Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Ischemic postconditioning for stroke treatment: current experimental advances and future directions
Time:2020-05-05
Number:11475
Author Affiliations

Conditioning Medicine 2020. 3(2):104-115.
Abstract
Ischemic postconditioning (IPostC) protects against brain injury induced by stroke and is a potential strategy for ischemic stroke treatment. Understanding its underlying mechanisms and potential hurdles is essential for clinical translation. In this review article, we will summarize the current advances in IPostC for stroke treatment and the underlying protective mechanisms. Strong evidence suggests that IPostC reduces brain infarct size, attenuates blood-brain barrier (BBB) damage and brain edema, and improves neurological outcomes. IPostC also promotes neurogenesis and angiogenesis at the recovery phase of ischemic stroke. The protective mechanisms involve its effects on anti-oxidative stress, anti-inflammation, and anti-apoptosis. In addition, it regulates neurotransmitter receptors, ion channels, heat shock proteins (HSP) 40/70, as well as growth factors such as BDNF and VEGF. Furthermore, IPostC modulates several cell signaling pathways, including the PI3K/Akt, MAPK, NF-κB, and the Gluk2/PSD95/MLK3/MKK7/JNK3 pathways. We also discuss the potential hurdles for IPostC’s clinical translation, including insufficient IPostC algorithm studies, such as therapeutic time windows and ischemia-reperfusion periods and cycles, as well as its long-term protection. In addition, future studies should address confounding factors such as age, sex, and pre-existing conditions such as hypertension and hyperglycemia before stroke onset. At last, the combination of IPostC with other treatments, such as tissue plasminogen activator (t-PA), merits further exploration.
Keywords: stroke; cerebral ischemia; ischemic postconditioning; oxidative stress; neuroinflammation
Abstract
Ischemic postconditioning (IPostC) protects against brain injury induced by stroke and is a potential strategy for ischemic stroke treatment. Understanding its underlying mechanisms and potential hurdles is essential for clinical translation. In this review article, we will summarize the current advances in IPostC for stroke treatment and the underlying protective mechanisms. Strong evidence suggests that IPostC reduces brain infarct size, attenuates blood-brain barrier (BBB) damage and brain edema, and improves neurological outcomes. IPostC also promotes neurogenesis and angiogenesis at the recovery phase of ischemic stroke. The protective mechanisms involve its effects on anti-oxidative stress, anti-inflammation, and anti-apoptosis. In addition, it regulates neurotransmitter receptors, ion channels, heat shock proteins (HSP) 40/70, as well as growth factors such as BDNF and VEGF. Furthermore, IPostC modulates several cell signaling pathways, including the PI3K/Akt, MAPK, NF-κB, and the Gluk2/PSD95/MLK3/MKK7/JNK3 pathways. We also discuss the potential hurdles for IPostC’s clinical translation, including insufficient IPostC algorithm studies, such as therapeutic time windows and ischemia-reperfusion periods and cycles, as well as its long-term protection. In addition, future studies should address confounding factors such as age, sex, and pre-existing conditions such as hypertension and hyperglycemia before stroke onset. At last, the combination of IPostC with other treatments, such as tissue plasminogen activator (t-PA), merits further exploration.
Keywords: stroke; cerebral ischemia; ischemic postconditioning; oxidative stress; neuroinflammation
1. Introduction
Stroke remains a leading cause of morbidity and mortality worldwide (Kuklina et al., 2012; Campbell et al., 2019), with ischemic stroke accounting for more than 85 percent of all stroke cases (Benjamin et al., 2018). Currently, the tissue plasminogen activator (t-PA) is the only FDA approved drug treatment used to dissolve blood clots due to ischemic stroke (Chapman et al., 2014). However, t-PA has a limited therapeutic time window of 4.5 hours, and treatment beyond this point increases the risk of hemorrhagic transformation 10-fold, a severe complication that could worsen stroke outcomes (Sussman and Connolly, 2013; Chen et al., 2017; Zhang et al., 2019). Recent advances showed that delayed endovascular thrombectomy improves functional outcomes from 6 to 16 and even 24h after ischemia, but this only applies to a small, select group of patients (Albers et al., 2018; Nogueira et al., 2018; Ma et al., 2019). Therefore, it is crucial to develop new therapeutic strategies for the treatment of ischemic stroke. As a result, scientists have been searching for drug candidates to target various ischemic cascades for stroke treatment. Although many drug candidates have been proven effective in attenuating brain injuries in animal stroke models, none have been successfully translated into clinical use (Hoyte et al., 2004). In facing these setbacks, which mainly target a single ischemic cascade or cell signaling pathway, scientists have explored whether endogenous protective mechanisms, such as pre- and postconditioning, could be employed for ischemic stroke treatment (Zhao, 2007).
Our group is among the first to demonstrate that ischemic postconditioning (IPostC) protects against ischemic brain injury (Zhao et al., 2006; Zhao, 2007). IPostC is defined in contrast to ischemic preconditioning (IPreC). While IPreC, which is induced by a sub-lethal ischemia, is conducted before stroke onset, IPostC is performed after stroke onset by a single or a series of mechanical interruptions of reperfusion. Classic postconditioning has evolved into a term that represents a broad range of stimuli or triggers, including pharmacological agents or brief episodes of ischemia conducted in a remote organ (Zhao, 2009; Xie et al., 2018). Remote IPostC shows neuroprotective effects in multiple cerebral vascular insult models, including the ischemic stroke model (Kitagawa et al., 2018; Liu et al., 2020) and the subarachnoid hemorrhage (SAH) model (Hu et al., 2018), It also accelerates hematoma resolution and improves neurological outcomes in the intracerebral hemorrhage (ICH) model via AMPK-dependent immune regulation (Vaibhav et al., 2018). In this brief article, we will exclusively review the protective effects of IPostC in ischemic stroke and its underlying protective mechanisms, and then discuss the potential problems and future directions in IPostC research.
2. IPostC neuroprotection effects
The protective effects of IPostC on ischemic injury and recovery are characterized by changes in infarction, neurological deficit, blood-brain barrier (BBB) leakage, brain edema, angiogenesis, and neurogenesis. Therefore, we will first summarize the protective effects of IPostC on these pathological events (Table 1).
Table 1. Summary of representative studies on postconditioning for ischemic stroke treatment.
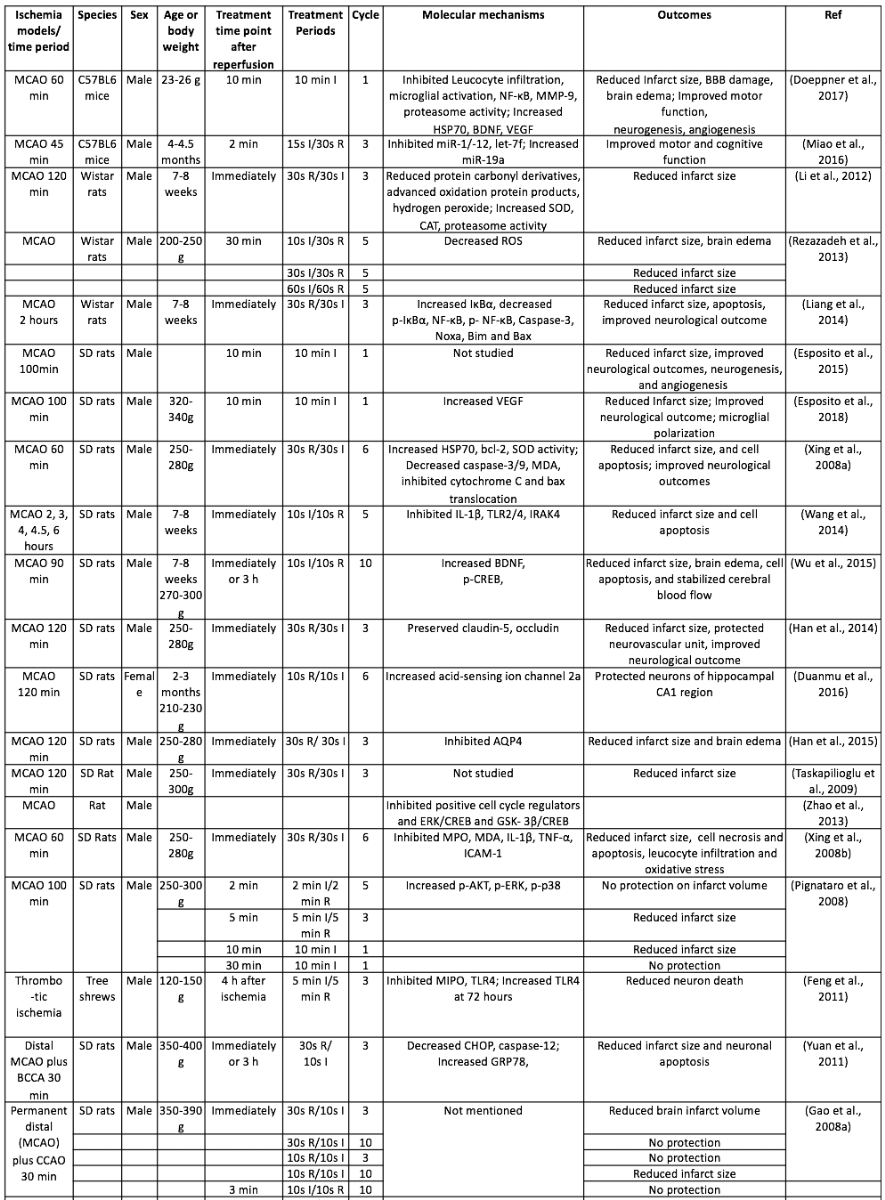
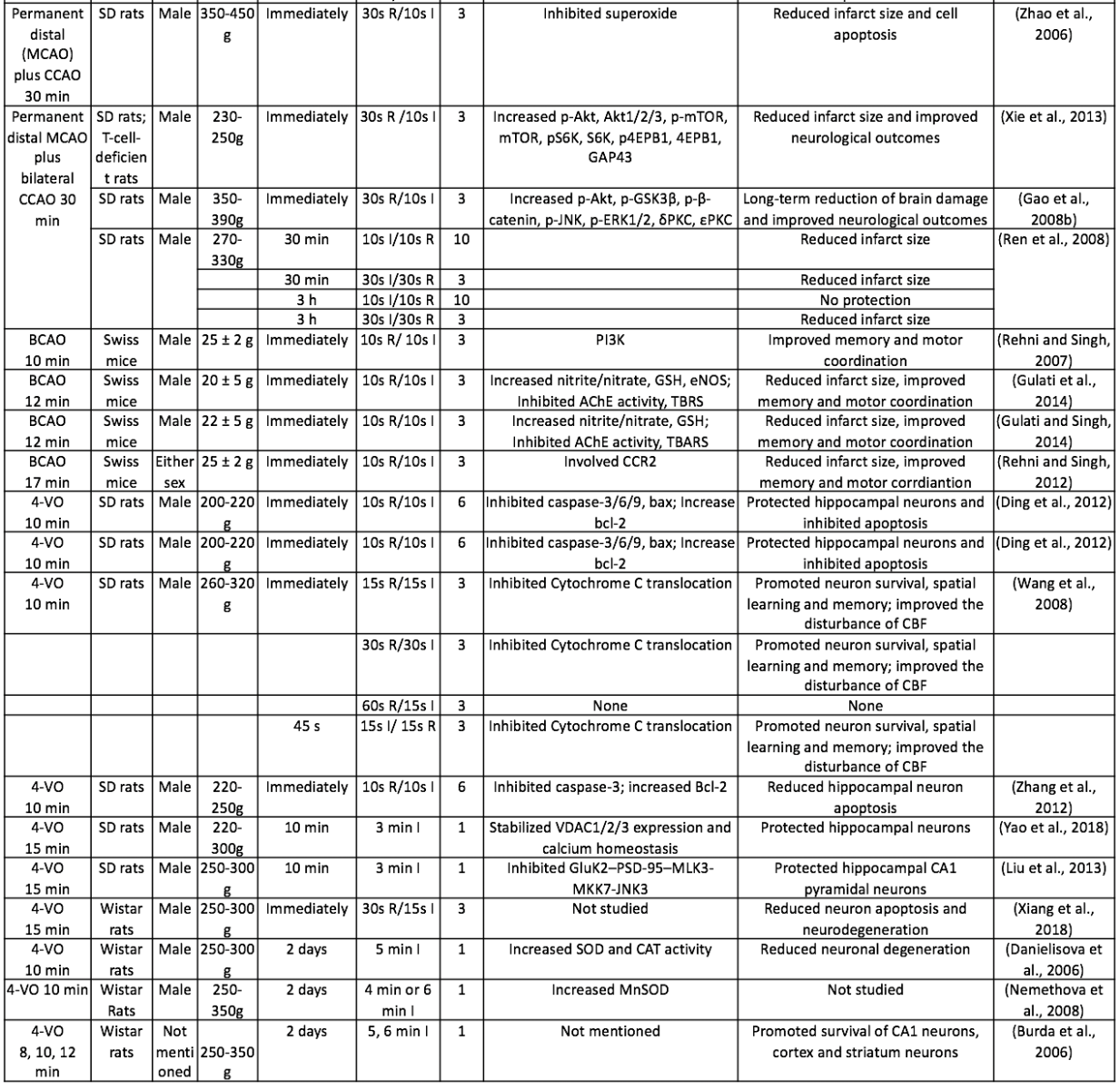
I, Ischemia; R, Reperfusion; 4-VO, four vessel occlusion; AQP4, Aquaporin 4; BCAO, bilateral carotid artery occlusion; BDNF, Brain-derived neurotrophic factor; CAT, Catalase; CCR2, C-C chemokine receptor type 2; CHOP, C/EBP-homologous protein; GAP43, Growth Associated Protein 43; Gluk2, Glutamatergic kainate receptor subunit 2; GSH, Glutathione; HSP70, heat shock protein 70; IRAK4, Interleukin-1 receptor-associated kinase 4; JNK3, c-Jun N-terminal kinase 3; MCAO, middle cerebral artery occlusion; MDA, Malondialdehyde; MLK3, Mixed-lineage protein kinase 3; MMP-9, Matrix metallopeptidase 9; MPO, Myeloperoxidase; mTOR, mammalian target of rapamycin; PSD-95, postsynaptic density protein 95; ROS, Reactive oxygen species; SOD, Superoxide dismutase; TBARS, Tihobarbituric acid reactive species; TLR4, Toll-like receptor 4; VDAC, Voltage-dependent anion channel; VEGF, Vascular endothelial growth factor
2.1 Brain infarction
We and others have reported that IPostC reduces brain infarction in focal and global cerebral ischemia in mice and rats (Xing et al., 2008b; Rehni and Singh, 2012; Gulati and Singh, 2014; Gulati et al., 2014; Han et al., 2014; Wang et al., 2014; Esposito et al., 2015; Wu et al., 2015; Doeppner et al., 2017). Most studies evaluated brain infarct sizes by using triphenyltetrazolium chloride (TTC) staining at 24h (Xing et al., 2008b, a; Taskapilioglu et al., 2009; Yuan et al., 2011; Rehni and Singh, 2012; Gulati and Singh, 2014; Gulati et al., 2014; Han et al., 2014; Han et al., 2015; Doeppner et al., 2017; Sun et al., 2018), 48h (Zhao et al., 2006; Ren et al., 2008; Xie et al., 2013; Wang et al., 2014), and 72h after ischemic onset (Xing et al., 2008b; Wu et al., 2015). Although one study suggests that early neuroprotection did not translate into long-term neuronal survival (Doeppner et al., 2017), we and others have shown that rapid IPostC, which was conducted within a few minutes after reperfusion, reduced infarct volume measured at 2 weeks or 1 month after ischemia (Gao et al., 2008; Esposito et al., 2015), suggesting long-term benefits of rapid IPostC.
We have found that ischemic severity determines the extent of protection that IPostC may afford, i.e. the longer the ischemic period, the less protection IPostC can achieve (Zhao et al., 2006). This finding is supported by another study, which shows that rapid IPostC did not reduce infarct sizes and brain edema when the ischemic period was extended to 4.5h (Wang et al., 2014).
We and others have evaluated the IPostC therapeutic time windows. We defined rapid IPostC when it is induced immediately or a few minutes after reperfusion onset, while delayed IPostC occurs when it is initiated a few hours after reperfusion onset. We found that delayed IPostC performed at 3h and 6h after ischemia/reperfusion robustly reduced infarction size in the rat stroke model (Ren et al., 2008). Compared with focal cerebral ischemia, the therapeutic time window in transient forebrain ischemia is longer, as delayed IPostC initiated as late as 2d after cerebral ischemia could still exert anti-oxidative stress effects and protect neurons (Burda et al., 2006; Danielisova et al., 2006; Nemethova et al., 2008).
Since preconditioning and postconditioning share some similar protective mechanisms, it is tempting to test whether preconditioning and postconditioning have synergistic effects. The combination of preconditioning with postconditioning showed greater neuroprotection (Wu et al., 2015). However, Taskapilioglu et al. (2009) showed contradictory results in that the combination of preconditioning and postconditioning did not yield further protection. Such inconsistent results could be due to the different experimental protocols employed in different studies.
In short, IPostC can reduce brain infarct size, and larger neuroprotection is achieved when IPostC is applied at an earlier time point of reperfusion. The exact therapeutic time window of IPostC varies in different stroke models, and whether preconditioning plus postconditioning has synergistic effects needs further investigation.
2.2 Neurological functions
Reductions in brain infarct sizes are often associated with neurological functional recovery. IPostC improves motor function, motor coordination, and short-term memory at the acute phase of ischemic stroke (Rehni and Singh, 2007; Xing et al., 2008b; Rehni and Singh, 2012; Gulati and Singh, 2014; Gulati et al., 2014; Han et al., 2014; Wu et al., 2015; Miao et al., 2016; Doeppner et al., 2017), attenuates neurologic deficit, and improves cognitive and memory function at the late phase of ischemic stroke (Rehni and Singh, 2007; Esposito et al., 2015; Miao et al., 2016). Notably, the improvement of neurological function at the acute phase by IPostC was abolished when the transient focal ischemia period was extended to 4.5h (Wang et al., 2014). We reported that delayed postconditioning at 6h after permanent focal ischemia provided long-term protection in attenuating motor asymmetry (Ren et al., 2008).
2.3 Blood-brain barrier damage and brain edema
The neuroprotective effects of IPostC are usually linked to the preservation of the BBB integrity and reduction in brain edema in stroke models (Han et al., 2014; Doeppner et al., 2017; Xiang et al., 2018). During the cerebral ischemia-reperfusion injury, activation of the oxidative stress-mediated matrix metalloproteinases (MMPs) disrupts the tight junction proteins and extracellular matrix components of BBB (Haorah et al., 2007; Gu et al., 2012; Chen et al., 2018). IPostC significantly inhibits the oxidative stress and MMP-9 activity in the ischemic brain (Liu et al., 2012; Doeppner et al., 2017), preserves the ultrastructure of the neurovascular unit, protects the tight junction protein, claudin-5 and occludin, the extracellular matrix laminin, and fibronectin (Liu et al., 2012; Han et al., 2014). In addition, the protection of the BBB integrity is associated with the reduction of leukocyte infiltration and the attenuation of microglial activation (Doeppner et al., 2017). Therefore, IPostC protects the BBB possibly by inhibiting neuroinflammation and attenuating the oxidative stress-mediated activation of MMP-9, and preserving tight junction proteins and extracellular matrix.
The formation of brain edema during ischemic stroke could be due to both cytotoxic and vasogenic changes (Heo et al., 2005). Therefore, BBB protection could contribute to the attenuation of brain edema. Evidence showed that IPostC reduced brain edema in focal ischemia models, as calculated by brain swelling or wet-dry weight methods (Rezazadeh et al., 2013; Han et al., 2015). In addition, the reduction of brain edema is associated with the inhibition of membrane water channel aquaporin-4 (AQP4), which regulates water flux in the ischemic brain (Han et al., 2015).
2.4 Neurogenesis and angiogenesis
Neurogenesis and angiogenesis play important roles in brain functional recovery after stroke, which are promoted by IPostC. In a middle cerebral artery occlusion (MCAO) model, IPostC significantly enhanced neurogenesis and angiogenesis 2 weeks after ischemia, as indicated by the double staining of doublecortin/BrdU and collagen-IV/Ki67 in the ischemic brain (Anrather and Hallenbeck, 2013). In contrast, in another study using the mouse MCAO model, IPostC itself did not promote endogenous neurogenesis or angiogenesis when measured 28d after cerebral ischemia (Doeppner et al., 2017), but IPostC could facilitate the intracerebral transplant of neural progenitor cells and promoted cell survival in the post-ischemic brain (Doeppner et al., 2017). Therefore, IPostC may provide a favorable microenvironment for the ischemic brain repair process.
3. Molecular mechanisms involved in IPostC’s protection
IPostC triggers multiple molecular mechanisms, including its anti-oxidative, anti-inflammatory effects, anti-apoptotic effects, and its regulation of the neurotransmitter and ion channels that maintain calcium balance, as well as the induction of growth factors and heat shock proteins (Table 1, Figure 1). These cascades link to multiple kinase signaling pathways, including the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt), mitogen-activated protein kinase (MAPK), and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways (Table 1, Figure 2), which are detailed below.
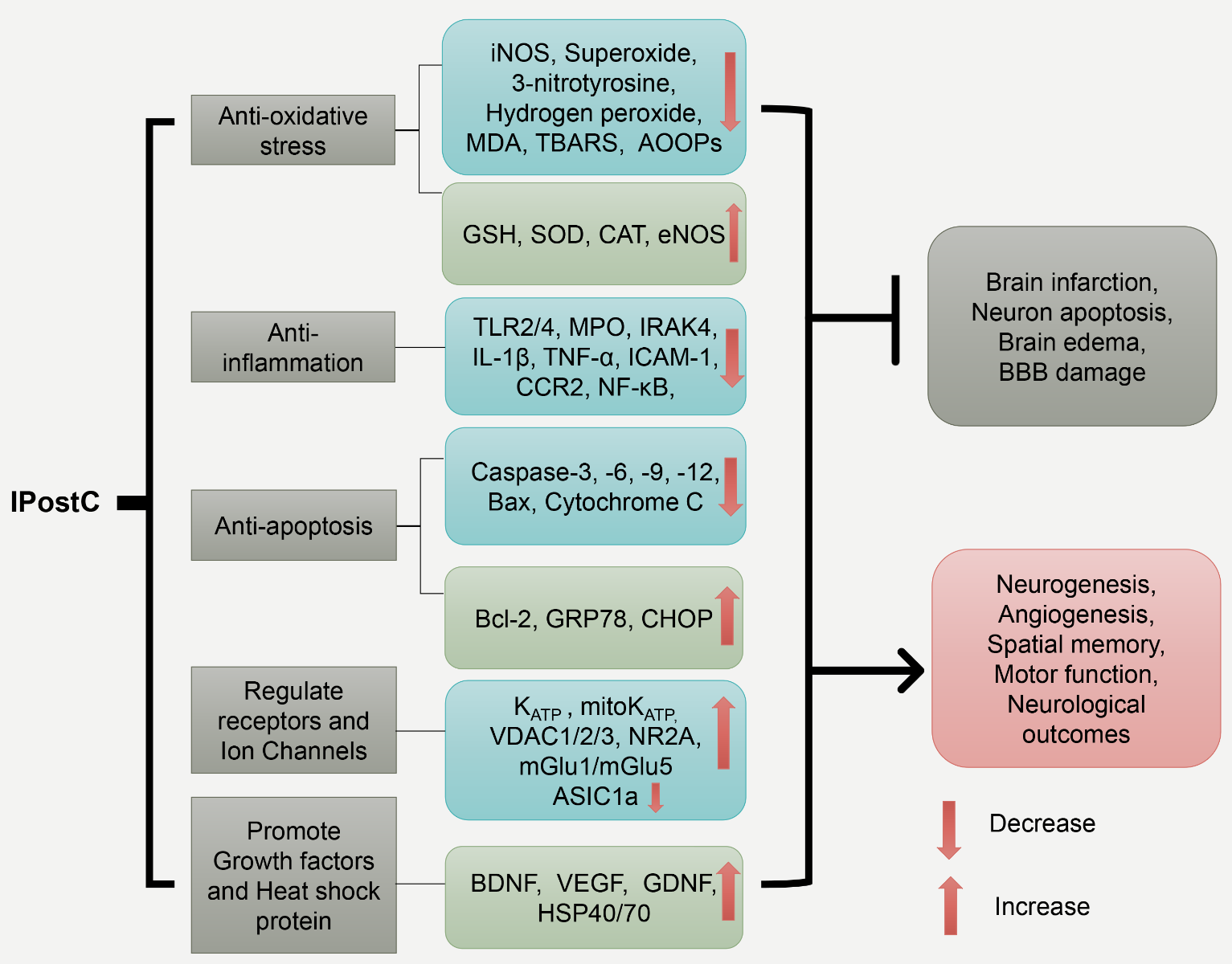
In a new window | Download PPT
Figure 1: The schematic represents IPostC’s major protective mechanisms. The five major protective mechanisms mediated by IPostC are listed in the first row, and their corresponding molecules are shown in the second row. Together, these molecular events lead to the inhibition of brain infarction, neuronal apoptosis, edema, and BBB leakage, while promoting neurogenesis and angiogenesis related to functional recovery. Inducible nitric oxide synthase (iNOS); malondialdehyde (MDA); thiobarbituric acid reactive species (TBARS); advanced oxidation protein products (AOOPs); Glutathione (GSH); Superoxide dismutase (SOD); Catalase (CAT); endothelial nitric oxide synthase (eNOS); toll-like receptor 2/4 (TLR2/4); Myeloperoxidase (MPO); interleukin-1 receptor-associated kinase 4 (IRAK4); Interleukin 1 beta (IL-1β); Tumor necrosis factor α (TNF-α); Intercellular Adhesion Molecule 1 (ICAM-1); C-C chemokine receptor type 2 (CCR-2); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); glucose-regulated protein 78 (GRP78); C/EBP-homologous protein (CHOP); mitochondrial potassium ATP-dependent channel (mitoKATP); voltage-dependent anion channel protein (VDAC); glutamate receptor ε1 (NR2A); metabotropic glutamate receptor 1/5 (mGlu1/mGlu5); Acid-sensing ion channel 1a (ASIC1a); brain-derived neurotrophic factor (BDNF); vascular endothelial growth factor (VEGF); glial cell-derived neurotrophic factor (GDNF); heat shock protein 40/70 (HSP40/70); blood-brain barrier (BBB).
3.1 Anti-oxidative actions
IPostC protects against oxidative stress. In both global and focal cerebral ischemia/reperfusion models, IPostC increases the activities of the endogenous antioxidant enzymes, superoxide dismutase (SOD) and catalase (CAT) (Danielisova et al., 2006; Nemethova et al., 2008; Xing et al., 2008b; Li et al., 2012), and inhibits inducible nitric oxide synthase (iNOS) expression (Wei et al., 2015). As a result, IPostC reduces oxidative products, including superoxide (Zhao et al., 2006), 3-nitrotyrosine (a footprint of peroxynitrite) (Zhao et al., 2014a), and hydrogen peroxide levels. IPostC also prevents lipid peroxidation and protein oxidation as indicated by the reduction of malondialdehyde (MDA), protein carbonyl derivatives, and advanced oxidation protein products (AOPPs) (Xing et al., 2008a, b; Li et al., 2012; Wei et al., 2015). In addition, IPostC reduces thiobarbituric acid reactive species (TBARS) levels, a lipid peroxidation marker, and improves glutathione levels (Gulati and Singh, 2014; Gulati et al., 2014; Doeppner et al., 2017; Xiang et al., 2018). Therefore, IPostC’s anti-oxidative effects may contribute to its neuroprotective effects against cerebral ischemia/reperfusion injury.
3.2 Regulation of neurotransmitter receptors and ion channels
The regulation of ion channels and neurotransmitter receptors by IPostC also contributes to its neuroprotection by maintaining ion stasis (Fan et al., 2017). For IPostC’s effects on the ion channels, Pateliya et al. (2008) showed that IPostC reduces brain infarct size by promoting the opioid receptor and subsequently opening KATP channels, and that the opioid receptor antagonist naloxone or the KATP channel blocker, glibenclamide, significantly abolishes IPostC’s protection. IPostC also protects against ischemic brain damage by modulating the mitochondrial potassium ATP-dependent channel (mitoKATP), and by inhibiting the mitochondrial permeability transition pore (MPTP) opening, subsequently preventing hyperpolarization (Robin et al., 2011). In addition, IPostC inhibits the transcription and expression of the acid-sensing ion channel, ASIC1a, a proton-gated cation channel activated during cerebral ischemia, leading to an excessive calcium entry in the temporoparietal cortex after transient focal ischemia (Pignataro et al., 2011). The down-regulation of ASIC1a by IPostC was dependent on the Akt signaling, as the Akt inhibitor, LY-294002, reversed ASIC1a modulation (Pignataro et al., 2011). Moreover, IPostC stabilizes the expressions of functional mitochondrial voltage-dependent anion channel proteins (VDAC1, VDAC2, and VDAC3), which are essential for calcium homeostasis (Yao et al., 2018).
The N-methyl-D-aspartate (NMDA) receptors are also important neuroprotection mediators of IPostC against cerebral ischemic injury (Celso Constantino et al., 2014). An in vivo study showed that the activation of the NMDA receptor subunit, NR2A, mediates the neuroprotective effects of IPostC by promoting activation of pro-survival signaling such as the extracellular signal regulated kinase (ERK) and cAMP response element binding proteins (CREB), and inducing brain-derived neurotrophic factor (BDNF) expression, which is blocked by the NR2A antagonist NVP-A (Zhang et al., 2015). An ex vivo study using rat organotypic hippocampal slices showed that metabotropic glutamate receptor 1/5 (mGlu1/mGlu5) contributes to IPostC’s protection, as the antagonist of mGlu1/mGlu5 abolished the protection (Scartabelli et al., 2008). This is strengthened by the fact that the mGlu1 and mGlu5 agonist 3,5-dihydroxyphenylglycine (DHPG) protects organotypic hippocampal slices from oxygen and glucose deprivation (OGD)-induced cell damage, which is dependent on Akt activation. Therefore, neurotransmitters and ion channel regulations, as well as the maintenance of ion homeostasis are important factors for IPostC-induced protection against ischemic brain injury.
3.3 Anti-apoptotic effects and the potential role of autophagic effects
IPostC’s anti-apoptotic effects have been reported by several ischemic stroke studies (Zhao et al., 2006; Xing et al., 2008a; Yuan et al., 2011; Ding et al., 2012; Zhang et al., 2012; Liang et al., 2014; Wang et al., 2014). IPostC significantly inhibits hippocampal neuronal apoptosis by inhibiting pro-apoptotic caspase-3 activation and promoting anti-apoptotic Bcl-2 expression (Zhang et al., 2012). In addition, IPostC down-regulates caspase-3/6/9 and pro-apoptotic Bax expression, and increases Bcl-2 in the hippocampal CA1 region (Ding et al., 2012). Moreover, IPostC also inhibits Bax translocation into the mitochondria and prevents the release of cytochrome C into the cytosol in a focal cerebral ischemia model (Xing et al., 2008b). Similarly, in a global ischemia model, IPostC inhibits cytosolic cytochrome C release, thus preventing subsequent neuronal cell death, and improves the learning and memory deficit (Wang et al., 2008). Furthermore, IPostC attenuates endoplasmic reticulum (ER)-stress-mediated apoptosis as indicated by the inhibition of C/EBP-homologous protein (CHOP), caspase-12, and increased levels of glucose-regulated protein 78 (GRP78) in the ischemic brain (Yuan et al., 2011). It is suggested that the PI3K/Akt pathway is necessary for such protection, as its inhibitor, LY294002, significantly abolishes IPostC’s effects on ER-stress related molecules and apoptosis (Yuan et al., 2011).
Autophagy also plays an important role in IPostC-mediated neuroprotection. One study reported that IPostC induces autophagy, which subsequently inhibits apoptosis (Sun et al., 2018). The neuroprotective effects of IPostC are reversed by the autophagy inhibitor, 3-methyladenine (3-MA) (Sun et al., 2018). Controversially, another study showed that IPostC inhibited autophagy, which contributed to neuroprotection (Gao et al., 2012). 3-MA attenuated the ischemic insult (Gao et al., 2012). Such contradictory results indicate the complex roles of autophagy in ischemic stroke injury probably depend on the severity of the ischemic event. Indeed, mild autophagy may protect the ischemic brain while excessive autophagy could cause damage (Feng et al., 2017). Therefore, IPostC may play an important role in balancing autophagy in the ischemic brain in different stroke models, and such roles need further verification.
3.4 Anti-neuroinflammatory effects
IPostC inhibits neuroinflammation in the post-ischemic brain. We reported that IPostC reduces leucocyte infiltration, including monocytes, CD4 T cells, CD8 T cells, and B cells, inhibits microglial activation in the ischemic brain, and attenuates systemic lymphopenia (Joo et al., 2013). Nevertheless, IPostC offered no protection against ischemic stroke in T cell-deficient nude rats, suggesting that IPostC protects the ischemic brain through T cell-mediated inflammation (Xie et al., 2013). We further found that IPostC inhibits T cell immunoglobulin and mucin domain containing-3 (Tim-3) expression and its ligand, galectin-9, which results in the elimination of type 1 helper T (Th1) cells (Wei et al., 2015). In addition to the lymphocytes, other studies report that IPostC also inhibits the innate immune response by down-regulating toll-like receptor-2/4 (TLR2/4) expression, thereby inhibiting myeloperoxidase (MPO) expression (Feng et al., 2011), and decreases the transcription and expression of interleukin-1 receptor-associated kinase 4 (IRAK4), subsequently down-regulating interleukin1 beta (IL-1β) in the ischemic brain (Wang et al., 2014). In addition, NF-κB is an essential downstream regulator of TLR’s activation in ischemic brain injury (Pradillo et al., 2009; Abdul et al., 2019). IPostC inhibits NF-κB expression and its translocation into the nucleus, and at the same time it promotes the expression of the NF-κB inhibitor, alpha (IκB-α) (Doeppner et al., 2017). In addition to inhibiting IL-1β, IPostC also decreases interleukin 6 (IL-6) (Kong et al., 2013), tumor necrosis factor alpha (TNF-α), and the intercellular adhesion molecule-1 (ICAM-1), the last being vital for leucocyte adhesion and infiltration (Xing et al., 2008a). Furthermore, C-C chemokine receptor type 2 (CCR-2) is involved in the neuroprotective effects of IPostC, as a selective CCR-2 antagonist abolished its neuroprotective effects (Rehni and Singh, 2012). Since CCR-2 is a pro-inflammatory monocyte marker, the role of monocytes in the neuroprotection of IPostC merits further investigation.
3.5 Increases in growth factors and heat shock proteins
IPostC promotes the expression of several growth factors in the ischemic brain, including BDNF and vascular endothelial growth factor (VEGF), which may contribute to neuronal repair and angiogenesis. For instance, IPostC enhances the concentrations of BDNF, glial cell-derived neurotrophic factor (GDNF), and VEGF in the ischemic brain with post-stroke neural progenitor cells (NPCs) transplantation, which is associated with enhanced neurogenesis and angiogenesis (Doeppner et al., 2017). Notably, the combination of early and delayed postconditioning further enhances BDNF expression in neurons and astrocytes in the ischemic brain, coupled with the induction of ERK and CREB phosphorylation, and further reduced infarct size (Wu et al., 2015). Similarly, IPostC induces neuronal VEGF expression in the ischemic penumbra and in cultured primary neurons, which binds to the VEGF receptor expressed on microglia, mediating microglial polarization into the anti-inflammatory M2 types (Esposito et al., 2018). Therefore, the release of VEGF may serve as a “help-me” signal for neurons to promote microglia and macrophages into beneficial functional types (Esposito et al., 2018).
Heat shock protein 40 and 70 (HSP40/HSP70) are important chaperones that assist in protein folding under physiological and stress conditions. IPostC enhances HSP70 expression and prevents HSP40 loss in ischemic CA1 tissue (Liang et al., 2012). The induction of HSP70 is vital for neuroprotection, and HSP70 knockdown reverses IPostC- induced reduction of infarction size and proteasome activation (Doeppner et al., 2017). Moreover, HSP70 induction by IPostC is associated with the inhibition of cytochrome c release, preventing cell apoptosis in a focal cerebral ischemia model (Xing et al., 2008b). Similarly, an in vitro study showed that postconditioning increases HSP70 expression and inhibits apoptosis of cultured primary cortical neurons exposed to OGD treatment (Zhao et al., 2014b). Therefore, the induction of HSPs is involved in IPostC neuroprotection possibly by attenuating neuronal cell apoptosis.
3.6 Regulation of kinase signaling pathways
Several protein kinase cell signaling pathways are involved in mediating the neuroprotective effects of IPostC in stroke models, which will be detailed in the following sections.
3.6.1 The PI3K/Akt pathway
Our group has studied the protective efficacy of IPostC and its associated mechanisms. We found that IPostC increases Akt activity and phosphorylation, which contributes to the protective effects of IPostC. The PI3K inhibitor, LY294002, reduces Akt activity, which partially suppresses the protective effects on infarct size reduction (Gao et al., 2008). Other research groups confirmed our results showing that PI3K/Akt inhibitors significantly reduced neuroprotective effects of IPostC (Rehni and Singh, 2007; Pignataro et al., 2008; Liu et al., 2013). The increase in Akt activity is associated with glycogen synthase kinase 3 beta (GSK3β) phosphorylation changes shortly after cerebral ischemia, and changes in the β-catenin phosphorylation pattern in the ischemic core and penumbra (Gao et al., 2008).
Akt directly and indirectly activates mammalian target of rapamycin (mTOR) signaling by enhancing its downstream molecules, including eukaryotic initiation factor 4E-binding protein (4E-BP1) and ribosomal protein S6 kinase (S6K), and promoting cell differentiation, growth, and survival (Martelli et al., 2010; Sabbah et al., 2011). We reported that IPostC increases phosphorylated Akt (p-Akt), phosphorylated mTOR (p-mTOR), phosphorylated S6K (p-S6K), and phosphorylated 4EBP1 (p-4EBP1) in the mTOR pathway and increases the presynaptic growth associated protein 43 (GAP43) protein levels (Xie et al., 2013). On the other hand, the administration of the mTOR inhibitor, rapamycin, abolishes the long-term protective effects of IPostC on reducing brain infarct size, and reduces GAP43 protein levels. Thus, the Akt-mediated mTOR pathway is important for the neuroprotection induced by IPostC (Xie et al., 2013).
Other groups have further explored the crosstalk between Akt and the GluK2–PSD-95–MLK3-JNK3 pathway. Postsynaptic density protein-95 (PSD-95) interacts with the glutamatergic kainate receptor subunit 2 (GluK2), and their downstream signaling molecules, such as mixed lineage kinase 3 (MLK3) and c-Jun N-terminal kinase 3 (JNK3) are important in mediating neuronal loss during cerebral ischemia (Pei et al., 2006; Hu et al., 2009). A recent study shows that IPostC suppresses the formation of the GluK2–PSD-95–MLK3-JNK3 signaling complex in the ischemic brain. These effects are abolished by Akt inhibition (Liu et al., 2013), suggesting that Akt signaling crosstalks with the GluK2–PSD-95–MLK3-JNK3 signaling pathway in ischemic brain injury (Figure 2).
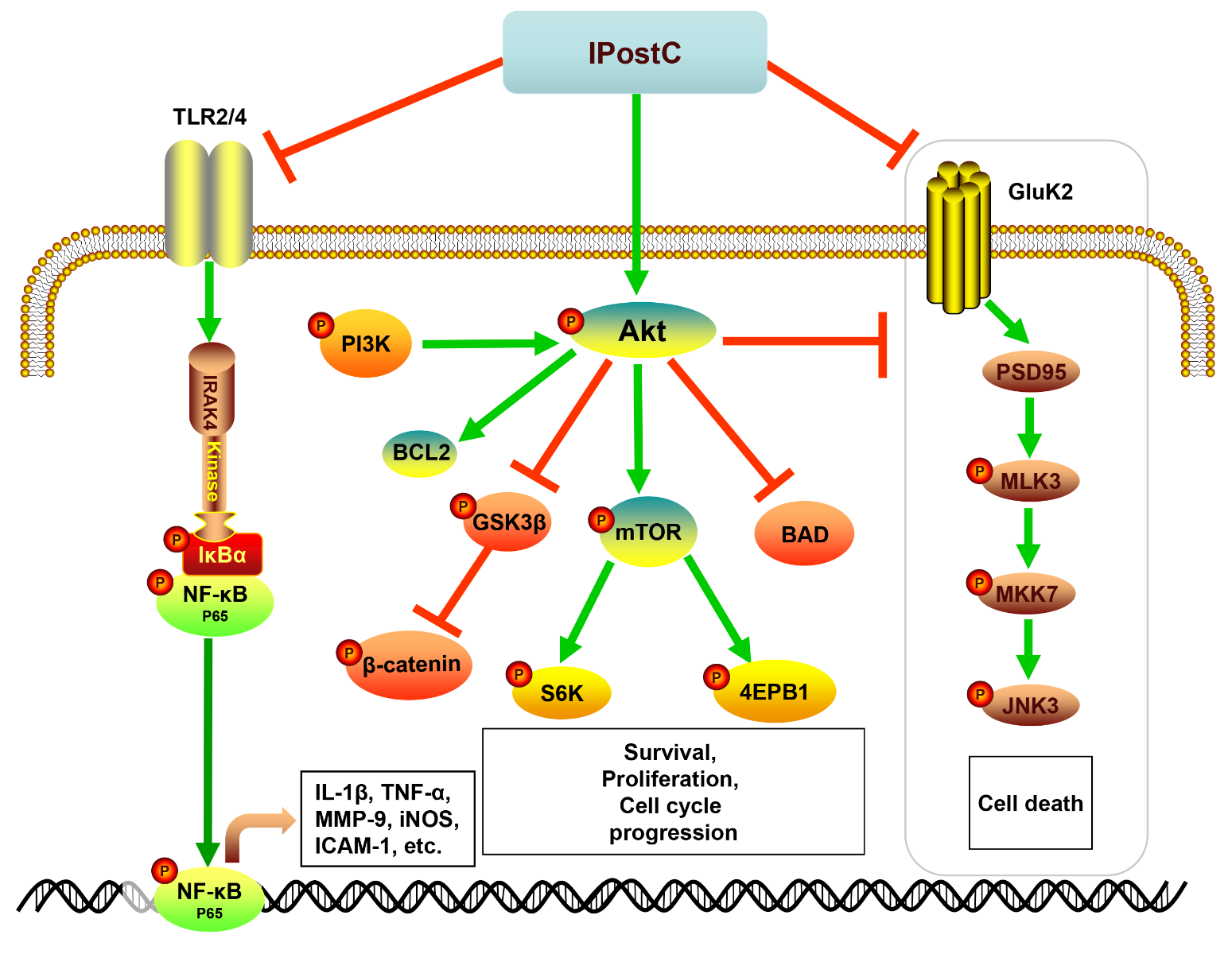
In a new window | Download PPT
Figure 2: The diagram illustrates the major cellular and molecular signaling pathways activated by IPostC for neuroprotection. IPostC inhibits both the TLR2/4/NF-κB pathway and the GluK2/PSD-95/MLK3/MKK7/JNK3 signal complex to inhibit cell death, while promoting the PI3K/Akt pathway for neuronal survival. Toll-like receptor 2/4 (TLR2/4); interleukin-1 receptor-associated kinase 4 (IRAK4); nuclear factor kappa-light- chain-enhancer of activated B cells (NF-κB); NF-κB inhibitor α (IκB-α); protein kinase B (AKT); B-cell lymphoma 2 (Bcl-2); Bcl-2-associated death promoter (BAD); mammalian target of rapamycin (mTOR); ribosomal protein S6 kinase (S6K); eukaryotic initiation factor 4E-binding protein (4E-PB1); glutamatergic kainate receptor subunit 2 (Gluk2); postsynaptic density protein 95 (PSD95); mixed-lineage kinase 3 (MLK3); mitogen-activated protein kinase kinase 7 (MKK7); c-Jun N-terminal kinase 3 (JNK3).
3.6.2 The MAPKs signaling pathway
The MAPKs, including ERK1/2 and p38 MAPK, play important roles in regulating cell survival and cell death after cerebral ischemia (Lai et al., 2014). Yet their roles in IPostC are not fully understood. While one study showed that IPostC increased the phosphorylation of ERK1/2 and p38 MAPK in the ischemic cortex (Pignataro et al., 2008), another study showed contradictory results that IPostC reduced the phosphorylation of ERK1/2 and CREB in the ischemic brain (Zhao et al., 2013). Nevertheless, the ERK1/2 inhibitor, U0126, or p38 MAPK inhibitor, SB203580, did not abolish the neuroprotective effects of IPostC but did block the protective effects of IPreC (Pignataro et al., 2008). These results suggest that activation of ERK1/2 and p38 MAPK may not be necessary for IPostC’s neuroprotection. However, given these uncertain research results, dedicated studies are warranted to further verify the roles of ERK1/2 and p38 MAPK in the neuroprotection of IPostC.
3.6.3 The NF-κB pathway and PKC pathway
The NF-κB pathway is essential for mediating neuroinflammation after stroke. IPostC reduces NF-κB phosphorylation and inhibits its translocation from the cytosol to the nucleus (Liang et al., 2014; Doeppner et al., 2017). In addition, IPostC enhances the expression of the NF-κB inhibitor, IκBα, but inhibits its phosphorylation, increasing its ability to inhibit NF-κB (Liang et al., 2014). In the same study, the authors showed that the inhibition of NF-κB was associated with the reduction of pro-apoptotic proteins, including Noxa, Bim, Bax, and caspase-3, subsequently attenuated brain cell apoptosis, reduced infarct size, and improved neurological outcomes (Liang et al., 2014). Recent studies suggest that the activated TLR2/4 receptor is one of the upstream signaling pathways that activate NF-κB in ischemic injury (Liu et al., 2017; Wu et al., 2018). IPostC inhibited TLR2/4 and IRAK4 (Wang et al., 2014), which may contribute to the inhibition of NF-κB signaling for neuroprotection (Figure 2).
Another important kinase is protein kinase C (PKC). Delta protein kinase C (δPKC) cleavage or membrane translocation increases its activity and promotes neuronal death (Raval et al., 2005; Shimohata et al., 2007b), while an increase in εPKC levels promotes neuronal survival (Shimohata et al., 2007a). Our study showed that IPostC blocks δPKC cleavage and promotes the phosphorylation of εPKC, suggesting that the PKCs pathway is involved in IPostC’s neuroprotection (Gao et al., 2008).
4. Problems and prospects
Despite extensive IPostC studies in the past decade, many problems remain. We will now discuss the major problems and hurdles that may restrict IPostC’s clinical translation and future research directions.
4.1 IPostC algorithms
IPostC algorithms have great impacts on preventive outcomes (Wang et al., 2008; Lee et al., 2018). As summarized in Table 1, in most studies, when IPostC was induced immediately or shortly after reperfusion, it offered significant neuroprotection. Nevertheless, we have reported that the neuroprotection still existed when the IPostC was initiated 3h post-stroke (Ren et al., 2008). Notably, in a global cerebral ischemia model that produced delayed hippocampal neuron death, IPostC initiated as late as 2 days after ischemia still exerted neuroprotective effects, suggesting a wide therapeutic time window of IPostC in a global ischemia model (Burda et al., 2006). In addition to the therapeutic time window, other IPostC parameters are also important, including the duration and cycle numbers of occlusion and reperfusion, but the most optimal algorithms remain unknown. Future studies may further address these issues in different stroke models.
4.2 Long-term protection
It is important to know how long IPostC’s protection may last (Zhao, 2011). One report suggests that IPostC provided only temporary neuroprotection, but not when measured 28d after transient focal ischemia (Doeppner et al., 2017). Nevertheless, we and others have shown that IPostC offered long-term protection against infarction and neurological dysfunctions (Ren et al., 2008), and attenuated delayed neuron cell death in the hippocampus (Xiang et al., 2018). Such contradictory results may be explained by the different ischemia models (focal and global ischemia), animal strains (mouse and rat), or the protocols involved in different studies, which may be compared in future studies by using different models and different animal species and strains.
4.3 Sex
Stroke outcomes differ between males and females (Zhang et al., 2019). Sexual dimorphism exists in inflammatory responses, including microglial activation and polarization, and T cell response after stroke (Ahnstedt and McCullough, 2019; Kerr et al., 2019). Nevertheless, most studies were conducted in male rodents, and few studies have validated the protective effects of IPostC in female rodents. Although two studies have reported the protective effects of IPostC in female stroke models (Rehni and Singh, 2012; Duanmu et al., 2016), they only focused on neurons and neurological functions, and the underlying mechanisms are not known. Taken together, it is necessary to investigate the effects of IPostC in female stroke models, and compare them with males.
4.4 Age
Age is another important factor that affects stroke outcomes (Chen et al., 2010; Zhang et al., 2019). Aging could promote inflammatory responses, which exacerbate ischemic injury (Ritzel et al., 2018), and be a predictor of poor neurological outcomes and mortality (Kammersgaard et al., 2004; Saposnik et al., 2008). Unfortunately, all of the studies listed in Table 1 used young rodents in their experiments. We must keep in mind that the successful application of IPostC in young rodents is not equivalent to studies conducted in the aged group. Indeed, in the myocardial infarction model, postconditioning did not provide benefits to the elderly group while it protected the young age group (Somers et al., 2011). Therefore, it is important to verify whether IPostC protects against stroke and whether the known IPostC algorithms are still applicable in the aged group.
4.5 Confounding factors
Stroke patients usually have preexisting conditions such as hypertension, hyperlipidemia, and hyperglycemia, which greatly affect stroke outcomes (Luitse et al., 2013; Cipolla et al., 2017). Nevertheless, most studies were conducted in healthy animals without these confounding factors, and it remains unclear whether IPostC could exert beneficial effects against ischemic stroke in animals with one or more of these factors. These issues need to be addressed in future studies to promote IPostC’s clinical translation.
4.6 Combination therapy
Given that t-PA is the only FDA approved drug for ischemic stroke, it is important to test whether IPostC can prolong t-PA’s therapeutic time window and reduce t-PA-induced complications of hemorrhagic transformation. We have shown that delayed postconditioning could reverse infarction exacerbated by delayed t-PA treatment (Ren et al., 2008), suggesting that delayed IPostC could be a potential adjuvant therapy with t-PA. Further studies are needed to address whether delayed IPostC could prevent hemorrhagic transformation and improve neurological outcomes. Moreover, the combination of IPostC with other strategies are also important. For example, a study showed that IPostC could provide a favorable microenvironment for facilitating the homing of the transplanted neural stem cells and promoted NPC-mediated neurogenesis and angiogenesis (Doeppner et al., 2017). Therefore, future studies may test whether IPostC can be combined with other treatments to create novel strategies for stroke therapies.
5. Summary
In summary, IPostC is conducted by brief cycles of occlusion-reperfusion after ischemia, altering the hydrodynamics of reperfusion. The therapeutic effects of IPostC have been validated in various cerebral ischemia-reperfusion models with different animal strains, and different treatment protocols. IPostC reduces brain infarct, attenuates BBB damage, and brain edema in the acute phase, promotes neurogenesis and angiogenesis in the recovery phase, and subsequently improves neurological outcomes. Additionally, IPostC’s protection is linked to cellular and molecular mechanisms that inhibit oxidative stresse, inflammatory responses, apoptotic pathways, as well as modulating ion channels and heat shock proteins, and promoting growth factors such as VEGF and BDNF. Furthermore, IPostC modulates multiple kinase signaling pathways, including the PI3K/Akt/mTOR, the NF-κB, and the GluK2–PSD-95–MLK3-JNK3 pathways. Therefore, IPostC seems to have network regulators that modulate signaling pathways for attenuating ischemic brain damage (Anrather and Hallenbeck, 2013). Despite these studies, many critical issues have not been clarified. Future studies may further optimize IPostC algorithms, evaluate long-term outcomes, assess whether potential confounders such as age and sex affect IPostC, and investigate the potential of combining IPostC with other therapies to maximize its neuroprotection.
Funding
This work was supported by a National Institutes of Health Grant (R01NS064136C) (H.Z.).
Acknowledgments
The authors wish to thank Ms. Felicia F. Beppu at the Department of Neurosurgery, Stanford University School of Medicine for language editing.
References
Hansen Chen1
1Department of Neurosurgery, School of Medicine, Stanford University, CA, 94305 USA.
Jiangang Shen2
2School of Chinese Medicine, The University of Hong Kong, Hong Kong S.A.R, P. R China.
Heng Zhao1
1Department of Neurosurgery, School of Medicine, Stanford University, CA, 94305 USA.
Corresponding author:
Heng Zhao
Email: hzhao@stanford.edu
In a new window | Download PPT
Figure 1: The schematic represents IPostC’s major protective mechanisms. The five major protective mechanisms mediated by IPostC are listed in the first row, and their corresponding molecules are shown in the second row. Together, these molecular events lead to the inhibition of brain infarction, neuronal apoptosis, edema, and BBB leakage, while promoting neurogenesis and angiogenesis related to functional recovery. Inducible nitric oxide synthase (iNOS); malondialdehyde (MDA); thiobarbituric acid reactive species (TBARS); advanced oxidation protein products (AOOPs); Glutathione (GSH); Superoxide dismutase (SOD); Catalase (CAT); endothelial nitric oxide synthase (eNOS); toll-like receptor 2/4 (TLR2/4); Myeloperoxidase (MPO); interleukin-1 receptor-associated kinase 4 (IRAK4); Interleukin 1 beta (IL-1β); Tumor necrosis factor α (TNF-α); Intercellular Adhesion Molecule 1 (ICAM-1); C-C chemokine receptor type 2 (CCR-2); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); glucose-regulated protein 78 (GRP78); C/EBP-homologous protein (CHOP); mitochondrial potassium ATP-dependent channel (mitoKATP); voltage-dependent anion channel protein (VDAC); glutamate receptor ε1 (NR2A); metabotropic glutamate receptor 1/5 (mGlu1/mGlu5); Acid-sensing ion channel 1a (ASIC1a); brain-derived neurotrophic factor (BDNF); vascular endothelial growth factor (VEGF); glial cell-derived neurotrophic factor (GDNF); heat shock protein 40/70 (HSP40/70); blood-brain barrier (BBB).
In a new window | Download PPT
Figure 2: The diagram illustrates the major cellular and molecular signaling pathways activated by IPostC for neuroprotection. IPostC inhibits both the TLR2/4/NF-κB pathway and the GluK2/PSD-95/MLK3/MKK7/JNK3 signal complex to inhibit cell death, while promoting the PI3K/Akt pathway for neuronal survival. Toll-like receptor 2/4 (TLR2/4); interleukin-1 receptor-associated kinase 4 (IRAK4); nuclear factor kappa-light- chain-enhancer of activated B cells (NF-κB); NF-κB inhibitor α (IκB-α); protein kinase B (AKT); B-cell lymphoma 2 (Bcl-2); Bcl-2-associated death promoter (BAD); mammalian target of rapamycin (mTOR); ribosomal protein S6 kinase (S6K); eukaryotic initiation factor 4E-binding protein (4E-PB1); glutamatergic kainate receptor subunit 2 (Gluk2); postsynaptic density protein 95 (PSD95); mixed-lineage kinase 3 (MLK3); mitogen-activated protein kinase kinase 7 (MKK7); c-Jun N-terminal kinase 3 (JNK3).
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11475 | 29 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA