Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Krüppel-like transcription factors in vascular biology and cerebrovascular diseases
Time:2019-11-02
Number:14780
Author Affiliations

Conditioning Medicine 2019. 2(5):192-203.
Abstract
Krüppel-like factors (KLFs) belong to the zinc finger family of transcription factors, which currently contain 18 known protein members. Increasing evidence has shown that KLFs play critical roles in many vascular biological processes including development, proliferation, migration, apoptosis, inflammation, and pluripotency. Also, KLFs have been implicated in regulating the pathogenesis of various cerebrovascular diseases, such as ischemic stroke. In this review, we summarize the important roles of this transcription factor family in vascular biology and cerebrovascular diseases.
Abstract
Krüppel-like factors (KLFs) belong to the zinc finger family of transcription factors, which currently contain 18 known protein members. Increasing evidence has shown that KLFs play critical roles in many vascular biological processes including development, proliferation, migration, apoptosis, inflammation, and pluripotency. Also, KLFs have been implicated in regulating the pathogenesis of various cerebrovascular diseases, such as ischemic stroke. In this review, we summarize the important roles of this transcription factor family in vascular biology and cerebrovascular diseases.
1. Introduction
Krüppel-like factors (KLFs) refer to members of the zinc finger family of transcription factors. To date, 18 mammalian KLF family members have been identified and classified as KLF1 through KLF18, corresponding to the approximate order in which the genes were described (Pearson et al., 2008; Pei and Grishin, 2013). The first mammalian Krüppel-like factor, KLF1 or EKLF (Erythroid Krüppel-like Factor), was originally cloned in red blood cells, which is critical in β-globin gene formation and erythrPearsonocyte growth (Miller and Bieker, 1993). The newest member added to the KLF family, KLF18, was identified by using similarity searches and gene synteny analysis (Pei and Grishin, 2013). The KLF18 gene is located in the chromosome neighboring KLF17 and is probably the consequence of duplication of the KLF17 gene (Pei and Grishin, 2013). It is estimated that the KLF18 protein is conserved across many species, as are other KLF family members, although there is no reported expression data for KLF18 at present, which likely suggest that it either has severely constricted expression patterns or might have turned into a meaningless gene in surviving placental mammals. The nomenclature of KLFs is derived from chromatin, which is homologous with the DNA-binding domain of the Drosophila melanogaster krüppel protein. A deficiency in this protein in Drosophila embryos can lead to abnormal thoracic and abdominal segmentation. Interestingly, krüppel is a German word meaning “cripple” (Nusslein-Volhard and Wieschaus, 1980). Besides, KLF proteins contain zinc-finger structures and share homology with Sp1 (stimulatory protein 1), the first identified and characterized mammalian transcription factor, and are classified as part of the Sp1/KLF family (Kadonaga et al., 1987).
In recent decades, focus has been on KLF family members because of their involvement in cell differentiation, proliferation, and development in many systems or organs (Kaczynski et al., 2003). Numerous studies have reported that several KLFs (KLF2, KLF4, KLF5, KLF6, and KLF11) are actively involved in regulating complex vascular processes such as development, differentiation, inflammation, thrombosis, angiogenesis, and atherosclerosis (Suzuki et al., 2005; Atkins and Jain, 2007; Lu et al., 2013; Yin et al., 2013; Novodvorsky and Chico, 2014). Cerebrovascular diseases remain a prominent cause of morbidity and mortality worldwide, which result when an area of the brain is affected by transient or permanent ischemia or bleeding, yet therapeutic intervention are limited (Mozaffarian et al., 2015). Cerebrovascular diseases include ischemic stroke, transient ischemic attack, and hemorrhagic stroke (Cai et al., 2016). The pathophysiology of cerebrovascular diseases is complicated and involves an abnormal interaction between the vessel wall and other components of the brain. Herein, this review focuses on the current role of KLF family members in vascular biology and their distinct involvement of the KLFs in the pathogenesis of various cerebrovascular diseases.
2. Krüppel-like transcription factors: Basic structure, Classification, and Function
As one type of DNA-binding transcription factor, KLFs belong to a subgroup of the zinc finger family, which have a conserved protein structure among primates and rodents. A recent study on the evolution of the KLF/Sp transcription factor family showed that the genesis of the KLF/Sp family even predates the separation of the Metazoan (Presnell et al., 2015). All KLF family members have three highly conserved zinc finger structures in the C-terminus and three zinc finger motifs that contain 81 amino acids in total (Suske et al., 2005). Furthermore, along with the heavy constraints of the three-finger units is the highly conserved interfinger domains that act as a single unit. These three fingers prefer to bind to GC-rich sites in the promoter region of target genes. DNA binding sites are similar among the KLF proteins due to the highly conserved zinc finger structure (Dang et al., 2000). It is worth noting that the zinc finger region of several KLFs may be significant for nuclear location in addition to its role in DNA binding (Shields and Yang, 1997; Pandya and Townes, 2002). In contrast to the very similar carboxyl-terminal ends, the amino-terminal regions of KLFs are highly divergent and contain trans-activation domains and/or trans-repression domains to allow KLFs to interact with different co-activators, co-repressors, and modifiers. The diversity of amino-terminal regions provides the functional versatility and specificity for different KLF family members.
KLF family members can be classified into several distinct groups based on the diversity of their amino-termini and differences in their transcription activities (Black et al., 2001; Pearson et al., 2008). Generally, by binding to different promoters and recruiting coregulators-individual KLF family members can either activate or repress transcriptional progress. Several KLF members are ubiquitously expressed, whereas other members are tissue-restricted, suggesting the possibility of both exclusive and redundant functions for each. The expression patterns of KLFs can change markedly during development (McConnell and Yang, 2010). The biological functions of KLFs are very extensive. KLF proteins play an important role in morphogenesis, some of them regulate mammalian cell physiology ubiquitously, others show cell-specific functional characteristics. It has been reported that these transcription factors have vital effects on complex cellular processes such as cell proliferation (Sun et al., 2001), differentiation (Hodge et al., 2006), metabolism (Prosdocimo et al., 2014), apoptosis (Huang et al., 2008), and pluripotency (Yamanaka, 2007). They are also involved in many physiological processes, including hematopoiesis (Nuez et al., 1995; Perkins et al., 1995), cardiac remodeling (Shindo et al., 2002), angiogenesis (Bhattacharya et al., 2005), gluconeogenesis (Gray et al., 2007), adipogenesis (Wu and Wang, 2013), neurite outgrowth and axon regeneration (Moore et al., 2011), and nerve regeneration (Wang et al., 2017). The biochemical mechanisms of KLFs involve binding with distinct interaction partners, such as histone acetyltransferases, C-terminal binding protein, and SIN3 transcription regulator family member A. These co-regulatory proteins then modify the transcriptional activity of KLF family members by means of phosphorylation, acetylation, ubiquitination, and SUMOylation (McConnell and Yang, 2010).
Cumulative studies have demonstrated that KLFs participate in various human diseases and their dysregulation contributes to the pathogenesis of many organic lesions. It has been shown that KLFs are involved in many human diseases related to abnormal growth and differentiation (Kaczynski et al., 2003). For example, in cardiovascular diseases, KLFs are important modulators of cardiac fibrosis, cardiac hypertrophy, arrhythmogenesis, thrombosis, restenosis, and atherosclerosis (McConnell and Yang, 2010; Prosdocimo et al., 2014; Prosdocimo et al., 2015). KLFs also play critical roles in several pathological processes in cancer, including epithelial-mesenchymal transition (EMT), invasion, and metastasis (Tetreault et al., 2013; Limame et al., 2014). In addition, several KLF family members are involved in maintaining glucose homeostasis; therefore they could be targeted to treat type 2 diabetes (T2D) (Gutierrez-Aguilar et al., 2007). In particular, mutations in the KLF11 gene lead to maturity onset diabetes of the young type 7 (Neve et al., 2005). Accumulative numbers of publications have also demonstrated their contributions to several other human diseases such as neurodegenerative diseases, inflammatory conditions and so on (McConnell and Yang, 2010; Yin et al., 2013; Yin et al., 2015). Taken together, KLF transcription factors exhibit crucial effects on virtually all systems or organs in the human body. This review will address the significant role of KLFs in modulating vascular wall biology and discuss the prospective therapeutic effects of KLFs in cerebrovascular diseases. (Kaczynski et al., 2003).
3. Expression, physiological function and pathophysiology of KLFs in the vasculature
The vasculature or vascular wall, which is mainly composed of vascular endothelial cells (ECs) and smooth muscle cells (SMCs), is critically involved in the biological response to injury or inflammation. Vascular wall cells play an important role in preserving the structure and function of blood vessels under normal and injury conditions (Black et al., 2001). If vascular homeostasis is broken in response to trauma or inflammation, it will show morphological and functional changes including narrowing, regeneration, stiffening, blockage, and even expansion and rupture, which directly leads to many vessel-related diseases such as atherosclerosis, restenosis, angiogenesis, hypertension, thrombosis, and aneurysms.
KLFs are selectively located in various vascular cells (Figure 1) and altered expression of KLFs is involved in the regulation of vascular-related diseases. As transcription factors, KLFs trans-activate or trans-repress downstream target genes to influence vascular disease-related signaling pathways and thus change the characteristic morphology and function of vascular ECs, vascular smooth muscle cells (VSMCs) and other vascular cells.
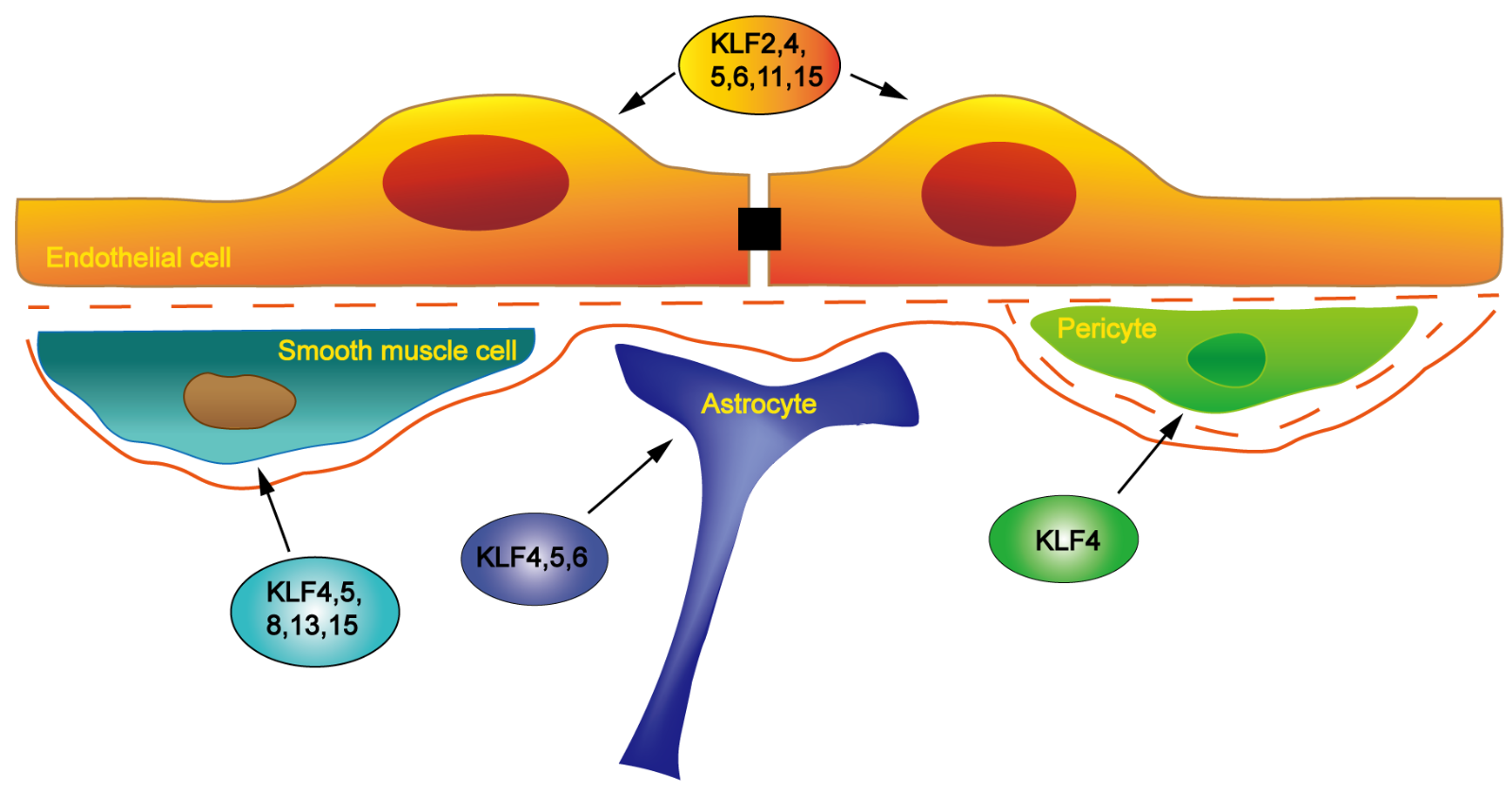
In a new window | Download PPT
Figure 1: Schematic representation of the expression of KLF family members in different vascular cells. KLF2, KLF4, KLF5, KLF6, KLF11, and KLF15 have been shown to participate in the regulation of EC structure and function. KLF4, KLF5, KLF8, KLF13, and KLF15 have been implicated in the regulation of VSMC growth, differentiation, and phenotype-related gene expression. KLF4 is found in pericytes. KLF4, KLF5, and KLF6 are distributed in astrocytes and their expression can be induced in reactive astrocytes in a mouse experimental stroke model.
3.1 KLFs in endothelial cell biology and pathologies
ECs that line blood vessels can integrate and transmit physiological external stimuli. They are needed to regulate many important physiological processes, including selective permeability of the blood-brain barrier (BBB), protective blood coagulation, and homing of immune cells (Gimbrone et al., 2000). The critical role of ECs in the formation and function of the BBB has recently garnered more attention. The barrier, which is mainly composed of capillary ECs, astrocytic perivascular end-feet, and basal lamina, has basic roles in the brain, such as supplying essential nutrients and mediating discharge of waste products (Abbott, 2005). By regulating ion transporters and channels, the BBB limits specific ionic and fluid exchanges between the brain and blood to produce an optimal brain medium for maintaining neuronal function (Abbott, 2004).
In some pathological conditions, barrier function is damaged. For example, several disorders appear to be related to the disturbance of endothelial-glia interaction. In cerebral ischemia, it has been reported that qualitative increases in transport across the barrier occurs (Pluta et al., 1991). The dysfunction of the endothelial cell is critical to many vascular diseases including atherosclerosis and thrombosis not only in experimental observations but also in clinical pathology (Libby et al., 2002).
There have been a series of studies implicating KLFs in regulating endothelial structure and function. Up to now, at least 6 KLF family members, KLF2, KLF4 ,KLF5, KLF6, KLF11 and KLF15, have been shown to participate in the regulation of ECs (Kuo et al., 1997; Yet et al., 1998; Kojima et al., 2000; Botella et al., 2002; Helbing et al., 2010; Yin et al., 2013). KLF2 is highly expressed in lung tissue and was initially termed lung krüppel-like factor. As a transcriptional regulator, KLF2 inhibits NF-κB, which is implicated in endothelial proinflammatory pathways (SenBanerjee et al., 2004). Besides the NF-κB pathway, two other important proinflammatory transcription factors cyclic-AMP-dependent transcription factor-1 (ATF1) (Fledderus et al., 2007) and C-Jun N-terminal kinase (JNK) (Boon et al., 2010) can also be regulated by KLF2. Thus, KLF2 decreases proinflammatory activity and can further prevent immune cell attachment and rolling in monolayer vascular ECs (SenBanerjee et al., 2004; Lin et al., 2006). In contrast, KLF2 recruits p300 away from p65 to increase the expression of endothelial nitric oxide synthase (eNOS), which is an important endothelial anti-inflammatory molecule (Dekker et al., 2006). Flavonoids, a class of low-molecular-weight polyphenols, regulate the expression of eNOS through the transcription factor KLF2 (Martinez-Fernandez et al., 2015). KLF2 also has effective antiangiogenic ability. The molecular mechanism of this effect has been partly attributed to the capacity of KLF2 to bind to the vascular endothelial growth factor receptor 2 (VEGFR2) promoter and to compete with SP1 to inhibit the expression of VEGFR2 (Bhattacharya et al., 2005). Interestingly, high concentration of uric acid (UA) inhibits angiogenesis induced by VEGF-A expression, which is down-regulated by KLF2 in cultured ECs (Yu et al., 2015). Recently, a class of small non-coding RNAs, called microRNAs, has been reported to be involved in the EC biology. Indeed, miR-126 (microRNA-126), an EC-specific microRNA, regulates endothelial cell proliferation through VEGF-dependent angiogenesis (Fish et al., 2008). Of note, the transcription of miR-126 is regulated by KLF2 and this microRNA was reported to promote flow-induced angiogenesis by promoting the spred-1 and PI3KR2/p85 pathways, which are down regulators of VEGF signaling (Fish et al., 2008; Wang et al., 2008). It has been concluded that KLF2 is a regulator of endothelial activation in response to inflammatory stimuli.
KLF4, also known as gut-enriched krüppel-like factor, and epithelial zinc-finger protein, was originally identified in gut and skin epithelium (Garrett-Sinha et al., 1996; Shields et al., 1996). Similar to KLF2, KLF4 is expressed in endothelial cells and is induced by shear stress (Hamik et al., 2007). ECs display another protective phenotype by increasing expression of KLF4 (Yoshida et al., 2008). It has been reported that KLF4 is an important down-regulator of the Wnt signaling pathway (Evans et al., 2010). Through this pathway, KLF4 regulates vascular endothelial (VE)-cadherin expression, which is a crucial determinant of endothelial barrier function (Cowan et al., 2010). Thus, KLF4 may function to maintain normal adherens junctions (AJs) of developing blood vessels. During cardiac ischemia, KLF4 makes the AJ barrier more resistance to inflammatory stimuli and serves to prevent vascular leakage (Cowan et al., 2010). By reducing KLF4 levels and functional KLF4-histone deacetylase (HDAC) association, the transcription of VEGF is inadequately suppressed. The KLF4-HDAC complex may act as a molecular switch to regulate VEGF expression dynamically and may be involve in the increase in VEGF and VEGF-mediated angiogenesis (Ray et al., 2013). KLF4 inhibits endothelial proliferation and angiogenesis by increasing microRNA-15a (miR-15a) in ECs (Zheng et al., 2013). In ECs, drugs that protect the vascular, such as statins, elevate vascular protection-related mRNA levels of eNOS and thrombomodulin through transcriptional regulation of KLF2 and KLF4 (Sen-Banerjee et al., 2005).
Endothelial KLF5 is upregulated by insulin, which mediates the activation of mammalian target of rapamycin (mTOR), oxidative stress, and protein kinase C (PKC) pathways. KLF5 overexpression has been shown to seriously damage insulin-regulated ECs migration and blood vessel formation. Dysfunction of endothelial KLF5 is involved in vascular dysfunction in T2D partly through this signaling pathway (Caradu et al., 2018).
According to its special structure, KLF6 is also called GC-rich sites-binding factor, or Zf9. KLF6, which is expressed by vascular ECs, can promote the transactivation of urokinase type plasminogen activator (uPA), which leads to activation of transforming growth factor β (TGF-β). TGF-β is implicated in the regulation of EC quiescence. It is suggested that TGF-β plays an important role in vascular injury response, including atherosclerosis and restenosis (Grainger et al., 1994; Ross, 1999). The interaction between KLF6 and Sp1 or Sp2 is necessary for the activation and migration of ECs (Garrido-Martin et al., 2013).
Cloned as a Sp1-like transcription factor, KLF11 is highly expressed in ECs. By direct binding to NF-κB, KLF11 works as a potent suppressor of endothelial inflammatory activation by down-regulating NF-κB (Fan et al., 2012; Glineur et al., 2013). Our previous research found that KLF11 deficiency aggravates ischemic stroke in a mouse middle cerebral artery occlusion (MCAO) model (Tang et al., 2018). KLF11 accelerates peroxisome proliferator-activated receptor-gamma (PPARγ)-mediated inhibition of pro-apoptotic miR-15a in cerebral vascular ECs (Yin et al., 2013). In addition it has been shown that miR-15a/16-1 inhibition could improve ischemic heart and brain injury (Hullinger et al., 2012; Yang et al., 2017). Also, KLF11 is involved in cholesterol metabolism by arresting Sp1/sterol-responsive element-binding protein-depended caveolin-1 transcription in ECs (Cao et al., 2005).
KLF15 and bone morphogenetic protein binding endothelial regulator (BMPER) are inhibited by endothelin-1 in ECs, indicating that KLF15 is involved in regulating the function of endothelin-1 in blood vessel formation as a trans-regulator of BMPER, which is a key modulator of BMP signaling (Helbing et al., 2010).
3.2 KLFs in smooth muscle biology and pathologies
Besides ECs, VSMCs are another important components of the blood vessel wall. Indeed, the primary physiological function of VSMCs is to regulate vessel tone by contraction and relaxation. Mature VSMCs exist in a quiescent condition, which have a differentiated, contractile phenotype and a low rate of proliferation (Owens et al., 2004). In response to vascular injury, VSMCs change from quiescent and differentiated cells to dedifferentiated, proliferated, migrated and re-expressed cells (Ross, 1993, 1999; Owens et al., 2004; Pislaru and Simari, 2005). This feature of VSMCs causes the formation and development of vascular occlusive diseases such as atherosclerosis, restenosis, and transplant arteriopathy in humans (Ferns et al., 1991). Up to now, the molecular mechanisms that regulate the VSMC phenotype are poorly explored and need to be further elucidated (Wei et al., 2005). Among all KLF members, KLF4, KLF5, KLF8, KLF13, and KLF15 have been implicated in the regulation of VSMC growth, differentiation, and phenotype-related gene expression (Adam et al., 2000; Gray et al., 2002; Shindo et al., 2002; Martin et al., 2003; Ha et al., 2017).
KLF4 expression is increased from baseline to a high level within 1-2 h and returns to normal after 24 h in mice models of vascular injury (Liu et al., 2005). The TGF-β control element (TCE) of KLF4 is the binding site for the TGF-β-dependent factor in cultured VSMCs. The TCE-dependent VSMC differentiation may provide a molecular mechanism for VSMC phenotypic plasticity in response to vascular injury response (Owens, 1995). Moreover, potent growth factors of VSMCs such as platelet-derived growth factor-BB (PDGF-BB) and TGF-β1 can regulate KLF4 expression through transcription factor Sp1 to inhibit VSMC differentiation (King et al., 2003; Deaton et al., 2009). In addition, all-trans retinoic acid promotes VSMCs differentiation and inhibits their proliferation by downregulating the expression of KLF4 (Wang et al., 2015). In terms of mechanism, KLF4 represses the expression of myocardin, a critical transcriptional co-activator involved in VSMC differentiation (King et al., 2003), and mediates the elongation of long-chain fatty acid family member 6-induced VSMC phenotypic switch (Sunaga et al., 2016). Therefore, KLF4 is a critical promoter for VSMCs to swift from the contractile to the secretory phenotype. In contrast, TGF-β, a positive regulator of VSMCs differentiation from the secretory to the contractile phenotype, reduces KLF4 expression through inhibition of miR-143/145 (Davis-Dusenbery et al., 2011). Overexpression of miR-145 and the addition of klf4 siRNA significantly attenuated the VSMC proliferation induced by hyperglycemia (Shyu et al., 2015). Recent studies have also found that the smad and p38 MAPK pathways are also involved in KLF4 promotion of VSMC differentiation (Li et al., 2010).
KLF5 mediates the tumor necrosis factor α (TNF-α)-induced phenotypic conversion of VSMCs by transactivating VSMC differentiation marker genes such as the SM22α promoter (Adam et al., 2000). The Ras-MAPK pathway is implicated in the induction of KLF5, so inhibition of MAPK blocks TNF-α-induced transcriptional activity of KLF5 in VSMCs (Kim et al., 2015). KLF5 expression can be directly activated by TGF-β triggering the mTOR pathway, thus regulating the differentiation of SMCs (Zhu et al., 2017). The antiproliferative effect of rosiglitazone was attributed to a mechanism that reduces KLF5 expression and crosstalk between PPAR-γ and PKC/ERK/Egr in angiotensin II (Ang II)-stimulated VSMCs (Liu et al., 2010). KLF5 is also involved in the imbalance between proliferation and apoptosis of pulmonary artery SMCs and regulates the development of pulmonary arterial hypertension (Courboulin et al., 2011). Similar to KLF5, KLF13 (also known as BTEB 3) regulates the differentiation of smooth muscle by transactivating the minimal promoter for the SM22α gene (Martin et al., 2003).
Several studies have shown that the expression of KLF8 is significantly increased in the contractile phenotype of VSMCs, which is in contrast to KLF5. By regulating the transcriptional activity of KLF8, KLF4 suppresses the transcriptional activity of KLF5, thereby enhancing the contractile marker protein of VSMCs. The VSMC phenotype is orchestrated by the cross-regulation of KLF4, KLF8, and KLF5 (Ha et al., 2017).
KLF15 expression is robust under physiological conditions but strongly attenuated in response to pro-proliferative stimuli or vascular damage (Lu et al., 2010). In contrast to KLF4 and KLF5 in VSMCs, KLF15 inhibits proliferation in response to PDGF-BB in vitro. Genetic deletion of klf15 promoted VSMC proliferation and migration in cultured VSMCs and KLF15 knockout mice exhibited enhanced neointimal formation after vascular injury, resulting in serious arteriopathy (Lu et al., 2010), which is the major feature of aortic aneurysm formation after Ang II stimulation (Haldar et al., 2010). Indeed, KLF15 expression is significantly decreased in atherosclerotic tissue and KLF15 deficiency in vascular smooth muscle exacerbates inflammatory vasculopathy leading to atherosclerosis (Lu et al., 2013). KLF15 is also an important regulator of insulin-sensitive glucose transporter 4 expression (Gray et al., 2002), which is involved in VSMCs contraction (Banz et al., 1996).
3.3 KLFs in other vascular cell biology and pathologies
As important components of the blood barrier, pericytes and astrocytes sustain blood-brain barrier function by interacting with ECs and increasing the integrity of the tight junction (Lai and Kuo, 2005; Edelman et al., 2006; Bell et al., 2010) or preventing endothelial transcytosis (Armulik et al., 2010; Gursoy-Ozdemir et al., 2012). Some pericytes have the ability to contract like VSMCs after stroke, potentially leading to the “no-reflow” phenomenon after cerebral ischemia, in which blood cannot return to capillaries successfully even if recanalization of a large vessel is established in the brain (Hall et al., 2014). Because of their multipotential identity, pericytes have garnered more attention in the field of regenerative medicine. KLF4 is reported to be expressed in pericytes (Cantoni et al., 2015). Astrocytes also exacerbate “no-reflow” because it can rapidly shrink, compress the vessel lumen, and decrease blood reperfusion after stroke (Ito et al., 2013). It is unclear whether KLF members in pericytes or astrocytes play a role in the regulation of the no-reflow status.
The characteristic response of astrocytes to vascular injury is called stressful astrogliosis, which is involved in the pathogenesis of cerebral ischemia (Pekny and Nilsson, 2005). Some KLF transcription factors have been shown to participate in this pathological process. For example, KLF4 was increased in astroglia in response to ischemic brain injury. The involvement of KLF4 in the astroglia reaction after global cerebral ischemia was also investigated (Park et al., 2014). The differential genomic analysis revealed that KLF5 and KLF6 were induced in reactive astrocytes in the mouse MCAO model (Zamanian et al., 2012), indicating a possible role of KLF5 and KLF6 in the astroglia response to the injured brain.
4. KLFs and cerebrovascular diseases
Cerebrovascular diseases remain a major cause of long-term disability in the United States and 6.6 million Americans have had a stroke. It is estimated that the total direct stroke-related medical costs will increase from $71.6 billion to $184.1 billion between 2020 and 2030 (Writing Group et al., 2016). It is becoming apparent that KLF signaling pathways play a central regulatory role in the pathogenesis of cerebrovascular diseases (Figure 2). Therefore, KLF transcription factors may become novel candidates for therapeutic drug discovery against cerebrovascular diseases (Shi et al., 2013; Yin et al., 2013; Yin et al., 2015; Tang et al., 2018).

In a new window | Download PPT
Figure 2: Schematic representation of distinct roles of KLF transcription factors in the pathogenesis of various cerebrovascular diseases. KLF2 and KLF11 are important transcription factors offering neurovascular protection in ischemic stroke. KLF14 may regulate the formation or progression of ischemic stroke by influencing lipoprotein metabolism. Inhibition of KLF4, KLF5, or KLF6 activities in astrocytes can alleviate stressful astrogliosis after cerebral ischemia. KLF2 is related to SAH treatment. KLF2 and KLF4 have opposite regulatory effects on cerebral atherosclerosis in different kinds of cells. KLF14 and KLF15 have protective effects on cerebral atherosclerosis. KLF2 may inhibit the formation and development of cerebral aneurysms (CAs). KLF4 and KLF5 are highly expressed in CAs and may exacerbate the lesion of CAs. KLF2 and KLF4 may be involved in the regulation of cerebral amyloid angiopathies (CAAs). KLF2 and KLF4 are possibly involved in the vascular dementia (VaD) and its early manifestation vascular cognitive impairment (VCI). Inhibition of KLF2 and KLF4 related signaling pathways suggests a potential therapeutic approach to treat cerebral cavernous malformation (CCM) diseases. The green color indicates those KLFs that may have therapeutic effects against cerebrovascular diseases, the red color indicates those KLFs that may promote the pathogenesis of the diseases, and the yellow color indicates those KLFs with undetermined roles in the diseases.
4.1 KLFs and cerebral ischemia
Ischemic stroke accounts for more than 80% of cerebrovascular diseases and comprises a sequence of biochemical events that eventually result in neuronal death in the brain (Love, 2003). So far, different pathophysiological mechanisms such as oxidative stress and inflammation have been identified during ischemic stroke, but comprehensive understanding of ischemic stroke remains a daunting task (Pei et al., 2015). As previously mentioned, KLF members have been shown to be critical regulators in many human diseases. To date, numerous studies have shown that several KLF members (KLF2, KLF4, KLF5, KLF6, KLF11, and KLF14) play important roles in the pathogenesis of cerebral ischemia (Shi et al., 2013; Yin et al., 2013; Park et al., 2014; Yin et al., 2015; Tang et al., 2018).
Using Affymetrix GeneChip arrays to profile gene expression in populations of reactive astrocytes, it has been demonstrated that KLF5 and KLF6 are induced in fluorescence activated cell sorting-isolated reactive astrocytes in mouse brains following MCAO (Zamanian et al., 2012), suggesting a potential role of KLF5 and KLF6 in the regulation of the astroglial reaction in the cerebral ischemia. KLF4 expression is also specifically induced in astroglia by ischemic injury both in vivo and in vitro, implying that KLF4 functions as a transcriptional regulator following cerebral ischemia (Park et al., 2014). Therefore, inhibition of KLF4, KLF5, or KLF6 in astrocyte can alleviate stressful astrogliosis after cerebral ischemia (Yin et al., 2015).
It has been suggested that KLF2 is an important transcription factor in the protection against ischemic stroke. KLF2 inhibits thrombus formation by decreasing the expression of endothelial thrombotic factors (Nayak et al., 2014). Shi et al. (2013) found that BBB permeability and cerebral infarction were aggravated in the absence of the klf2 gene and were relieved via KLF2 overexpression, which is direct evidence of a protective role of KLF2 against cerebral ischemia (Shi et al., 2013). In addition, some clinical research found that the KLF2-VEGF pathway is involved in the therapeutic effects of UA in ischemic stroke patients with hypertension (Vila et al., 2019).
Our research team found for the first time that KLF11 is the direct transcriptional target of PPARγ, which is a known critical regulator in neurological diseases. KLF11 enhances PPARγ transcriptional suppression of the pro-apoptotic miR-15a, resulting in endothelial protection after ischemic stimuli in vivo and in vitro (Yin et al., 2013). These results provide novel insights about how PPARγ and KLF11 function in the cerebral vasculature and novel molecular mechanism of PPARγ-mediated vascular protection following cerebral ischemia. Recently, we also demonstrated that KLF11 expression is significantly decreased in the cerebral cortex of mice following focal cerebral ischemia. Of note, KLF11 genetic deficiency results in larger brain infarction, increased BBB permeability/leakage, higher water content, worsen neurobehavioral performance, and less cerebral blood flow perfusion in mouse ischemic brain regions after MCAO. Further, we have shown that KLF11 plays anti-inflammatory roles in mouse brains following ischemic stroke. These results suggest that KLF11 itself plays critical protective roles in ischemic stroke as well (Tang et al., 2018).
As a critical regulator of lipoprotein metabolism, KLF14 is implicated in the pathogenesis of atherosclerosis-related diseases such as ischemic stroke and myocardial infarction (Chen et al., 2012).
4.2 KLFs and intracerebral hemorrhage and subarachnoid hemorrhage
Approximate 80% of subarachnoid hemorrhage (SAH) is caused by the rupture of an intracranial aneurysm. Other causes of SAH include vascular malformation and vasculitis, which accounts for 5 to 10% of stroke patients in the United States (Johnston et al., 1998; Rincon et al., 2013). SAH affects younger patients, which results in a greater loss of productive life (Johnston et al., 1998). Of those patients that survive SAH, half of them suffer from long-term neuropsychological diseases leading to decreased quality of life (Taufique et al., 2016). The risk of recurrent bleeding is increased in patients with cerebral amyloid angiopathy or cerebral hemorrhage (van Etten et al., 2016). Scutellarin, a flavonoid extracted from the traditional Chinese herb Erigeron breviscapus, has been reported to ameliorate vasospasm associated with upregulation of KLF2-eNOS pathway in the rats SAH model (Li et al., 2016).
4.3 KLFs and cerebral atherosclerosis
The major pathological change in cerebral atherosclerosis is the activation and recruitment of monocytes/macrophages that respond to an excessive accumulation of modified lipids in the brain vessel wall, followed by an adaptive immune response from lymphocytes (Hansson, 2005). Interaction of blood leukocytes with ECs and VSMCs plays a prominent role in the pathogenesis of cerebral atherosclerosis and its complications (Levin and Santell, 1991). Cerebral atherosclerosis may result in thromboembolism with or without hypoperfusion, leading to transient or permanent cerebral ischemia. KLF2 and KLF4 can be pharmacologically manipulated to provide an antithrombotic effect without altering the risk of bleeding. Some pharmacological agents including resveratrol (Gracia-Sancho et al., 2010), statins (Parmar et al., 2005), and antimyeloma agent bortezomib (Nayak et al., 2014) have been reported to upregulate both KLF2 and KLF4, which provide a mechanistic basis for the previously observed thrombo-protective effect of these drugs (Gomez and Owens, 2012). By enhancing activities of KLF2 and KLF4, endothelial Grb2-associated binder 1 protects the vasculature against Ang II-dependent vascular inflammation and atherosclerosis in apolipoprotein E (ApoE) knockout mice with abnormal lipid metabolism (Higuchi et al., 2012). In contrast, VSMC-specific deletion of KLF4 could reduce atherosclerosis by reducing the number of VSMC-derived macrophages and increasing plaque stability compared to wild-type controls (Shankman et al., 2015). These results indicate that KLF2 and KLF4 represent opposite regulatory effects against cerebral atherosclerosis on different kinds of cells.
Although KLF14 has only recently been identified, it plays critical roles in the regulation of normal lipid metabolism processes, such as adipocyte differentiation and proliferation, lipid uptake and fat catabolism (Xie et al., 2017). Moreover, many risk factors of native atherosclerosis, including dysfunction in homeostatic regulation, inflammation (Wei et al., 2017), dysfunction (Sarmento et al., 2015), obesity (Anunciado-Koza et al., 2016), insulin resistance (Yang et al., 2015), diabetes (Huang et al., 2013) and others, have been reported to associate with altered KLF14 gene expression. TGF-β could reduce EC and VSMC proliferation in the earliest stages of atherogenesis and make plaques more stable (Redondo et al., 2007). TGF-β could induce the expression of KLF14, which trans-represses TGF-β receptor II expression in the transfected human pancreatic epithelial cancer cell line, PANC-1. It has also been reported that KLF14 regulate cell homeostasis in cerebral atherosclerosis (Truty et al., 2009). Similar to KLF14, KLF15 is decreased in human atherosclerotic lesion and VSMC-specific KLF15 gene deficiency aggravates the progression of atherosclerosis in APOE knockout mice by increasing proinflammatory activation of VSMCs (Lu et al., 2013), implying that KLF15 has a protective effect against cerebral atherosclerosis. These two members of the KLF family may become potential therapeutic targets for cerebral atherosclerosis.
4.4 KLFs and cerebral aneurysm
Cerebral aneurysms (CAs) occur in 3% to 5% of the general population with high mortality and morbidity (Brisman et al., 2006). The formation and growth of CAs include endothelial dysfunction induced by hemodynamic stress, followed by an inflammation reaction in blood vessels involving macrophages and VSMCs, and degradation of the extracellular matrix by matrix metalloproteinases (MMPs), which finally lead to aneurysm rupture (Chalouhi et al., 2013). Early diagnosis and treatment of the aneurysm can block its rupture and prevent sequelae of the initial rupture (Tanaka et al., 2013).
NF-κB induced by hemodynamic stress in ECs plays a prominent role in the formation of CAs. Inhibition of NF-κB in mice can dramatically block the formation of an aneurysm (Aoki et al., 2007). It has been demonstrated that KLF2 is a transcriptional inhibitor of NF-κB, and may inhibit the formation and development of CAs (SenBanerjee et al., 2004).
Some studies have also shown a relationship between KLF4 and cerebral aneurysm. For example, KLF4 is highly expressed in the vessel wall and VSMC-specific deletion of KLF4 effectively relieves aortic aneurysms in mice (Salmon et al., 2013). As a downstream target of TNF-α, KLF4 can inhibit the contractile phenotype of VSMCs and increase the expression of proinflammatory/matrix-remodeling genes, including MCP-1, MMPs, VCAM1, and IL-1β (Ali et al., 2013). These findings suggest that KLF4 plays an important role in regulating the pathogenesis of CAs.
It has been reported that KLF 5 is highly expressed in large or giant unruptured CAs in patient samples. The specific role and the underlying mechanism of KLF5 in CAs need to be further studied (Nakajima et al., 2012).
4.5 KLFs and cerebral amyloid angiopathy
The major pathological change in cerebral amyloid angiopathy (CAA) includes accumulation of β-amyloid in the vascular wall of cerebral arteries, arterioles, and capillaries. The deposited material is the breakdown product of amyloid precursor protein amyloid-beta (Aβ) fragments with different amino acid lengths, such as Aβ40 and Aβ42 (Revesz et al., 2009). Later, Aβ deposits in all layers of the vascular wall, leading to inflammatory responses and loss of VSMCs (Keable et al., 2016), resulting in fibrinoid necrosis and microaneurysm formation. Owing to increased vessel fragility and subsequent rupture, CAA has become the second leading cause of intracerebral hemorrhage (Yeh et al., 2014; Yamada, 2015). CAA is closely associated with dysfunction of ApoE (Verghese et al., 2011). Although there is no direct evidence for the role of KLFs in the regulation of CAA, the characteristics of the disease suggests that several KLFs, such as KLF2 and KLF4, may be involved.
4.6 KLFs and vascular dementia
Vascular dementia (VaD) is recognized as the second most common type of dementia, which can be attributed to global or focal effects of vascular brain diseases. VaD is generally considered the functional consequences of reduced blood flow to the brain (Esiri et al., 1997). Acutely or chronically, reduced blood flow would certainly jeopardize cognitive ability (Zhang et al., 2014; Venkat et al., 2015). White matter lesions or subcortical leukoencephalopathy with an incomplete brain infraction is a special pathological change associated with dementia (Deramecourt et al., 2012). Cerebrovascular pathological changes and ischemia hypoperfusion are often accompanied by BBB lesion, especially in the hippocampus (Montagne et al., 2015; Ueno et al., 2016). The BBB defends the brain environment against various pathogenic agents in the peripheral circulation, and its dysfunction may lead to the development of VaD (Srinivasan et al., 2016). The involvement of the BBB in VaD pathogenesis implies that functional restoration of the BBB should be considered as a therapeutic strategy.
KLF2 and KLF4 in cerebral ECs might play a pivotal role in the process of anti-inflammation, anti-apoptosis, axon regeneration, and BBB integrity, suggesting that KLFs may be involved in VaD and its early manifestation in vascular cognitive impairment (VCI) (Fang et al., 2017; Li et al., 2017).
4.7 KLFs and other cerebrovascular disorders
Cerebral cavernous malformation (CCM) is a common cerebrovascular disease that can occur sporadically or hereditarily. It is one of the major causes of stroke, cerebral hemorrhage, and neurological deficits in the early stage of life (Fischer et al., 2013). The major pathological characteristic of CCM lesions is the morphological destruction of ECs and disruptions of the EC-EC or EC-pericyte interaction (Choi et al., 2018).
Loss of function or mutations in the CCM1, CCM2, or CCM3 gene cause human familial CCM (Fischer et al., 2013). Numerous studies have shown that the CCM protein complex is a suppressor of the MEKK3 kinase cascade. Genetic deletion of CCM genes activates the MEKK3 kinase cascade and increases the expression of downstream genes such as KLF2/4 (Zhou et al., 2016). Cell division cycle 42 (Cdc 42) is also engaged in the CCM signaling network to restrain the MEKK3-MEK5-ERK5-KLF2/4 pathway (Castro et al., 2019). These results suggest that a potential therapeutic approach to treat CCM disease is to pharmacologically inhibit the MEKK3-KLFs signaling pathway.
5. Summary and future prospects
In this review, we summarize the important roles of KLF transcription factors in vascular cell biology and cerebrovascular diseases. Although conserved zinc finger domains dictate that KLF family members express proteins with similar structures, they vary in expression types in different tissues and exhibit significant pathobiological function in diverse cerebrovascular diseases. Considering all of the evidence, we have come to the conclusion that KLFs significantly influence the pathological process of cerebrovascular diseases by regulating the biological function of various vascular cells including ECs, VSMCs, pericyte, and astrocyte. In addition to KLF2 and KLF4, the newly discovered KLF11, KLF14, and KLF15 have gradually received more attention in the past 10 years, and have been the focus in the treatment of cerebrovascular diseases.
Recently, increasing pharmacologic tools have been developed to intervene in different KLF signaling pathways. Some pharmacological reagents including resveratrol (Gracia-Sancho et al., 2010), statins (Parmar et al., 2005), antimyeloma agent bortezomib (Nayak et al., 2014), UA (Vila et al., 2019), dietary flavonoids (Martinez-Fernandez et al., 2015), and scutellarin (Li et al., 2016) have been reported to regulate KLF2 or KLF4 signal pathways. Moreover, microRNA antagomirs or mimetics have also been used as molecular approaches to regulate individual KLF factors and their related signal cascades. For instance, KLF17 is post-transcriptionally inhibited by a microRNA-9 (miR-9) mimic in hepatocellular carcinoma (Sun et al., 2013). The expression of KLF12 is deceased by a miR-14-3p mimic and increased by a miR-14-3p antagomir in endometrial stromal cells (Zhang et al., 2019), and a miR-145 mimic is able to inhibit the expression of KLF4 in VSMCs (Shyu et al., 2015). In addition, the development of small-molecule chemical compounds and siRNAs targeting KLF family members has also recently become an important field of drug research (Flandez et al., 2008; Lyssiotis et al., 2009; Khedkar et al., 2015; Yu and Kim, 2018).
With the help of advanced techniques such as genetic manipulation, genomic editing, and proteomics, more comprehensive and in-depth understanding of KLFs’ roles in vascular cell biology and cerebrovascular diseases will be conducive to develop novel therapeutic approaches to effectively reduce the risk of cerebrovascular diseases, as well as improve prognosis and decrease complications.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Acknowledgments
This work was supported by the National Institutes of Health Grant NS086820.
References
Jing Zhang1
1Pittsburgh Institute of Brain Disorders & Recovery, Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Xuelian Tang1
1Pittsburgh Institute of Brain Disorders & Recovery, Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Xuejing Zhang1
1Pittsburgh Institute of Brain Disorders & Recovery, Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Chao Zhou1
1Pittsburgh Institute of Brain Disorders & Recovery, Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA.
Ke-Jie Yin1,2*
1Pittsburgh Institute of Brain Disorders & Recovery, Department of Neurology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA. 2Geriatric Research, Education and Clinical Center, Veterans Affairs Pittsburgh Healthcare System, Pittsburgh, PA, 15261, USA.
Corresponding author:
Dr. Ke-Jie Yin
Email: yink2@upmc.edu
In a new window | Download PPT
Figure 1: Schematic representation of the expression of KLF family members in different vascular cells. KLF2, KLF4, KLF5, KLF6, KLF11, and KLF15 have been shown to participate in the regulation of EC structure and function. KLF4, KLF5, KLF8, KLF13, and KLF15 have been implicated in the regulation of VSMC growth, differentiation, and phenotype-related gene expression. KLF4 is found in pericytes. KLF4, KLF5, and KLF6 are distributed in astrocytes and their expression can be induced in reactive astrocytes in a mouse experimental stroke model.
In a new window | Download PPT
Figure 2: Schematic representation of distinct roles of KLF transcription factors in the pathogenesis of various cerebrovascular diseases. KLF2 and KLF11 are important transcription factors offering neurovascular protection in ischemic stroke. KLF14 may regulate the formation or progression of ischemic stroke by influencing lipoprotein metabolism. Inhibition of KLF4, KLF5, or KLF6 activities in astrocytes can alleviate stressful astrogliosis after cerebral ischemia. KLF2 is related to SAH treatment. KLF2 and KLF4 have opposite regulatory effects on cerebral atherosclerosis in different kinds of cells. KLF14 and KLF15 have protective effects on cerebral atherosclerosis. KLF2 may inhibit the formation and development of cerebral aneurysms (CAs). KLF4 and KLF5 are highly expressed in CAs and may exacerbate the lesion of CAs. KLF2 and KLF4 may be involved in the regulation of cerebral amyloid angiopathies (CAAs). KLF2 and KLF4 are possibly involved in the vascular dementia (VaD) and its early manifestation vascular cognitive impairment (VCI). Inhibition of KLF2 and KLF4 related signaling pathways suggests a potential therapeutic approach to treat cerebral cavernous malformation (CCM) diseases. The green color indicates those KLFs that may have therapeutic effects against cerebrovascular diseases, the red color indicates those KLFs that may promote the pathogenesis of the diseases, and the yellow color indicates those KLFs with undetermined roles in the diseases.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14780 | 52 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA