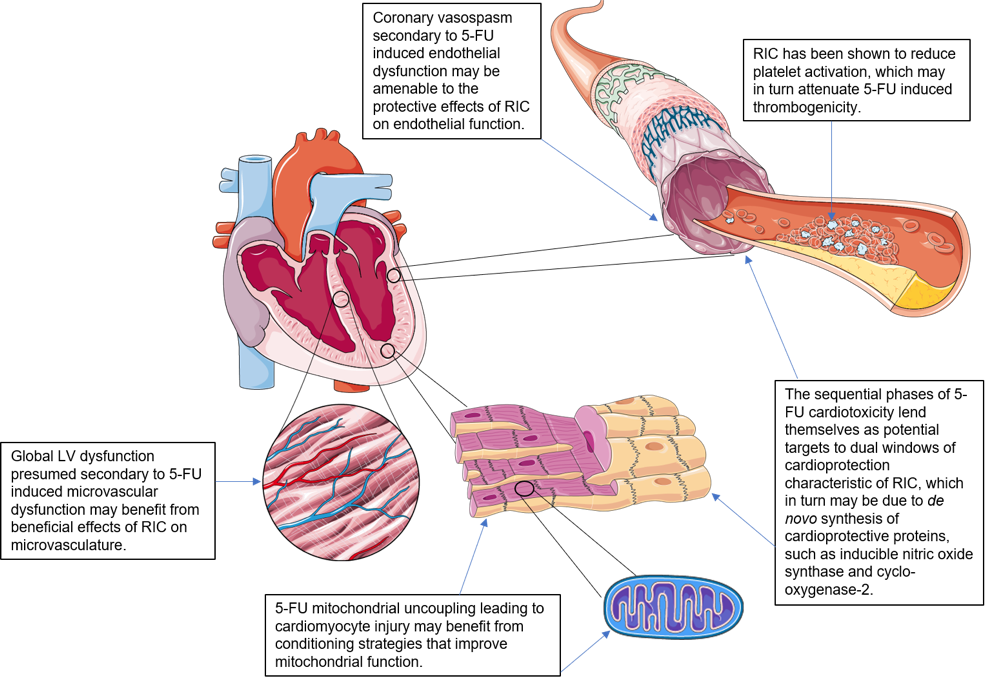
In a new window | Download PPT
Figure 1: Potential cellular targets in 5-fluorouracil cardiotoxicity amenable to RIC cardioprotection.
*Image components courtesy of Servier Medical Art. Figure 1 (with annotations) is original work by first author with use of said image components.
Potential cellular targets in 5-fluorouracil cardiotoxicity amenable to RIC cardioprotection
1. Two phases of 5-FU cardiotoxicity amenable to two windows of RIC cardioprotection
While coronary vasoconstriction may be observed in patients during or immediately after 5-FU injection, clinical features of toxicity do not typically manifest until after the end of an infusion or even hours to days later (Grem 2000; Jensen et al., 2010, Basselin et al., 2011). 5-FU is also rapidly cleared from the bloodstream after bolus administration with a half-life of 15 to 20 minutes, therefore a direct effect of the drug seems unlikely to be the only cause of cardiotoxicity. There is also a higher incidence of angina with administration through continuous infusion compared to bolus infusion (Sudhoff et al., 2004; Saif and Shah 2009). The delayed phase of 5-FU cardiotoxicity could be explained in part by the generation of toxic breakdown products of 5-FU. FBAL (a metabolite of 5-FU) is further catabolized into highly-cardiotoxic fluoroacetate. Metabolic pathways leading to FBAL generation may be a significant pathophysiologic component of 5-FU cardiotoxicity (Marsh et al., 2002; Guo et al, 2003; Yip et al., 2003).
Gross evidence of myocarditis has been demonstrated in rabbits exposed to 5-FU (Becker et al., 1999) where left ventricular hypertrophy, myocardial necrosis, thickening of intramyocardial arterioles, and disseminated apoptosis in myocardial and endothelial cells have been demonstrated. The use of a high single dose of 5-FU in this study was intended to differentiate the acute toxic effects of 5-FU, which resulted in thrombogenesis and spasm due to endothelial lesions, from delayed cardiotoxicity after four injections at 7-day intervals, which lead to apoptosis of myocardial and endothelial cells without evidence of spasm. These results also support an alternative mechanism for 5-FU cardiotoxicity beyond vasospasm and ischemia.
The sequential phases of 5-FU cardiotoxicity lend themselves as potential targets to dual windows of cardioprotection characteristic of RIC. The protective effects of RIC on endothelial function do not display tachyphylaxis, suggesting that RIC may confer long-term cytoprotective effects against acute ischemia and reperfusion. It is currently thought that a single limb RIC stimulus confers 2 windows of protection, the first occurring immediately and lasting 2-3 hours, and the second window of preconditioning (SWOP), appearing 12-24 hours later and lasting 2-3 days (Hausenloy and Yellon, 2010). RIC appears to extend the window of protection to 8 days. The SWOP may be due to de novo synthesis of cardioprotective proteins, such as inducible nitric oxide synthase and cyclo-oxygenase-2 (Hausenloy and Yellon, 2010). This memory effect could be explained by epigenetic changes in the vasculature, which can extend the protective effect beyond conventional SWOP.
2. Endothelial and vascular smooth muscle dysfunction and the potential role of RIC
Immediately following the intravenous administration of 5-FU, coronary artery vasospasm has been directly visualized during coronary angiography (Heistad et al., 1984; Lopez et al., 1989; Luwaert et al., 1991), as has brachial artery vasoconstriction (38,85). Arterial vasospasm can be related to endothelial dysfunction (an endothelial-dependent mechanism) or primary vascular smooth muscle dysfunction (an endothelial-independent mechanism) (Sara et al., 2018). Endothelial dysfunction is the reaction of the vasculature to a range of insults and clinical circumstances (Reddy et al., 1994; Bonetti et al., 2003), and represents the initial stage of atherosclerosis. It is characterized by an abnormal vasodilatory response to increased flow/shear stress or endothelial-dependent vasodilating agents such as acetylcholine (Vita et al., 1990; Hasdai et al., 1996; Suwaidi et al., 2000; Schwartz et al., 2010). In normal physiology, acetylcholine induces vasodilation through the release of nitric oxide from endothelial cells, which induces vascular muscle cell relaxation, and in turn vessel dilatation through the cyclic-guanosine monophosphate (cGMP) pathway (Hasdai et al., 1997). Any damage to endothelial cells disrupts this process and upon acetylcholine administration, paradoxical vasoconstriction occurs instead (Sara et al., 2018). In the coronary arteries, endothelial function is assessed by invasive pharmacologic provocation during coronary angiography with excessive vasoconstriction representing endothelial dysfunction (Hasdai et al., 1998; Pyke and Tschakovsky 2005; Dalzell and Samuel 2009; Schwartz et al., 2010). Endothelial-independent vascular smooth muscle dysfunction leads to vasoconstriction in the presence of a functionally intact endothelium, and can also be assessed with invasive pharmacologic provocation using nitroglycerin (Hasdai et al., 1998).
RIC has been demonstrated to ameliorate endothelial dysfunction. The mechanisms responsible for these vascular effects are unclear but may relate to shear stress adaptations, augmentation of endothelium-dependent vasodilation and production of nitric oxide (Kimura et al., 2007), circulation of vasoactive mediators such as nitric oxide (Kimura et al., 2007), and systemic antioxidant and anti-inflammatory effects. Kharbanda et al (2002) reported that RIC in one arm attenuated the endothelial dysfunction (assessed by flow-mediated dilatation) induced by a sustained episode of limb ischemia and reperfusion in the contralateral arm. Luca et al (2013) demonstrated improved endothelial function following acute ischemia and reperfusion in healthy volunteers that received 7 days of daily RIC. In the study by Pryds et al (2017), RIC applied daily for 28 days in chronic ischemic heart failure patients was shown to improve global longitudinal strain (GLS) in patients with the highest NT-proBNP plasma levels and lowered systolic blood pressure.
The beneficial effects of RIC on both NT-proBNP and GLS may relate to less myocardial wall stress, caused by reduction in afterload (as evidenced by lowered systemic blood pressure), and this may due to the release of known vasodilatory mediators of RIC such as adenosine and nitric oxide (Pryds et al., 2017). These beneficial vasodilatory effects could be conferred on patients receiving 5-FU through RIC administration peri- and during chemotherapy. Indeed, RIC activation of pathways producing vasodilatory mediators such as adenosine and nitric oxide can potentially bypass/override the deleterious effect of dysfunctional endothelium in vasoconstriction by delivering vasodilatory mediators direct to vessel smooth muscle lining, facilitating cGMP-mediated muscle relaxation, and vessel dilatation.
3. Dysfunctional coronary microvasculature with vasospasm and the potential role of RIC
Echocardiography has demonstrated global akinesia of the left ventricular myocardium in 5-FU associated cardiotoxicity not corresponding to segmental myocardial distribution of the major coronary arteries (de Forni et al., 1992). The discordance between echocardiographic and angiographic findings could undermine the epicardial arterial vasospasm theory in patients receiving 5-FU. However, microvascular vasospasm could be postulated to explain global, non-segmental akinesia. Endothelial-dependent and endothelial-independent dysfunction also affects the coronary microvasculature, often in the absence of affecting the epicardial vessels (Kinhult et al., 2001) where it leads to global versus segmental ischemia. Since the coronary microvasculature cannot be directly visualized, its function is assessed through measurements of coronary blood velocity and flow with intravascular Doppler techniques and also with pharmacologic provocation at coronary angiography (Hasdai et al., 1998).
Beneficial effects on vascular and endothelial function have been reported in the brachial artery and forearm microcirculation in healthy volunteers following daily RIC for 7 days, suggesting vascular effects of RIC which extend from conduit arteries to the skin microvasculature (Jones et al., 2014). It would be of clinical interest to explore if these beneficial effects can also be conferred onto coronary microvasculature, particularly in the case of diffuse myocardial ischemia induced by 5-FU administration. RIC-induced upregulation of vasodilatory mediators would be expected to enhance vascular smooth muscle relaxation and vessel dilatation at the microvascular level in 5-FU associated coronary arterial dysfunction. Interestingly, the vascular effects induced by RIC were shown to still be present 8 days after the end of the intervention, suggesting a ‘memory’ effect (Jones et al., 2014) that extends beyond the usual 2-3 days observed with the SWOP in terms of its cardioprotective effect (Marber et al., 1993).
4. 5-FU induced thrombogenicity and role of RIC induced fibrinolysis
Damaged endothelium exposes tissue factors, initiating platelet aggregation that is further propagated by the release of von Willebrand factor and fibrin aggregation, resulting in thrombi. 5-FU may lead to thrombotic occlusive disease, and studies of rabbit endothelium exposed to 5-FU have shown areas of platelet aggregation and fibrin formation (Yudkin et al., 1999; Jensen et al., 2012). Regulating the initiation of thrombus formation is an additional aspect of endothelial function and studies have characterized abnormal endothelial function by identifying altered levels of endothelium-derived markers such as von Willebrand factor and fibronectin (Spasojevic et al., 2005; 2008), suggesting a role of endothelium-associated thrombogenicity in 5-FU cardiotoxicity.
Pryds et al (2017) have investigated the effect of RIC applied daily on platelet function in chronic ischemic heart failure patients. RIC was shown to have no effect on platelet aggregation in response to collagen or arachidonic acid in heart failure patients or platelet turnover, which differs from the effects of a single limb RIC stimulus which was reported to reduce platelet activation (Lanza et al., 2016; Pedersen et al., 2011). However, RIC did increase fibrinolysis in both heart failure and control patients, suggesting it may reduce the risk of thrombosis (Pryds et al., 2017). This effect may be of benefit in 5-FU associated thrombogenicity.
5. Direct myocardial cellular damage by 5-FU and the potential role of RIC on mitochondria
Direct cardiomyocyte and vascular cell damage could also contribute to 5-FU-induced cardiotoxicity. Animal studies have demonstrated dose-dependent pathological changes to cardiomyocytes (Dickson et al., 1999) and endothelial cells (Lamberti et al., 2012), which could be a representation of the initial insult and subsequent ‘reaction to injury’ that leads to endothelial dysfunction in response to 5-FU. These changes are thought to be caused by induction of apoptosis with an absence of necrosis as opposed to that seen with direct cytotoxicity (Matsubara et al., 1980), as is the mechanism in neoplastic cells. Other animal models have demonstrated specific biochemical changes in cardiomyocytes, including increased oxygen consumption, depletion of high-energy phosphate compounds, and citrate accumulation (Tamatsu et al., 1984; Millart et al., 1992; Durak et al., 2000) occurring independently of changes in blood and oxygen supply. This is thought to be secondary to reduced aerobic efficiency caused by 5-FU-related mitochondrial uncoupling (Tamatsu et al., 1984), which in turn leads to hypoxic cell injury.
Characteristics of mitochondrial dysfunction include changes in the mitochondrial membrane potential, a reduction in the adenosine triphosphate (ATP) level and the inhibition of mitochondrial oxygen consumption (Pieczenik and Neustadt 2007). Excessive formation of reactive oxygen species (ROS) contributes to mitochondrial dysfunction (Litvinova et al., 2015). In particular, superoxide anion generated by the mitochondria, namely by complexes I and III of the electron transport chain (ETC), is the precursor of most ROS and a mediator in oxidative chain reactions (Litvinova et al., 2015). Dismutation of superoxide produces hydrogen peroxide, which in turn may be partially reduced to hydroxyl radicals, causing more damage to various mitochondrial and cellular components (Turrens 2003). Free radical damage to mitochondria may lead to decreased affinity of mitochondrial proteins for substrates or coenzymes (Liu et al., 2003).
Previous studies have suggested an association of RIC with improved mitochondrial function (Kleinbongard et al., 2018). In mitochondria of isolated perfused rat hearts after RIC in vivo, there was preserved mitochondrial respiration after ischemia/reperfusion (Ferko et al., 2014). In mitochondria taken from atrial tissue of patients undergoing cardiac surgery with RIC, there was also preserved respiration when the atrial tissue was obtained after aortic cross-clamping, but not when obtained before aortic cross-clamping (Slagsvold et al., 2014a; 2014b). The different conditioning strategies including preconditioning, remote preconditioning, and postconditioning target mitochondria and can improve their function (Boenglet et al., 2018), with potential to ameliorate direct cellular toxicity secondary to mitochondrial dysfunction conferred by 5-FU.
Conclusion and future directions
There remains a pressing need to discover novel and effective therapies that can prevent or ameliorate 5-FU associated cardiotoxicity. The proposed mechanisms underlying cardiotoxicity induced by 5-FU are multifactorial and include vascular endothelial damage followed by thrombus formation, ischemia secondary to coronary artery vasospasm, direct toxicity on myocardium and thrombogenicity (Chong and Ghosh 2019). There is a promising overlap between proposed RIC cardioprotective mechanisms with 5-FU-associated cardiotoxic cellular pathways. Therefore, further studies are needed to investigate the therapeutic potential of RIC for preventing 5-FU coronary artery spasm/cardiotoxicity.
Funding
Jun Chong is supported by the Singapore Ministry of Health’s National Medical Research Council under its Collaborative Centre Grant scheme seed fund (NMRC/CGAug16C006) and Research Training Fellowship. Jonathan Yap is supported by National Institutes of Health grant F31 HL139082. Derek Hausenloy was supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
Conflict of interests
None.
References
Jun Chong1,2,3
1Barts Heart Centre, St Bartholomew’s Hospital, London, United Kingdom. 2National Heart Centre Singapore, Singapore. 3Cardiovascular and Metabolic Disorders Program, Duke-National University of Singapore, Singapore.
Andrew FW Ho3,4,5
3Cardiovascular and Metabolic Disorders Program, Duke-National University of Singapore, Singapore. 4National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 5Department of Emergency Medicine, Singapore General Hospital.
Jonathan Yap6
6Center for Cardiovascular Research, John A. Burns School of Medicine, University of Hawaii, USA.
Heerajnarain Bulluck7
7Norfolk and Norwich University Hospital, Norwich, UK.
Derek J Hausenloy2,3,8-11
2National Heart Centre Singapore, Singapore. 3Cardiovascular and Metabolic Disorders Program, Duke-National University of Singapore, Singapore. 8The National Institute of Health Research University College London Hospitals Biomedical Research Centre, London, United Kingdom. 9Yong Loo Lin School of Medicine, National University Singapore, Singapore. 10The Hatter Cardiovascular Institute, University College London, London, UK. 11Tecnologico de Monterrey, Centro de Biotecnologia-FEMSA, Nuevo Leon, Mexico.
Corresponding author:
Dr. Jun Hua Chong
Email: jcho5751@hotmail.com
In a new window | Download PPT
Figure 1: Potential cellular targets in 5-fluorouracil cardiotoxicity amenable to RIC cardioprotection.
*Image components courtesy of Servier Medical Art. Figure 1 (with annotations) is original work by first author with use of said image components.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11892 | 22 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA