Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Dietary Restriction and Epigenetics: Part I
Time:2020-01-06
Number:14312
Author Affiliations

Conditioning Medicine 2019. 2(6): 284-299.
Abstract
Biological aging occurs concomitantly with chronological aging and is commonly burdened by the development of age-related conditions, such as neurodegenerative, cardiovascular, and a myriad of metabolic diseases. With a current global shift in disease epidemiology associated with aging and the resultant social, economic, and healthcare burdens faced by many countries, the need to achieve successful aging has fueled efforts to address this problem. Aging is a complex biological phenomenon that has confounded much of the historical research effort to understand it, with still limited knowledge of the underlying molecular mechanisms. Interestingly, dietary restriction (DR) is one intervention that produces anti-aging effects from simple organisms to mammals. Research into DR has revealed robust systemic effects that can result in attenuation of age-related diseases via a myriad of molecular mechanisms. Given that numerous age-associated diseases are often polygenic and affect individuals differently, it is possible that they are confounded by interactions between environmental influences and the genome, a process termed ‘epigenetics’. In part one of the review, we summarize the different variants of DR regimens and their corresponding mechanism(s) and resultant effects, as well as in-depth analysis of current knowledge of the epigenetic landscape.
Keywords: dietary restriction, epigenetics, aging, caloric restriction, intermittent fasting, fasting-mimicking diet, DNA methylation, histone modifications, histone remodelling, microRNAs
Abstract
Biological aging occurs concomitantly with chronological aging and is commonly burdened by the development of age-related conditions, such as neurodegenerative, cardiovascular, and a myriad of metabolic diseases. With a current global shift in disease epidemiology associated with aging and the resultant social, economic, and healthcare burdens faced by many countries, the need to achieve successful aging has fueled efforts to address this problem. Aging is a complex biological phenomenon that has confounded much of the historical research effort to understand it, with still limited knowledge of the underlying molecular mechanisms. Interestingly, dietary restriction (DR) is one intervention that produces anti-aging effects from simple organisms to mammals. Research into DR has revealed robust systemic effects that can result in attenuation of age-related diseases via a myriad of molecular mechanisms. Given that numerous age-associated diseases are often polygenic and affect individuals differently, it is possible that they are confounded by interactions between environmental influences and the genome, a process termed ‘epigenetics’. In part one of the review, we summarize the different variants of DR regimens and their corresponding mechanism(s) and resultant effects, as well as in-depth analysis of current knowledge of the epigenetic landscape.
Keywords: dietary restriction, epigenetics, aging, caloric restriction, intermittent fasting, fasting-mimicking diet, DNA methylation, histone modifications, histone remodelling, microRNAs
1.0 Introduction
Over recent decades, human life expectancy has improved significantly due to better medical care, hygiene, food abundance, and lower child mortality rate (Zheng et al., 2014; Brown, 2015; Crimmins, 2015; Lang and Rupprecht, 2019). However, an increase in the length of an individual’s life may not correspondingly result in added quality. Indeed, poor aging occurs when an increase in the number of years of survival is accompanied by the development of age-related conditions, such as cardiovascular, neurodegenerative, as well as metabolic diseases. Such a shift in global disease epidemiology associated with aging is now resulting in an increasing worldwide prevalence of disability, and is a burgeoning concern for many countries facing associated healthcare, social, and economic burdens (Niccoli and Partridge, 2012; Jaul and Barron, 2017; Franceschi et al., 2018; Kehler, 2019). As such, the ideal paradigm of achieving successful aging by reaching old age in good health has fuelled research into new approaches to ameliorate the development and manifestation of age-related diseases (Katz and Calasanti, 2015; Tesch-Römer and Wahl, 2017). Nevertheless, the complexity of the biological process of aging is such that our understanding of the underlying molecular mechanisms remains poor and so interventional strategies to improve lifespan and counter the development of age-associated diseases have been limited.
People around the globe have practiced voluntary abstinence from food since antiquity, including among many religious groups such as Buddhists, Hindus, Jews, Muslims, and Christians, where restriction of food practice is incorporated into traditions and rituals (( Ri et al., 2012 )). Despite such prevalent observation of these practices in these groups, the effects energy restriction has on the human body remains quite poorly understood. It has been recently proposed that energy restriction could exert an evolutionary influence whereby our early ancestors were often challenged with extended periods of food restriction due to famine, and so procurement of food was accompanied by considerable physical activity. Thus, the fundamental needs for survival in terms of physical and mental maintenance without the consumption of food for extended periods may have shaped physiological, behavioural, and cognitive adaptations that have been inherited by modern humans in our genetic framework. Indeed, the current relative abundance of food coupled with our sedentary modern lifestyle, may be poorly compatible with the programmed genetic activity we have inherited. This may disrupt biochemical processes within the body, leading to biochemical derangements and ultimately contributing to the manifestation of a myriad of age-related diseases (Chakravarthy and Booth, 2004; Martin et al., 2010; Bake et al., 2014; Mattson et al., 2014, 2018; Lanktree and Hegele, 2017; Wayhart and Lawson, 2017) (Figure 1a). Indeed, extensive literature has shown unrestricted excessive energy intake provides the impetus for the development of age-related diseases (Uauy and Díaz, 2005). However, it is naïve to posit that starvation is a key to reverse the onset and development of chronic diseases because a balanced diet is critical for the proper maintenance of healthy physiological and metabolic functions, and individuals experiencing undernutrition often suffer from numerous health problems (Andersson and Bryngelsson, 2007; Skerrett and Willett, 2010). Interestingly, adoption of a dietary restriction (DR) regimen has garnered recent popularity, and it is indeed possible to voluntarily abstain from certain or total nutrients without compromising nutritional intake or energy balance. DR can promote beneficial effects on health and longevity, consistent with a potential for attenuation of age-related diseases through various molecular mechanisms (Harvey-Berino, 1999; Koubova and Guarente, 2003; Rogina and Helfand, 2004; Fontana et al., 2004; Guarente and Picard, 2005; Qin et al., 2006; Haigis and Guarente, 2006; Giudicessi and Ackerman, 2008; Larson-meyer et al., 2009; Cruzen and Colman, 2009; Qiu et al., 2010b; Duan and A. Ross, 2010; Manzanero et al., 2011; Bake et al., 2014; Colman et al., 2014; Olivo-Marston et al., 2014; Pifferi et al., 2018; Mitchell et al., 2019).
Epigenetics has gained widespread attention as a means to better understand how an invariant genome can in fact be programmed to adapt to different environmental influences. Twin studies have provided fascinating findings whereby two individuals with almost an identical genome who are subjected to different environmental influences may acquire different traits, which may in turn affect disease susceptibility or resistance (Poulsen et al., 2007; Bell and Spector, 2011; Dempster et al., 2011; Tan et al., 2015) (Figure 1b). Thus, despite our genetic code being ‘hard-wired’, it appears that our genome is not absolutely static but there is instead robust interaction with environmental influences through the heritability of diverse epigenetic marks. As aging is defined as a gradual and progressive decline in normal biological functions (Calvanese et al., 2009; Saldanha and Watanabe, 2015), this phenomenon may coincide with the progressive accumulation of diverse combinations of epigenetic signatures throughout life in response to a myriad of environmental influences. Such a process could underlie the development of diseases and various health outcomes plaguing the global population, especially in people with increasing age. We suggest that it is plausible that many polygenic diseases may be confounded by environmental influences or stochastic variables (Kim et al., 2019). Indeed, numerous studies provide evidence that epigenetics may play a critical role in influencing biological aging and promoting the onset of age-related diseases (Calvanese et al., 2009; Saldanha and Watanabe, 2015). As a result, a better understanding of the interaction between our genetic framework and our environment using an ‘epigenetics perspective’ may be valuable. Although we cannot control our exposure to many environmental factors, our dietary intake is one factor that can be controlled to a large extent. DR is indeed considered an acceptable intervention by gerontologists and researchers in countering aging (Kirk, 2001; Masoro, 2006; Minor et al., 2010), and it is quite possible that DR acts partly or even substantially via epigenetic mechanisms. In the first part of the review on DR, we will introduce current knowledge of both DR and epigenetic signatures.

In a new window | Download PPT
Figure 1: Environmental Influences and Genetic Framework. (a) As our early ancestors were often challenged with periods of famine. And their procurement of food was accompanied by regular physical activity, they were presumably faced with periods of obligate food restriction and physical activity. The fundamental need for survival in terms of physical and mental maintenance without the consumption of food for extended periods appears to have shaped physiological, behavioural, and cognitive adaptations, and may thus have become inherited by modern humans within our genetic framework. As a result, the relative abundance of food coupled with our sedentary lifestyle today may be less compatible with the activity our evolutionarily programmed genes. As such, our modern lifestyle may disrupt our biochemical processes, and contribute to a myriad of age-related diseases. Thus, perhaps to some extent mimicking the environment of our ancestors may help to ameliorate the onset and development of diseases. (b) Twins possess high similarities in their genetic framework, yet when subjected to different environmental influences may acquire different traits that affect susceptibility to disease.
2.0 Dietary Restriction
DR is defined as a voluntary abstinence from consumption of a selected or entire nutrient composition without compromising energy balance or inducing malnutrition (Masoro, 1998; Robertson and Mitchell, 2013). As shown in Table 1, variants of DR regimens have emerged over the years, and can be broadly classified as either caloric restriction (CR) or intermittent fasting (IF) (Lee and Longo, 2016), as well as the new ‘fasting mimicking diet’ (FMD) (Wei et al., 2018).
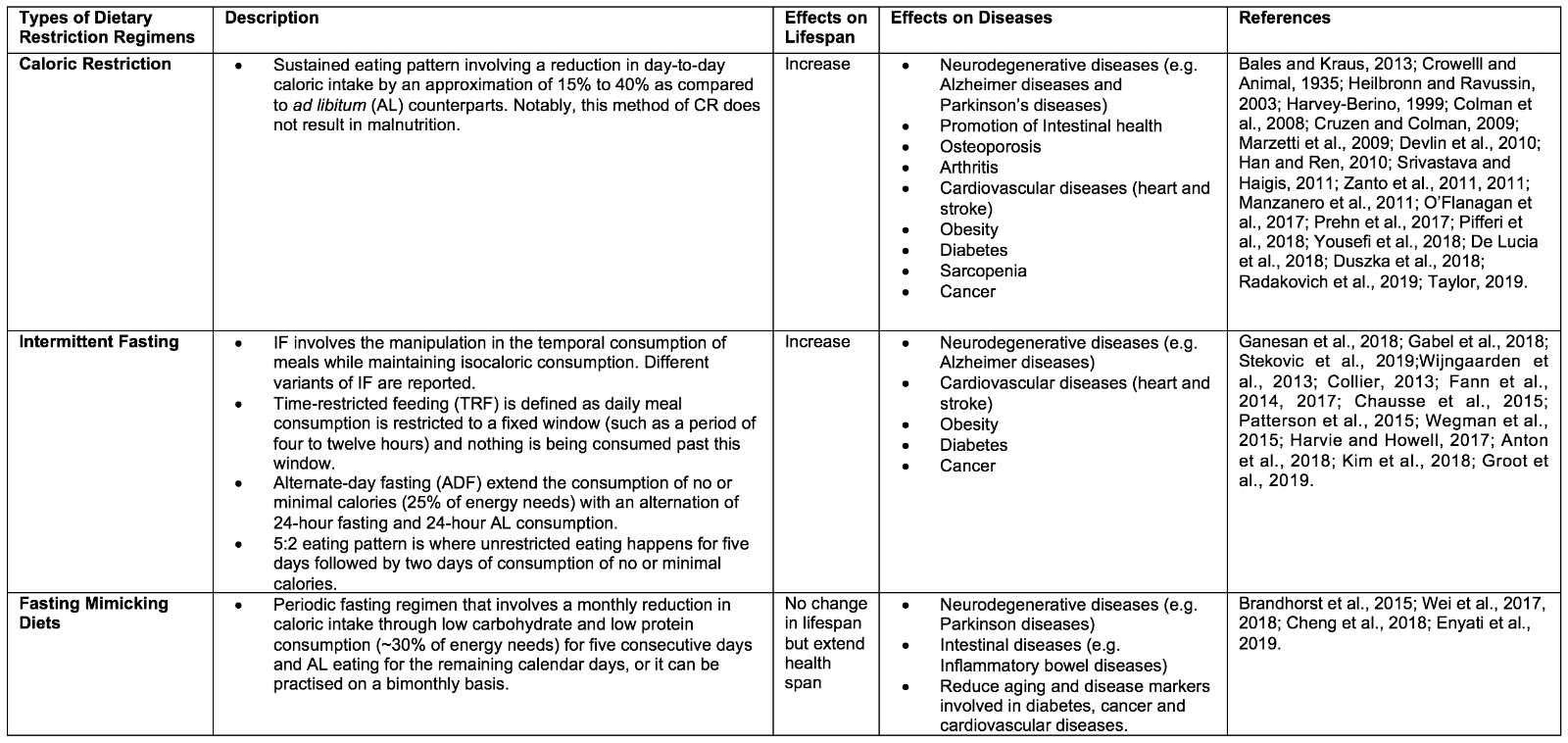
2.1 Caloric Restriction
CR is defined as a sustained eating pattern involving a reduction in day-to-day caloric intake by 15% to 40% as compared to ad libitum (AL). Notably, this method of DR does not result in malnutrition (Bales and Kraus, 2013) . The ideology of CR spans many centuries, with notable figures promoting CR including Hippocrates, to whom is ascribed the notion: “Let food be thy medicine and medicine be thy food” (Square, 2002). Experimental studies that began much later first found CR to be able to extend lifespan and improving reproductive performance in CR as opposed to AL rats (CM et al., 1935). These experimental findings drove a renaissance of nutrition research, in which CR was found to positively impact longevity and health across multiple life forms, from simple organisms (such as yeast Saccharomyces cerevisiae, nematode Caenorhabditis elegans, and fruit fly Drosophila melanogaster) to vertebrates (such as mice Mus musculus and primates Macaca mulatta) (Heilbronn and Ravussin, 2003). Besides, CR has been shown to provide protection against numerous diseases, including slowing brain atrophy and preserving cognition, and protecting against neurodegenerative diseases (e.g. Alzheimer and Parkinson’s diseases), intestinal dysfunction, obesity, osteoporosis, arthritis, cardiovascular disorders (e.g. heart and stroke), diabetes, and sarcopenia, as well lowering the risk and progression of cancer (Harvey-Berino, 1999; Luchsinger et al., 2002; Colman et al., 2008; Cruzen and Colman, 2009; Marzetti et al., 2009; Devlin et al., 2010; Han and Ren, 2010; Longo and Luigi Fontana, 2010; Manzanero et al., 2011; Srivastava and C. Haigis, 2011; González et al., 2012; O’Flanagan et al., 2017; Prehn et al., 2017; Pifferi et al., 2018; Yousefi et al., 2018; De Lucia et al., 2018; Duszka et al., 2018; Radakovich et al., 2019; Taylor, 2019).
2.2 Intermittent Fasting
IF involves adjusting the temporal intake of food while maintaining isocaloric consumption overall (Ganesan et al., 2018). IF also has deep historical roots, with observance being reported in many religions for either spiritual reasons or physical benefits (Trepanowski and Bloomer, 2010; Patterson et al., 2015). IF is an umbrella term that encompasses different variants in the frequency of meal consumption. Time-restricted feeding (TRF) and alternate-day fasting (ADF) are examples of IF in which the timing of meals is varied. For TRF, daily meal consumption is restricted to a fixed window (such as a period of four to twelve hours) with nothing consumed outside of this window. ADF, on the other hand, extends the period of no or minimal calorie consumption (25% of energy needs), alternating between 24 hours of fasting and 24 hours of AL consumption. Another popular variant of IF is the 5:2 eating pattern, whereby during a 7 day period, unrestricted eating happens on five days in addition to two non-consecutive days of consumption of no or minimal calories (Gabel et al., 2018; Stekovic et al., 2019). Like CR, IF has been found to induce longevity across many animal models, as well as improving metabolic health. For instance, IF has been reported to influence the circadian clock, intestinal microbiota niche, and metabolic regulation controlling insulin sensitivity, lipid metabolism, hormonal changes, and inflammatory responses. This may lead to an improvement of metabolic function and may explain the development of resistance to cardiovascular diseases (e.g. heart attack and stroke), neurodegenerative diseases (e.g. Alzheimer disease), obesity, type II diabetes mellitus, and cancer (Wijngaarden et al., 2013; Collier, 2013; Fann et al., 2014, 2017; Chausse et al., 2015; Patterson et al., 2015; Wegman et al., 2015; Harvie and Howell, 2017; Anton et al., 2018a; Kim et al., 2018; De Groot et al., 2019).
2.3 Fasting Mimicking Diet
Our current social and economic environment typically enables instant gratification from food-seeking to meet hunger, and this “obesogenic” model has made it more difficult for individuals to alter their eating patterns. The recently introduced FMD appears to be a combination of CR and IF, and it has arisen due to the lack of compliance in individuals to adhere to either CR or IF for a prolonged period. FMD is a form of periodic fasting that involves a monthly reduction in caloric intake through low carbohydrate and low protein consumption (~30% of energy needs) for five consecutive days and AL eating for the remaining calendar days, and can be practiced on a monthly or bimonthly basis. Notably, FMD has been shown to promote the extension of health span but not lifespan in rodents. FMD is also effective in enhancing neurocognitive function, and reducing a plethora of aging and disease markers, such as diabetes, cancer, and cardiovascular diseases (Brandhorst et al., 2015; Cheng et al., 2017; Wei et al., 2017, 2018; Rangan et al., 2019).
2.4 Mechanism of Dietary Restriction
From findings in less complex, short-lived organisms through to mammals, it is generally accepted that DR can be an effective non-genetic and non-pharmacological intervention against aging through increased longevity and delayed onset and development of chronic diseases. Those findings from DR studies across diverse organisms suggest an innate evolutionary conservation of either genes or molecular players that respond positively to DR. Indeed, studies that have begun to decipher the mechanisms of DR effects have revealed a plethora of conserved biological pathways and proteins that are modulated in response to a low energy status (Figure 2).
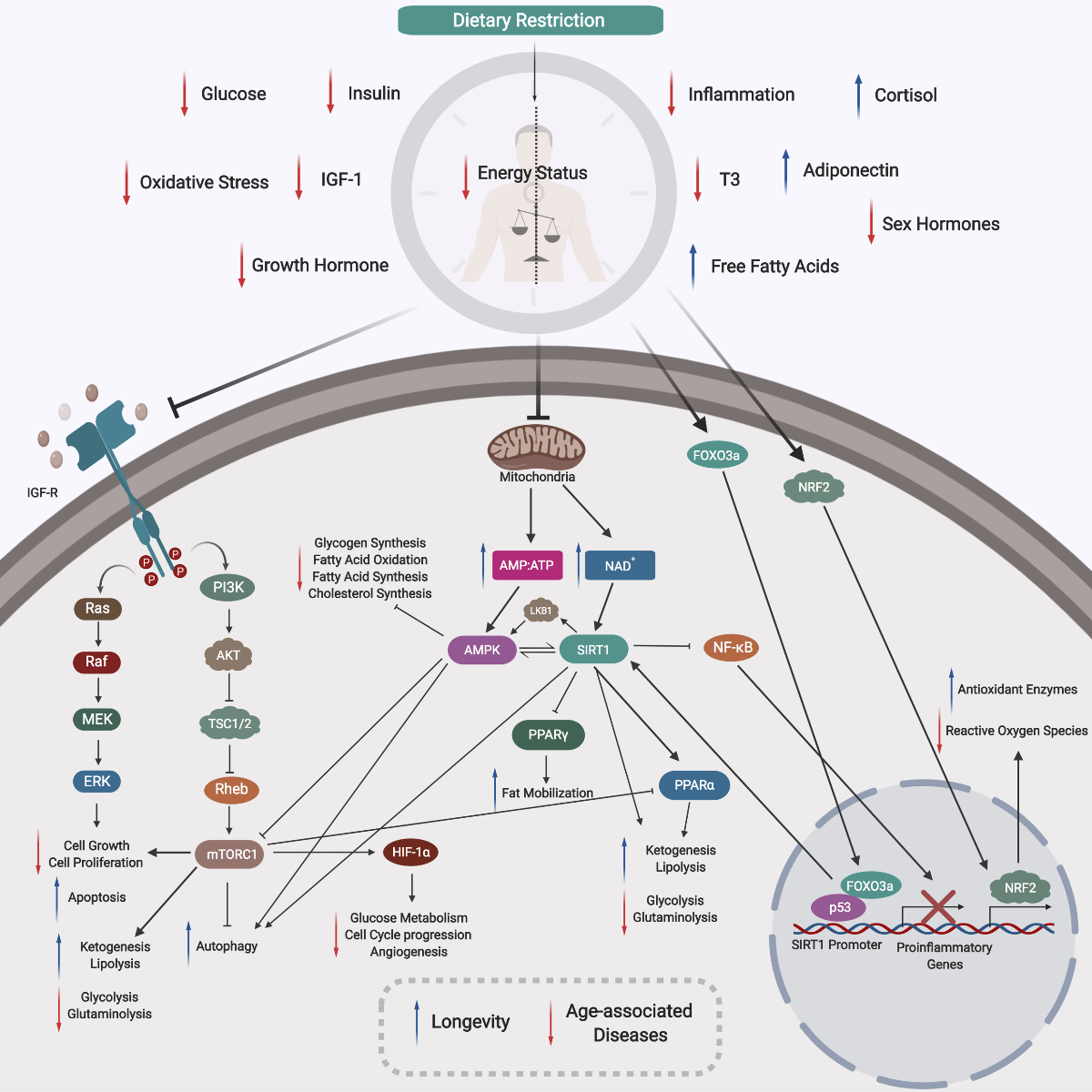
In a new window | Download PPT
Figure 2: Mechanism of Dietary Restriction. During DR there is a reduced energy status within the organism, resulting in corresponding decreases in blood glucose, insulin, IGF-1, growth hormones, sex hormones, and T3 thyroid hormones. Also, DR induces a reduction in both oxidative stress and inflammation. Following DR, there are increases in free fatty acids, adiponectin, and cortisol. A lower cellular energy status will lead to reduced mitochondrial activity and aerobic respiration, increasing the AMP:ATP ratio and NAD+ levels. Two major cellular nutrients and energy sensors, AMPK and SIRT1, will be then be activated, respectively. Activated AMPK will inhibit glycogen synthesis, ACC1, ACC2, and HMG-CoA to drive a reduction in fatty acid synthesis, oxidation, and cholesterol synthesis. Activated SIRT1 can enhance ketogenesis and lipolysis, and downregulate glycolysis and glutaminolysis. These effects may be also due to an activation of PPARα, reflecting direct and indirect functions of SIRT1. Activated SIRT1 can also repress the activity of PPARγ to modulate lipid metabolism and enhance fat mobilization in white adipose tissue during DR. Activated SIRT1 can inhibit NF-κB activity, inhibiting the expression of proinflammatory genes. SIRT1 activation is dependent on the increased interaction between FOXO3a and p53 at SIRT1 promoter sites. The physical interaction between FOXO3a and p53, and the subsequent activation of SIRT1, play important roles in repressing cell growth and proliferation. Notably, SIRT1 activation can activate LKB1, which will further activate AMPK, thus creating a positive feedback loop. Reduced levels of insulin, glucose and growth hormone downregulates insulin and IGF-R signaling pathways, inactivating the RAS/MAPK axis, repressing cell growth and proliferation and promoting apoptosis. Repression of IGF-R signaling will include the PI3K pathway, and inhibit downstream mTORC1, promoting autophagy. Inhibition of mTORC1 can also occur via activated AMPK in response to DR. Decreased mTORC1 function can promote ketogenesis and lipolysis, and repress glycolysis and glutaminolysis. Moreover, the corresponding decreased function of mTORC1 also inhibit HIF-1α, which downregulates key biological processes such as glucose metabolism, angiogenesis, and cell cycle progression. The NRF2 pathway is also triggered by DR, which helps to increase production of protective antioxidant enzymes and mitochondrial biogenesis to reduce ROS activity. With lower levels of ROS, there will be reduced DNA damage and a maintained genome stability. Lower energy status will prime cells to low-intensity stress, and this hormetic mechanism will stimulate better management of stress, upregulate DNA repair genes, as well as shifting towards a preferred setting of maintenance and repair (not shown). Overall, DR can act via these mechanisms to counter age-associated diseases and induce longevity.
DR, dietary restriction; IGF-1, insulin growth factor 1; AMP, adenosine monophosphate; ATP, adenosine triphosphate; NAD+, nicotinamide adenine dinucleotide; AMPK, adenosine monophosphate kinase; SIRT1, sirtuin 1 deacetylase; ACC1 and ACC2, acetyl-coenzyme A carboxylase 1 and 2; HMG-CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A; PPARα and PPARγ, peroxisome proliferator-activated receptor gamma alpha and gamma; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; FOXO3a, forkhead box O3; LKB1, liver kinase B1; IGF-R, insulin growth factor receptor; RAS/MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; mTORC1, mammalian target of rapamycin complex 1; HIF-1α, hypoxia-inducible factor 1 alpha; NRF2, nuclear factor erythroid 2-related factor 2 ;ROS, reactive oxygen species
DR exerts pleiotropic cellular effects, inducing a reduced energy status within the organism, and a corresponding decrease in blood glucose, insulin, insulin-like growth factor 1 (IGF-1), growth hormones, sex hormones, and T3 thyroid hormones levels. Also, DR induces a reduction in both oxidative stress and inflammation. Moreover, following DR, an increase in free fatty acids, adiponectin, and cortisol can be observed (Ungvari et al., 2008; Redman et al., 2010; Abedelmalek et al., 2015; Lan et al., 2015; Kapahi et al., 2017).
A reduced cellular energy status will lead to lower mitochondrial activity and thus aerobic respiration, and corresponding increases in the adenosine monophosphate and adenosine triphosphate (AMP:ATP) ratio, and in nicotinamide adenine dinucleotide (NAD+) levels. In turn, two major cellular nutrients and energy sensors, adenosine monophosphate kinase (AMPK) and sirtuin 1 deacetylase (SIRT1), will be activated, respectively (Haigis and Guarente, 2006; Hatori et al., 2012; Marcinko and Steinberg, 2014). Activated AMPK will then inhibit glycogen synthesis, as well as acetyl-coenzyme A carboxylase 1 and 2 (ACC1 and ACC2) and 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) to reduce fatty acid synthesis, oxidation, and cholesterol synthesis (Motoshima et al., 2006; Thomson and Winder, 2009; Marcinko and Steinberg, 2014; Jeon, 2016; Foretz et al., 2018). Activated SIRT1 can enhance ketogenesis and lipolysis, and downregulate glycolysis and glutaminolysis (Pedersen et al., 2008; Chakrabarti et al., 2011; Chang, Hung-Chun; Guarente, 2012; Zhu et al., 2013). These effects may be also due to an activation of peroxisome proliferator -activated receptor alpha (PPARα), which suggests that SIRT1 can exert both direct and indirect functions. Activated SIRT1 can also repress the activity of peroxisome proliferator-activated receptor gamma (PPARγ), which will in turn modulate lipid metabolism and enhance fat mobilization in white adipose tissue during DR. Depending on the PPAR isoforms, it appears that SIRT1 can exert pleotropic effects on PPAR in response to DR (Purushotham et al., 2009; Hayashida et al., 2010; Picard et al., 2012; Bonzo et al., 2014; Khan et al., 2015). Furthermore, activated SIRT1 can also inhibit the activity of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), inhibiting the expression of proinflammatory genes (Gillum et al., 2011; Ghisays et al., 2015). SIRT1 activation is dependent on the increased interaction between forkhead box O3 (FOXO3a) and p53 at SIRT1 promoter sites during periods of nutrient deficiency, such as during DR. Thus, SIRT1 activation during DR can be regulated via the NAD+ or the FOXO3a axis. Furthermore, the physical interaction between FOXO3a and p53, as well as the subsequent activation of SIRT1, have been found to play key roles in repressing cell growth and proliferation (Hori et al., 2013; Yun et al, 2013; Zhang et al., 2014). Notably, SIRT1 activation can also activate liver kinase B1 (LKB1), which will further activate AMPK, thus creating a positive feedback loop (Wang et al., 2011; Laboratory, 2012).
When cells detect a corresponding drop in the levels of insulin, glucose, growth hormones, insulin, and insulin-like growth factor receptor (IGF-R) signaling pathways are downregulated (Straus and Takemoto, 1990; Cheng et al., 2017). As a result, DR can also inactivate the mitogen-activated protein kinase (RAS/MAPK) axis, to repress cell growth and proliferation and promote apoptosis (Morikawa et al., 2004; Fuentes et al., 2012). Repression of IGF-R signaling will also repress the phosphoinositide 3-kinase (PI3K) pathway and the downstream mammalian target of rapamycin complex 1 (mTORC1), promoting autophagy. Notably, inhibition of mTORC1 can also occur by activated AMPK in response to DR. Decreased mTORC1 function can promote ketogenesis and lipolysis and inhibit glycolysis and glutaminolysis, as in SIRT1-induced activation of PPARα (De Paula et al., 2017; Sabatini, 2017; Tulsian et al., 2018). Moreover, decreased function of mTORC1 inhibit hypoxia-inducible factor 1 alpha (HIF-1α), which in turn downregulates key biological processes such as glucose metabolism, angiogenesis, and cell cycle progression (Parsons, 2001; Laplante and Sabatini, 2013; Saxton and Sabatini, 2017). The nuclear factor erythroid-2-related factor 2 (NRF2) pathway is also triggered by DR and it contributes to increases in production of protective antioxidant enzymes and mitochondrial biogenesis and reduction of reactive oxygen species (ROS). With reduced ROS levels there will be less DNA damage and improved maintenance of genome stability (Kulkarni et al., 2014). Lower energy status will also prime cells to low-intensity stress, a hormetic mechanism that can facilitate better management of stress, upregulate DNA repair genes, and shift towards a preferred status of maintenance and repair (Kouda and Iki, 2010; Horne BD, Muhlestein JB, 2015).
Systemic effects on major organs are also known to occur in response to DR via these molecular and cellular adaptations. In the brain, DR has been reported to enhance cognitive function and neurotrophic factor release, serving to protect against neurotoxicity and improve stress resistance. DR can also promote neurogenesis and mitochondrial biogenesis, and reduce inflammation and oxidative stress within the brain (Qiu et al., 2010; Kynjai et al., 2019). Within the gut, DR promotes gut mobility, and induces a reduction in inflammation, cell proliferation, and energy uptake (Brennan et al., 2011; Ott et al., 2017; Id et al., 2018; Lian et al., 2018). On the other hand, in DR the pancreas comprises less fat and there is promotion of glucagon secretion (Anton et al., 2018b; Jiang et al., 2019). Moreover, DR upregulates lipolysis and ketogenesis within adipose tissue, increasing adiponectin synthesis and decreasing leptin secretion (Ding et al., 2012; Rogozina et al., 2012; Fabbiano et al., 2016). In skeletal muscle, oxidative metabolism is shifted preferentially towards increased anabolism, with a corresponding enhancement of insulin sensitivity, mitochondrial biogenesis, reduced body temperature, as well as stress adaptation and resistance in response to DR (Usuki et al., 2004; Hempenstall et al., 2012; Chen et al., 2015; Mitchell et al., 2015; Martins et al., 2018; Faitg et al., 2019). In the heart and vasculature, there is a reduced blood pressure and resting heart rate induced by DR, with increased parasympathetic tone, reduced heart rate variability, and improved cardiovascular stress adaptation (Weiss and Fontana, 2011; Stein et al., 2012; Shinmura, 2013; Nicoll and Henein, 2018). Finally, in response to DR the liver triggers gluconeogenesis, glycogenolysis, mitochondrial biogenesis, and enhanced insulin sensitivity (Hell et al., 1980; Weindruch et al., 1980; Hagopian et al., 2003; Browning et al., 2008; Kirk et al., 2010).
In summary, DR triggers a lower energy status within the organism, which in turn commonly induces robust metabolic switching in major organs (Webster et al., 1972; Millward et al., 1974; Camps et al., 1992; Trayhurn et al., 1995; Mattson and Wan, 2005; Longo and Mattson, 2014; Bujak et al., 2015; Mattson et al., 2017). The resulting systemic changes occur via molecular, cellular, and metabolic adaptations, and may promote longevity and delay the onset and development of many age-associated diseases (Figure 3).

In a new window | Download PPT
Figure 3: Effects of Dietary Restriction. DR can induce a myriad of systemic effects. In turn, a robust metabolic switching ensues, which results in molecular, cellular, and metabolic adaptations that delay the onset and development of many age-associated diseases and promote longevity.
3.0 Epigenetics
Epigenetics is an emerging field of knowledge relating to the complex interactions between genome and environment, defined as mitotically and meiotically heritable genetic changes without corresponding changes to the invariant DNA sequence (Panawala et al., 2017; Ng et al., 2018). These epigenetic effects on the genome occur via enzymatic modifications to either the DNA sequence directly or through histone proteins that form part of the hierarchical packing of the chromosomal DNA (Figure 4). Gene expression can then be modulated directly or indirectly via an overall modification towards the chromatin structure and accessibility. The landscape of epigenetic signatures across the genome is termed the ‘epigenome’, with these unique epigenetic tags functioning as distinct microdomains within the nucleus to differentially regulate patterns of gene expression (Qureshi and Mehler, 2010). The overall epigenome status within the nucleus can be also be considered as a ‘tug-of-war’, whereby each epigenetic modification can either promote or inactivate gene expression. The overall direction of gene expression is thereby dependent on the sum of these dynamic interactions. For didactic purposes, these epigenetic modifications can be broadly categorized to comprise DNA methylation, histone protein modifications, histone remodeling complexes, as well as involvement of microRNAs.
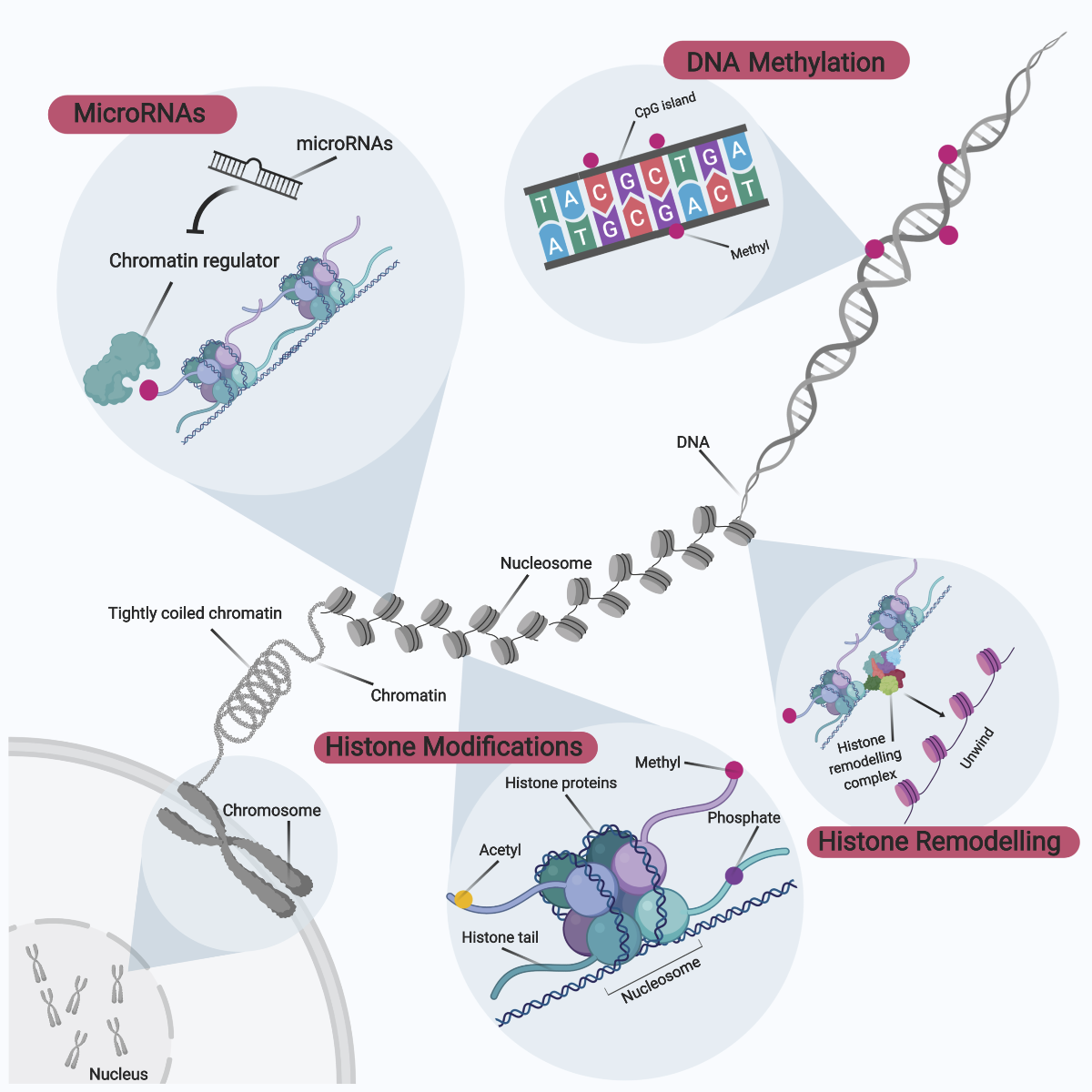
In a new window | Download PPT
Figure 4: Overview of Epigenetic Modifications. Epigenetic modifications can be broadly categorized into DNA methylation, histone modifications, histone remodeling, and microRNA involvement. These epigenetic modifications to the genome occur via enzymatic modifications to either the DNA sequence directly or through histone proteins that form part of the hierarchical packing of the chromosomal DNA. In turn, gene expression can be modulated directly or indirectly via an overall modification to chromatin structure and accessibility. The epigenetic signature landscape across the genome is collectively termed the ‘epigenome’, and unique epigenetic tags function as distinct microdomains within the nucleus to regulate differential patterns of gene expression.
3.1 DNA Methylation
DNA methylation is the principal epigenetic modification at the DNA sequence level (Figure 4 and 5). The double helical DNA structure is composed of a combinational sequence of four nucleotide bases; namely thymine, adenine, cytosine, and guanine. DNA methylation involves the principal tagging of a methyl group to carbon position five of the cytosine ring, and this process tends to occur at higher frequency in regions termed CpG dinucleotide islands. CpG islands are genomic regions spanning more than 500-base pairs of DNA and are often located at gene promoter regions which are enriched near the 5’ gene transcript. The characterization of CpG islands falls into two categories; the composition of both cytosine and guanine appears to occupy above 55% in these 500-base pairs region, or CG:GC observed frequency ratio to be at least 0.6. DNA methylation can also occur in other regions called ‘CpG shores’, defined as lower density CpG islands residing downstream by approximately 2 kb (Portela and Esteller, 2010a). Conventionally, DNA methylation was thought to occur only at CpG islands, but recent findings indicate its occurrence in CpG shores shown the complexity underlying this form of regulation. While cytosine nucleotides are scattered across the genome, DNA methylation distribution is asymmetrical and considered to be relatively rare, especially in the mammalian genome, accounting for only 1% of the genome size (Cooper et al., 2010). This is thought to be due to the fact that the addition of a methyl group to cytosine to form 5-methylcytosine is inherently genetically unstable. 5-methylcytosine cannot be excised from the genome or recognized by the DNA repair system. To counter this, 5-methylcytosine undergoes spontaneous deamination to yield a thymine, yet become prone to transition mutations. As a result, CpG islands have evolved to be depleted across the genome and account for very low proportions to decrease the risk of becoming mutational hotspots (Cooper et al., 2010).
DNA methylation is catalyzed by an important family of enzymes called DNA methyltransferases (DNMT) (Moore et al., 2013). The DNMT family consists of five members, of which only three (DNMT1, DNMT3a, and DMNT3b) possess enzymatic activity. DNMT3a and DNMT3b catalyze de novo methyl group transfer from S-adenosyl methionine to CpG sites, whereas DNMT1 is mostly important for the maintenance of the overall DNA methylation landscape across the genome (Kim et al., 2009; Feng et al., 2010). The conversion and maintenance of methionine and folate to S-adenosyl methionine are mediated by a group of enzymes called methylenetetrahydrofolate reductases (MTHFR). Besides DNA methylation, S-adenosylmethionine also plays important roles in DNA synthesis (Alluri et al., 2005; Daniels et al., 2007; Zhou et al., 2014). On the other hand, the process of DNA demethylation at 5-methylcytosines to 5-hydroxymethylcytosines is mediated by ten-eleven translocation (TET) enzymes in a locus-specific manner. However, these groups of enzymes have only recently become known in the field of epigenetics and understanding of their functions remains poor (Figure 5) (Xu and Wong, 2015).
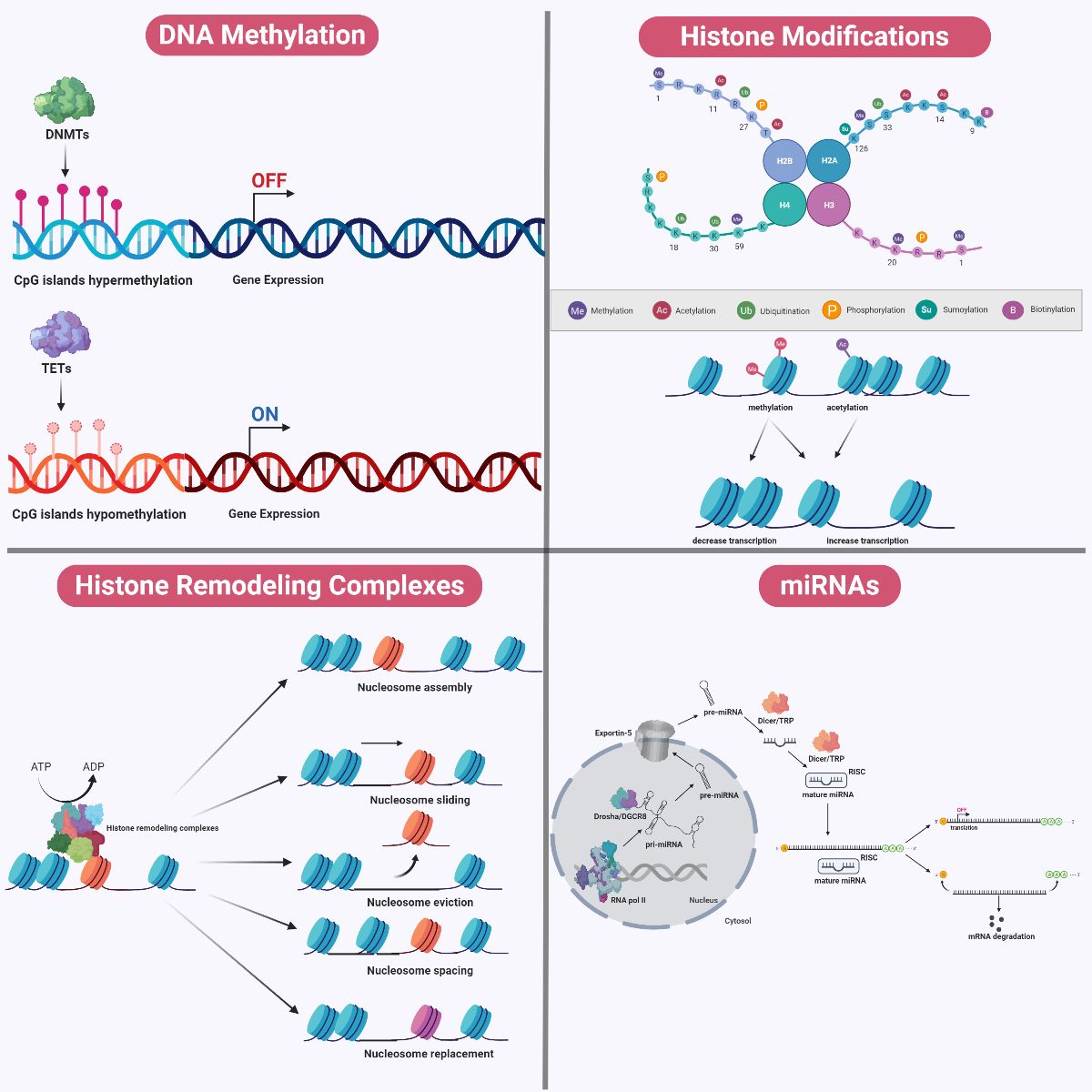
In a new window | Download PPT
Figure 5: Mechanisms of Epigenetic Modifications. During DNA methylation, a methyl group is added to carbon position five of the cytosine ring at either CpG dinucleotide islands or CpG shores. DNA methylation is catalyzed by DNMTs, whereas DNA demethylation is catalyzed by TETs. DNA methylation tends to be associated with gene silencing whereas DNA demethylation is associated with transcriptional activation. Chromosomal DNA within the eukaryotic genome is often interlaced with distinct packing and folding, resulting in a distinct organizational structure. At the fundamental level, a nucleosome structural core is first formed through wrapping of a 147 base-pair double helical DNA around an octamer of histone proteins consisting of pairs of H2A-H2B dimers and H3-H4 dimers. Between two nucleosome cores, DNA that is not wrapped around nucleosomes is referred to as ‘linker DNA’, which tends to be associated with H1 histone proteins. A plethora of post-translational modifications (such as acetylation, methylation, phosphorylation, sumoylation, biotinylation, and ubiquitination) can occur on the amino terminal tail protruding from each histone subunit within the nucleosome complex. These histone modifications can affect the relative packing of DNA around these nucleosome cores, and may determine the relative accessibility for the machinery to initiate replication and transcription. Chromatin structure can be modified by ATP-dependent histone remodeling complexes, using the energy from ATP hydrolysis to locally disrupt the interaction between DNA and histones. These remodelers mediate alterations to chromatin structure via nucleosome sliding, eviction, assembly, spacing, histone dimers eviction or replacement, and even entire histone replacement. In addition, miRNAs demonstrate complementary interaction with specific genes and modulate gene expression. Cellular miRNAs are synthesized in a sequential manner and occur in both the nucleus and the cytosol. Within the nucleus, the genome comprises numerous miRNA genes transcribed by RNA polymerase II to form a single large pri-miRNA transcript. Because this pri-miRNAs transcript consists of multiple miRNA loci, it undergoes further processing by a complex called Drosha/DGCR8, a class of RNaseIII enzymatic complex, to generate shorter hairpin-loop structurers termed ‘pre-miRNAs’. This pri-miRNA is exported into the cytosol by Exportin-5 in a RAN-GTP dependent manner. Within the cytosol, another complex belonging to another class of RNaseIII enzymes, termed ‘Dicer/TRBP’ exert its action. Dicer processes the pri-miRNAs into a duplex of mature miRNAs, which undergoes asymmetrical unwinding by the Dicer/TRBP complex to yield a single-stranded mature miRNA. This single-stranded form loads itself into a ribonucleoparticle to yield an RISC, which is the active form of the gene silencing complex. Together, the single-stranded mature miRNAs within the RISC complex will identify the 3’-UTR of targeted mRNAs. The mode of action of gene silencing mediated by miRNAs can occur in two ways, depending on its complementarity with the targeted mRNAs. miRNAs which bind to targeted mRNAs in perfect complementarity induce rapid deadenylation and decapping, leading to mRNA degradation. An imperfect complementarity binding of miRNAs to targeted mRNAs result in translational suppression.
DNMTs, DNA methyltransferase; TETs, ten-eleven translocation; pri-miRNAs, primary miRNA; pre-miRNAs, precursor miRNAs; RISC, RNA-induced silencing complex; 3’-UTR, 3’ untranslated region.
The effects of DNA methylation have often been linked to transcriptional inactivation via various mechanisms. DNA methylation can directly block the binding of specific DNA binding factors to transcriptional start sites and as a result halt transcription. In another instance, 5-methylcytosines within CpG sites are often recognized by protein families possessing conserved methyl-CpG-binding domains (MBD). Binding of these proteins may in turn recruit a combination of transcriptional corepressors, polycomb proteins, or even chromatin remodeling complexes to bring about transcriptional repression (Huck-Hui and Bird, 1999; Du et al., 2015). Besides playing a conventional role in the area of gene silencing, DNA methylation has been implicated in many biological processes such as genomic imprinting and stability, X chromosome inactivation, differentiation, and development (Moore et al., 2013; Wu and Zhang, 2014).
3.2 Histone Modifications and Remodeling
Chromosomal DNA within the eukaryotic genome is often interlaced with distinct packing and folding, resulting in distinct organizational structure (Figure 4 and 5). At the fundamental level, a nucleosome structural core is first formed through wrapping of a 147 base-pair double helical DNA around an octamer of histone proteins consisting of pairs of H2A-H2B dimers and H3-H4 dimers. This interaction is mediated by the positive charges within the rich pool of lysine and arginine residues in histone proteins, as well as the negatively charged sugar phosphate backbone of DNA. Between two nucleosome cores, DNA that is not wrapped around nucleosomes is referred to as linker DNA. This linker DNA tends to be associated with H1 histone proteins (Zhang and Reinberg, 2001; Boyanapalli and Kong, 2015). The globular structure of histone proteins within nucleosome complexes represents a form of steric hindrance that may impede the accessibility of DNA replication and transcriptional machinery to genomic sites. As a result, the relative packing of DNA around these nucleosome cores may determine the relative accessibility for the machinery to initiate replication and transcription. Therefore, at the genomic level, two defined areas of DNA can be classified depending on the relative packing of DNA; namely euchromatin and heterochromatin. Euchromatin regions are generally characterized by a more spacious chromatin structure which in turn exhibits higher transcriptional rates, whereas heterochromatin regions demonstrate transcriptional silence via tight packing of chromatin (Tamaru, 2010).
The relative transition between these two states is highly dependent on post-translational modifications that can occur on the amino terminal tail protruding from each histone subunit within the nucleosome complex. Histone modifications tend to be short-term and reversible and are readily modulated by external environmental changes (Cedar and Bergman, 2009; Handy et al., 2011; Qureshi, 2011). A myriad of post-translational modifications (such as acetylation, methylation, phosphorylation, sumoylation, biotinylation, and ubiquitination) catalyzed by a plethora of enzymes have been reported to occur at specific amino acid sites within each histone. Each histone modification in turn will determine a critical function, which translates to distinct cellular outcomes (Figure 5). As such, the sum of all histone modifications (termed ‘histone codes’) will determine the overall changes at the cellular level, such as chromatin assembly, transcriptional activation or repression, telomere dynamics, DNA repair, cell cycle, as well as apoptosis (Lennartsson and Ekwall, 2009; Portela and Esteller, 2010b). For didactic purposes, two well-studied examples of histone modifications, namely acetylation and methylation, will be discussed to more fully describe the mechanism and complexity underlying histone modifications.
Histone acetylation involves the covalent addition of an acetyl group to a histone tail lysine residue at a conserved epsilon-amino group and is often mediated by a family of enzymes called histone acetyltransferases (HATs). Upon histone acetylation, a synergistic interaction with histone remodeling complexes may be formed with HATs, which in turn weakens the interaction between DNA and histone proteins, thus promoting nucleosome sliding away from DNA, facilitating less packing of chromatin and higher accessibility of transcriptional machinery and activity (Figure 5). By contrast, the removal of acetyl groups from histones is mediated by histone deacetylases (HDACs), which may in turn recruit transcriptional repressors to downregulate transcription via promotion of tighter packing of chromatin (Legube and Trouche, 2003). Histone acetylation and deacetylation at various sites has been reported to be critical for a plethora of biological roles besides transcription, such as histone deposition, chromatin assembly, transcriptional elongation, telomeric silencing, and DNA repair (Tamburini and Tyler, 2005; Zhao et al., 2005; Gong and Miller, 2013; Church and Fleming, 2018).
On the other hand, methylation of histone involves the addition of methyl groups to histone amino terminal tails via the actions of histone methyltransferases (HMTs), with preferential addition to either lysine or arginine residues (Zhang and Reinberg, 2001; Chakravarty et al., 2017). However, effects arising from histone methylation are often diverse and complex, depending on the type or position of amino acid residues that are methylated, and the number of methyl groups added within each specific locus (Figure 5). For instance, at specific lysine residues, methyl groups can be added singly, twice or three times, whereas at specific arginine residues methyl groups can be added either once or twice. This complexity is confounded in that certain methylated residues, such as di-methylated arginine residues, often demonstrate stereo topological layout, either as symmetrical or asymmetrical, which may produce diverse functions (Chen et al., 2012; Chakravarty et al., 2017). Given that enormous complexity occurs as a result of the possible permutations and combinations of histone methylation tags, it is difficult to predict biological effects arising from this post-translational modification. Nevertheless, many studies have shown that histone methylation plays important roles in transcriptional activation and repression, transcriptional elongation, genomic imprinting, checkpoint response, and X chromosome inactivation (Greer and Shi, 2012; Chen and Zhu, 2016).
Chromatin structure within the eukaryotic genome can be modified not only by histone enzymatic modifications, but also through the actions of histone remodeling complexes (Figure 4 and 5). These complexes are referred to as ATP-dependent remodeling complexes, using the energy from ATP hydrolysis to locally disrupt the interaction between DNA and histones. The first histone remodeling complex gene was discovered during genetic screening of S. cerevisiae, and subsequently termed switching-defective 2 (SWI2) or sucrose non-fermenting 2 (SNF2). The SWI/SNF2 complex was found to mediate alterations in chromatin structure that led to transcriptional activation (Vignali et al., 2000). Besides the SWI/SNF2 family, three other families of remodelers have now been identified; namely the imitation-switch (ISWI), chromodomain helicase DNA-binding (CHD), and inositol-requiring 80 (INO80). These four identified families of remodelers share a high degree of conservation of an innate ATPase-helicase domain and can function as either a monomer (e.g. CHD1) or as a distinct macromolecular complex (e.g. ISWI). These complexes can be made up of different combinations of subunits, and may be accompanied by distinct microdomains. Depending on their composition, histone remodeling complexes can exert synergistic or antagonistic biochemical and genetic functions (Pillus, 2015). Moreover, these remodelers can mediate alterations in chromatin structure via nucleosome sliding, eviction, assembly, spacing, histone dimers eviction or replacement, and even entire histone replacement (Figure 5). Furthermore, histone modifications and histone remodeling complexes do not only function independently. It has been reported that complexes such as ISWI and CHD1 can also interact with histone modifiers and histone modifications, resulting in diverse functions. As such, the dynamic kinetics of nucleosome positions within the eukaryotic genome will impact key biological processes, such as regulation of gene expression, DNA replication and repair, and homologous recombination events (Vignali et al., 2000; Liu et al., 2012; Narlikar et al., 2013; Pillus, 2015; Tyagi et al., 2016; Zhang et al., 2016; De Castro et al., 2017; Stadler and Richly, 2017).
3.3 MicroRNAs
MicroRNAs (miRNAs) constitute an important class of noncoding RNA and are defined as short (18-25 nucleotides in length), single-strand molecules which exert their actions at the post-transcriptional level (Figure 4 and 5). miRNAs demonstrate complementary interaction with specific genes and can modulate gene expression. Cellular miRNAs are synthesized in a sequential manner and occur in both the nucleus and the cytosol. Within the nucleus, the genome comprises numerous miRNAs genes which will be transcribed to form a single large primary miRNA (pri-miRNAs) transcript by RNA polymerase II. Because this pri-miRNA transcript consists of multiple miRNA loci, it undergoes further processing by a complex called Drosha/DGCR8, a class of RNaseIII enzymatic complex, to generate shorter hairpin-loop structures termed as precursor miRNAs (pre-miRNAs). This pre-miRNA is exported into the cytosol by Exportin-5 in a RAN-GTP dependent manner. Within the cytosol, another complex belonging to another class of RNaseIII enzymes termed ‘Dicer/TRBP’ exerts its action. First, Dicer processes pre-miRNAs into a duplex of mature miRNAs, which undergoes asymmetrical unwinding by the Dicer/TRBP complex to yield a single-stranded mature miRNA. It will then load itself into a ribonucleoparticle to yield an RNA-induced silencing complex (RISC), which is the active form of the gene silencing complex (Figure 5) (Chuang and Jones, 2007; Moutinho and Esteller, 2017; Yao et al., 2019).
Together, the single-stranded mature miRNAs within the RISC complex identify the 3’ untranslated region (3’-UTR) of targeted mRNAs. Gene silencing by miRNAs can occur in two ways, depending on its complementarity with the targeted mRNAs. miRNAs which bind to targeted mRNAs with perfect complementarity induce rapid deadenylation and decapping, which leads to mRNA degradation. On the other hand, an imperfect complementarity binding of miRNAs to targeted mRNAs tends to result in translational suppression. While the activity of miRNAs tends to result in gene downregulation, miRNAs may also promote gene upregulation. For instance, a group of miRNAs can bind to 5’ untranslated region (5’-UTR) of targeted mRNAs to promote ribosomal protein expression, aiding in translation. Interestingly, given that miRNAs are short and can only recognize partial sequences within targeted mRNAs, miRNAs can perform its function simultaneously in a plethora of mRNAs and yet produce diverse outcomes in a cell-dependent manner (Figure 5). Besides modulating the level of gene expression, miRNAs can regulate key epigenetic remodelers such as DNMTs, TETs, HDACs, and HMTs. Notably, this epigenetic regulation by miRNAs is not one-sided, as the expression of miRNAs can be modulated by a myriad of factors, such as DNA methylation, histone modifications, and transcription factors (Figure 4 and 5). Given the complicated mode of action of miRNAs, it is not surprising that miRNAs play diverse roles in many biological processes such as cellular proliferation, differentiation, development, metabolic processes, and apoptosis (Chuang and Jones, 2007; Qureshi, 2011; Udali et al., 2013; Khoshnam et al., 2017; Moutinho and Esteller, 2017; Yao et al., 2019).
4.0 Conclusion
DR has a long historical root and has been practiced by many groups since antiquity. Research on the biological effects of DR and its underlying mechanisms has only become commonplace relatively recently, but has gained enormous interest as a potential ‘holy grail’ for extension of health and lifespan. Supporting evidence has come from studies of many organisms, and have identified a plethora of molecular players and signaling pathways induced or modulated by DR associated with profound metabolic effects that appear capable of counteracting the onset and development of disease. However, the nature of DR-induced effects is complex and often varied and non-translatable between different organisms. Given that DR is a non-pharmacological and non-genetic lifestyle intervention that be readily adopted by many individuals, it is appears that DR represents an environmental stressor that can truly influence an individual’s epigenetic landscape. Hence, it is paramount to also investigate the nature of epigenetic signatures, and the detailed relationship between both DR and epigenetics will be discussed in part two of this review.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Acknowledgments
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Singapore National Medical Research Council Research Grants (NMRC-CBRG-0102/2016 and NMRC/OFIRG/0036/2017) supported this work.
References
Gavin Yong-Quan Ng1
1Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
David Yang-Wei Fann1
1Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore.
Dong-Gyu Jo2
2School of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea.
Christopher G. Sobey3
3Department of Physiology, Anatomy & Microbiology, School of Life Sciences, La Trobe University, Bundoora, Victoria, Australia.
Thiruma V. Arumugam1,2,3
1Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore. 2School of Pharmacy, Sungkyunkwan University, Suwon, Republic of Korea. 3Department of Physiology, Anatomy & Microbiology, School of Life Sciences, La Trobe University, Bundoora, Victoria, Australia.
Corresponding author:
Thiruma V. Arumugam
Email: phstva@nus.edu.sg
In a new window | Download PPT
Figure 1: Environmental Influences and Genetic Framework. (a) As our early ancestors were often challenged with periods of famine. And their procurement of food was accompanied by regular physical activity, they were presumably faced with periods of obligate food restriction and physical activity. The fundamental need for survival in terms of physical and mental maintenance without the consumption of food for extended periods appears to have shaped physiological, behavioural, and cognitive adaptations, and may thus have become inherited by modern humans within our genetic framework. As a result, the relative abundance of food coupled with our sedentary lifestyle today may be less compatible with the activity our evolutionarily programmed genes. As such, our modern lifestyle may disrupt our biochemical processes, and contribute to a myriad of age-related diseases. Thus, perhaps to some extent mimicking the environment of our ancestors may help to ameliorate the onset and development of diseases. (b) Twins possess high similarities in their genetic framework, yet when subjected to different environmental influences may acquire different traits that affect susceptibility to disease.
In a new window | Download PPT
Figure 2: Mechanism of Dietary Restriction. During DR there is a reduced energy status within the organism, resulting in corresponding decreases in blood glucose, insulin, IGF-1, growth hormones, sex hormones, and T3 thyroid hormones. Also, DR induces a reduction in both oxidative stress and inflammation. Following DR, there are increases in free fatty acids, adiponectin, and cortisol. A lower cellular energy status will lead to reduced mitochondrial activity and aerobic respiration, increasing the AMP:ATP ratio and NAD+ levels. Two major cellular nutrients and energy sensors, AMPK and SIRT1, will be then be activated, respectively. Activated AMPK will inhibit glycogen synthesis, ACC1, ACC2, and HMG-CoA to drive a reduction in fatty acid synthesis, oxidation, and cholesterol synthesis. Activated SIRT1 can enhance ketogenesis and lipolysis, and downregulate glycolysis and glutaminolysis. These effects may be also due to an activation of PPARα, reflecting direct and indirect functions of SIRT1. Activated SIRT1 can also repress the activity of PPARγ to modulate lipid metabolism and enhance fat mobilization in white adipose tissue during DR. Activated SIRT1 can inhibit NF-κB activity, inhibiting the expression of proinflammatory genes. SIRT1 activation is dependent on the increased interaction between FOXO3a and p53 at SIRT1 promoter sites. The physical interaction between FOXO3a and p53, and the subsequent activation of SIRT1, play important roles in repressing cell growth and proliferation. Notably, SIRT1 activation can activate LKB1, which will further activate AMPK, thus creating a positive feedback loop. Reduced levels of insulin, glucose and growth hormone downregulates insulin and IGF-R signaling pathways, inactivating the RAS/MAPK axis, repressing cell growth and proliferation and promoting apoptosis. Repression of IGF-R signaling will include the PI3K pathway, and inhibit downstream mTORC1, promoting autophagy. Inhibition of mTORC1 can also occur via activated AMPK in response to DR. Decreased mTORC1 function can promote ketogenesis and lipolysis, and repress glycolysis and glutaminolysis. Moreover, the corresponding decreased function of mTORC1 also inhibit HIF-1α, which downregulates key biological processes such as glucose metabolism, angiogenesis, and cell cycle progression. The NRF2 pathway is also triggered by DR, which helps to increase production of protective antioxidant enzymes and mitochondrial biogenesis to reduce ROS activity. With lower levels of ROS, there will be reduced DNA damage and a maintained genome stability. Lower energy status will prime cells to low-intensity stress, and this hormetic mechanism will stimulate better management of stress, upregulate DNA repair genes, as well as shifting towards a preferred setting of maintenance and repair (not shown). Overall, DR can act via these mechanisms to counter age-associated diseases and induce longevity.
DR, dietary restriction; IGF-1, insulin growth factor 1; AMP, adenosine monophosphate; ATP, adenosine triphosphate; NAD+, nicotinamide adenine dinucleotide; AMPK, adenosine monophosphate kinase; SIRT1, sirtuin 1 deacetylase; ACC1 and ACC2, acetyl-coenzyme A carboxylase 1 and 2; HMG-CoA, 3-hydroxy-3-methyl-glutaryl-coenzyme A; PPARα and PPARγ, peroxisome proliferator-activated receptor gamma alpha and gamma; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; FOXO3a, forkhead box O3; LKB1, liver kinase B1; IGF-R, insulin growth factor receptor; RAS/MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; mTORC1, mammalian target of rapamycin complex 1; HIF-1α, hypoxia-inducible factor 1 alpha; NRF2, nuclear factor erythroid 2-related factor 2 ;ROS, reactive oxygen species
In a new window | Download PPT
Figure 3: Effects of Dietary Restriction. DR can induce a myriad of systemic effects. In turn, a robust metabolic switching ensues, which results in molecular, cellular, and metabolic adaptations that delay the onset and development of many age-associated diseases and promote longevity.
In a new window | Download PPT
Figure 4: Overview of Epigenetic Modifications. Epigenetic modifications can be broadly categorized into DNA methylation, histone modifications, histone remodeling, and microRNA involvement. These epigenetic modifications to the genome occur via enzymatic modifications to either the DNA sequence directly or through histone proteins that form part of the hierarchical packing of the chromosomal DNA. In turn, gene expression can be modulated directly or indirectly via an overall modification to chromatin structure and accessibility. The epigenetic signature landscape across the genome is collectively termed the ‘epigenome’, and unique epigenetic tags function as distinct microdomains within the nucleus to regulate differential patterns of gene expression.
In a new window | Download PPT
Figure 5: Mechanisms of Epigenetic Modifications. During DNA methylation, a methyl group is added to carbon position five of the cytosine ring at either CpG dinucleotide islands or CpG shores. DNA methylation is catalyzed by DNMTs, whereas DNA demethylation is catalyzed by TETs. DNA methylation tends to be associated with gene silencing whereas DNA demethylation is associated with transcriptional activation. Chromosomal DNA within the eukaryotic genome is often interlaced with distinct packing and folding, resulting in a distinct organizational structure. At the fundamental level, a nucleosome structural core is first formed through wrapping of a 147 base-pair double helical DNA around an octamer of histone proteins consisting of pairs of H2A-H2B dimers and H3-H4 dimers. Between two nucleosome cores, DNA that is not wrapped around nucleosomes is referred to as ‘linker DNA’, which tends to be associated with H1 histone proteins. A plethora of post-translational modifications (such as acetylation, methylation, phosphorylation, sumoylation, biotinylation, and ubiquitination) can occur on the amino terminal tail protruding from each histone subunit within the nucleosome complex. These histone modifications can affect the relative packing of DNA around these nucleosome cores, and may determine the relative accessibility for the machinery to initiate replication and transcription. Chromatin structure can be modified by ATP-dependent histone remodeling complexes, using the energy from ATP hydrolysis to locally disrupt the interaction between DNA and histones. These remodelers mediate alterations to chromatin structure via nucleosome sliding, eviction, assembly, spacing, histone dimers eviction or replacement, and even entire histone replacement. In addition, miRNAs demonstrate complementary interaction with specific genes and modulate gene expression. Cellular miRNAs are synthesized in a sequential manner and occur in both the nucleus and the cytosol. Within the nucleus, the genome comprises numerous miRNA genes transcribed by RNA polymerase II to form a single large pri-miRNA transcript. Because this pri-miRNAs transcript consists of multiple miRNA loci, it undergoes further processing by a complex called Drosha/DGCR8, a class of RNaseIII enzymatic complex, to generate shorter hairpin-loop structurers termed ‘pre-miRNAs’. This pri-miRNA is exported into the cytosol by Exportin-5 in a RAN-GTP dependent manner. Within the cytosol, another complex belonging to another class of RNaseIII enzymes, termed ‘Dicer/TRBP’ exert its action. Dicer processes the pri-miRNAs into a duplex of mature miRNAs, which undergoes asymmetrical unwinding by the Dicer/TRBP complex to yield a single-stranded mature miRNA. This single-stranded form loads itself into a ribonucleoparticle to yield an RISC, which is the active form of the gene silencing complex. Together, the single-stranded mature miRNAs within the RISC complex will identify the 3’-UTR of targeted mRNAs. The mode of action of gene silencing mediated by miRNAs can occur in two ways, depending on its complementarity with the targeted mRNAs. miRNAs which bind to targeted mRNAs in perfect complementarity induce rapid deadenylation and decapping, leading to mRNA degradation. An imperfect complementarity binding of miRNAs to targeted mRNAs result in translational suppression.
DNMTs, DNA methyltransferase; TETs, ten-eleven translocation; pri-miRNAs, primary miRNA; pre-miRNAs, precursor miRNAs; RISC, RNA-induced silencing complex; 3’-UTR, 3’ untranslated region.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14312 | 73 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA