Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
SAFE pathway and conditioning: from discovery to its effectiveness in the presence of cardiovascular risk factors
Time:2020-05-05
Number:10115
Author Affiliations

Conditioning Medicine 2020. 3(2):98-103.
Abstract
Although ischemic conditioning has been described as the most powerful strategy to protect against ischemia-reperfusion injury, its translation to the clinical setting has proved to be challenging, possibly due, at least in part, to the presence of different cardiovascular risk factors in patients, which are known to affect its efficacy. Ischemic conditioning confers cardioprotection via the activation of multiple prosurvival pathways, including the activation of the survivor activating factor enhancement (SAFE) path, which involves mediators of the innate immune system (i.e. tumor necrosis factor alpha) and key transcription factors such as the signal transducer and activator of transcription 3 (STAT3). Here, we describe the origin of the SAFE path as a key pathway in ischemic conditioning and we explore how the activation of this path may be affected in the presence of different cardiovascular risk factors.
Keywords: Prosurvival signalling pathways, SAFE, Tumor Necrosis Factor, signal transducer and activator of transcription 3, cardiovascular risk factors
Abstract
Although ischemic conditioning has been described as the most powerful strategy to protect against ischemia-reperfusion injury, its translation to the clinical setting has proved to be challenging, possibly due, at least in part, to the presence of different cardiovascular risk factors in patients, which are known to affect its efficacy. Ischemic conditioning confers cardioprotection via the activation of multiple prosurvival pathways, including the activation of the survivor activating factor enhancement (SAFE) path, which involves mediators of the innate immune system (i.e. tumor necrosis factor alpha) and key transcription factors such as the signal transducer and activator of transcription 3 (STAT3). Here, we describe the origin of the SAFE path as a key pathway in ischemic conditioning and we explore how the activation of this path may be affected in the presence of different cardiovascular risk factors.
Keywords: Prosurvival signalling pathways, SAFE, Tumor Necrosis Factor, signal transducer and activator of transcription 3, cardiovascular risk factors
The discovery of intrinsic cardioprotective signaling pathways in conditioning
Since its discovery in 1986, the phenomenon of ischemic conditioning has been the focus of intense research with more than 20,000 peer-reviewed publications (listed in PubMed) trying to understand this phenomenon and to translate it to the clinical arena (Cour and Lecour, 2019; Hausenloy et al., 2019). Unfortunately, as simple as ischemic conditioning may be applied (few episodes of ischemia/reperfusion prior to or after a sustained ischemic insult), these 34 years of research conducted worldwide have not been able to fully elucidate the mechanisms involved in this protective phenomenon, even though some important discoveries have been made over that time. In the 1990s, adenosine and protein kinase C were suggested as key components of the signaling cascade, their activation leading to the downstream activation of an unknown effector for cardioprotection (see Goto et al., 1996). The putative mitochondrial potassium adenosine triphosphate dependent channel was later proposed as the end effector of the cardioprotective signaling cascade, although the existence of this channel remained disputed for many years (Cohen et al., 2000; Paggio et al., 2019). The beginning of the 21st century was marked by research suggesting the involvement of the mitochondrial permeability transition pore (MPTP) whose identification has also revealed to be challenging (Downey et al., 2007). The discovery of the reperfusion injury salvage kinase (RISK) pathway also represented a major advance in the understanding of the cardioprotective signaling pathways, with the activation of both protein kinase B (Akt) and extracellular signal-regulated kinase (ERK) at the onset of reperfusion having proved to play a key role to limit infarct size following an ischemic conditioning stimulus (Schulman et al., 2002). In 2009, we described the activation of the survivor activating factor enhancement (SAFE) pathway as an alternative path to the RISK pathway for cardioprotection (Lecour, 2009a). The SAFE path is triggered by the activation of the immune system with tumor necrosis factor-alpha (TNF) initiating the activation of a myriad of prosurvival components including signal transducer and activator of transcription 3 (STAT3) (Lecour, 2009b).
The SAFE pathway and its origins
The research that led to the discovery of the SAFE pathway goes back to 2000 when data published by Douglas Mann’s group triggered our curiosity. Although TNF was known as a cytokine contributing to cardiac dysfunction in the context of heart failure, his group observed that mice with TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2) deficiency presented with a larger left ventricular infarct size compared to their littermate controls when subjected to an ischemia-reperfusion injury (Kurrelmeyer et al., 2000). These data suggested that TNF may trigger some cardioprotective intrinsic signaling pathways and this led us to hypothesize that endogenous TNF may play an important role as a signaling component involved in the cardioprotective effect of ischemic conditioning. Using an isolated heart model, we discovered that TNF was able, in a dose dependent manner, to limit infarct size in hearts subjected to an ischemia-reperfusion injury when given as a preconditioning stimulus (Lecour et al., 2002). Similar findings were confirmed using an in vivo model of ischemia-reperfusion injury (Deuchar et al., 2007). Most importantly both ischemic pre- and post-conditioning failed to reduce the infarct size in mouse hearts from TNF knockout mice (Smith et al., 2002; Lacerda et al., 2009). When TNF was given as a preconditioning mimetic in mouse hearts, cardioprotection occurred in TNFR1 deficient mice but failed to protect in TNFR2 deficient mice, therefore suggesting that the cardioprotective effect offered with TNF is mediated via TNFR2 (Lacerda et al., 2009). Trying to delineate downstream targets of TNF, we found that both ischemic preconditioning and pharmacological conditioning with TNF increased the activation of STAT3 and failed to protect cardiomyocytes in STAT3 deficient mice (Lecour et al., 2005b; Suleman et al., 2008), therefore pinpointing STAT3 as a downstream target of TNF for cardioprotection. Although STAT3 was already known as a downstream target of interleukin 6, (another key component of the immune system) via the activation of glycoprotein 130, its activation by TNF and TNFR2 at the time was very intuitive (Kurdi and Booz, 2007). Today, the exact mechanisms by which TNF may activate STAT3 still remain to be elucidated. The dimerization (and therefore activation) of STAT3 is mediated via nicotinamide adenine dinucleotide dehydrogenase 1 alpha subcomplex 13 (NDUFA13), which is a subunit of the mitochondrial complex I (Hu et al., 2017). Most importantly, the activation of STAT3 downstream of TNFR2 activates optic atrophy 1 protein (OPA 1), which promotes mitochondrial fusion (Nan et al., 2017). Other downstream targets include the activation of protein kinase C, free radicals, the mitochondrial potassium ATP dependent channel, and the inhibition of the proapoptotic protein Bcl-2 antagonist of cell death (BAD) (Lecour et al., 2002; Lecour et al., 2005a; Boengler et al., 2010; Boengler et al., 2013). Activated STAT3 translocates to the mitochondria and inhibits the opening of the MPTP, thus preventing mitochondrial leakage, and thereby improving the outcome of ischemia-reperfusion injury by maintaining cellular energy functions (Boengler et al., 2010). Very recently, we have also identified microRNAs as downstream targets of STAT3 for cardioprotection. Indeed, a microarray analysis performed in both control and STAT3 knockout mice suggested that miR-34b and miR-337 are downregulated downstream of STAT3 (Pedretti et al., 2019). Furthermore, in isolated cardiomyocytes, the expression of miR-34b and miR-337 was increased by hypoxia in isolated cardiomyocytes and reduced by cardioprotective strategies. In cardiomyocytes transfected with miRNA mimics, cardioprotective strategies failed to improve cell viability against hypoxia (Pedretti et al., 2019).
The activation of the SAFE path has been identified with different forms of conditioning including ischemic preconditioning (Lecour et al., 2005b), ischemic postconditioning (Lacerda et al., 2009), and remote ischemic conditioning (Tamareille et al., 2011). Crosstalk between the SAFE and the RISK paths has been suggested. In rodents subjected to an ischemia-reperfusion insult, a conditioning protocol failed to activate the key components of the RISK pathway Akt or ERK in the presence of a STAT3 inhibitor (Suleman et al., 2008; Tamareille et al., 2011; Somers et al., 2012). In a similar manner, inhibitors of Akt that blocked signling through the RISK pathway abolished the protection of an ischemic conditioning protocol, and this was associated with a failure to activate STAT3 (Suleman et al., 2008; Tamareille et al., 2011; Somers et al., 2012).
A particularity of the SAFE path compared to the RISK path is that it is activated both during the conditioning stimulus and at the onset of reperfusion following a conditioning protocol. Inhibitors of STAT3 activation such as AG490 abolished the cardioprotective effect of ischemic preconditioning when given either during the preconditioning stimulus (Suleman et al., 2008) or at the onset of reperfusion (Lecour et al., 2005b). Furthermore, STAT3 activation was observed at both time points (Lecour et al., 2005b; Suleman et al., 2008).
Table 1: Different conditioning stimuli confer cardioprotection via the activation of the survivor activating factor enhancement (SAFE) pathway. This path involves the activation of tumor necrosis factor alpha (TNF) and the signal transducer and activator of transcription 3 (STAT3) to promote cell survival.
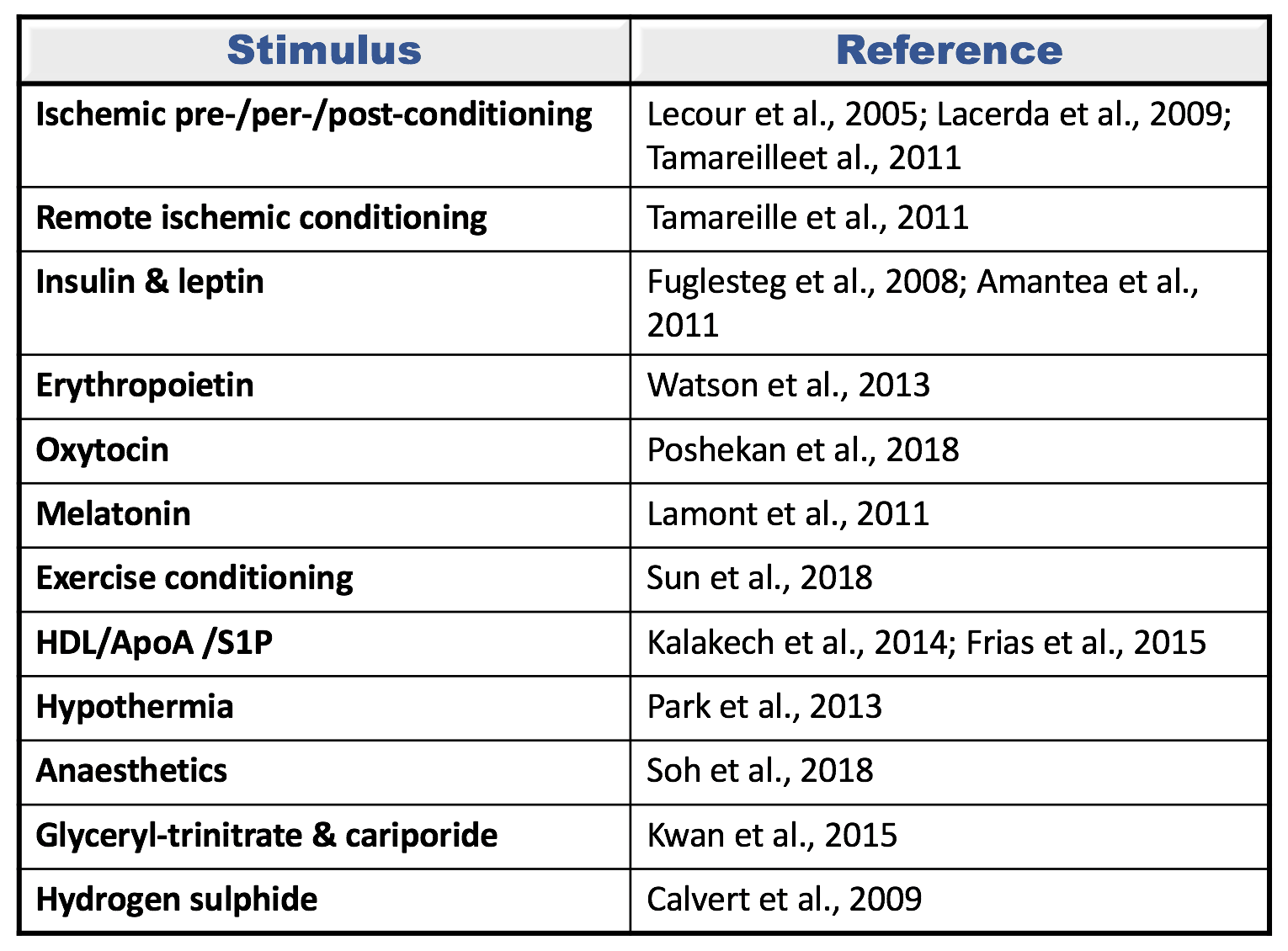
HDL: High density lipoprotein; ApoA: Apolipoprotein A; S1P: Sphingosine-1 phosphate; GH: Growth hormone.
Do all conditioning strategies activate the SAFE pathway?
The cardioprotective effect of ischemic preconditioning, ischemic postconditioning, and remote conditioning have all been identified as being dependent on the activation of the SAFE pathway (Lecour et al., 2005a; Lacerda et al., 2009; Tamareille et al., 2011). Similarly, multiple pharmacological pre- or post-conditioning drugs activate the SAFE pathway for cardioprotection and these include: glyceryl trinitrate, cariporide, growth hormones release hormone, erythropoietin, oxytocin, hydrogen sulphide, high density lipoproteins, apolipoprotein A, sphingosine-1 phosphate, anaesthetics, melatonin, and resveratrol (Lecour et al., 2005b; Fuglesteg et al., 2008; Calvert et al., 2009; Lacerda et al., 2009; Amantea et al., 2011; Lamont et al., 2011; Tamareille et al., 2011; Frias et al., 2012; Park et al., 2013; Penna et al., 2013; Watson et al., 2013; Kalakech et al., 2014; Brulhart-Meynet et al., 2015; Kwan et al., 2015; Sun and Mao, 2018) (see figure 1). Conditioning with exercise or hypothermia also activates the SAFE path (Park et al., 2013; Sun and Mao, 2018).
In a new window | Download PPT
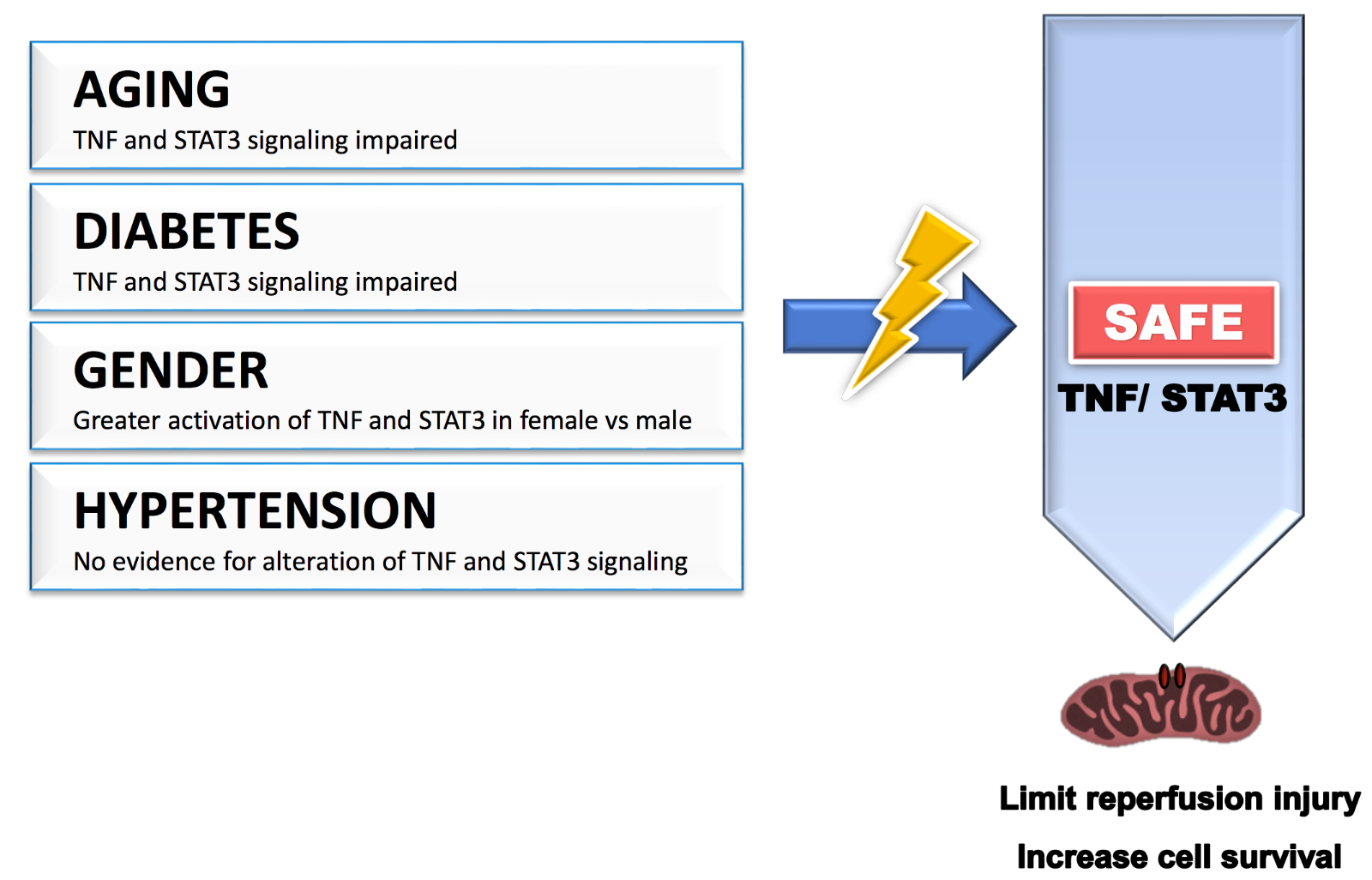
Figure 1: Cardiovascular risk factors such as age, gender, diabetes, and hypertension may affect the benefit of cardioprotective strategies by modulating the activation of the SAFE pathway. TNF: tumor necrosis factor alpha; STAT3: signal transducer and activator of transcription 3.
Does the SAFE pathway offer multi-organ protection?
Although initially discovered to protect against cardiac ischemia-reperfusion injury, there is clear evidence that activation of the SAFE path to promote cell survival can also occur in other organs such as the brain (Wang et al., 2010; Sakata et al., 2012; Niemi et al., 2016), liver (Lee et al., 2016), lungs (Luo et al., 2018), hindlimbs (Han et al., 2016), and kidney (Dube et al., 2017). Surprisingly, remote liver ischemic preconditioning protects against cerebral ischemia-reperfusion injury via mechanisms, which may be independent of STAT3 activation (Yang et al., 2020).
Do traditional risk factors affect the effectiveness of the SAFE pathway?
Ischemic heart disease is often caused by or is associated with multiple risk factors including age, gender, hypertension, and diabetes. Most of these risk factors are known to alter many intrinsic cardiac signaling pathways in both physiological and pathophysiological conditions, and multiple studies suggest that they negatively interfere with the effectiveness of ischemic conditioning, most likely by modifying the sensitivity of the activation of prosurvival signaling pathways (see review by Ferdinandy et al., 2014). There is now strong evidence in the literature supporting that the SAFE pathway is also affected by these risk factors.
Age
Experimental data support evidence that aging affects the efficacy of conditioning with a lack of protection observed with ischemic postconditioning and remote ischemic conditioning in older rodents (Somers et al., 2011; Adam et al., 2013; Behmenburg et al., 2017; Heinen et al., 2018). TNF-mediated cardioprotective signalling pathways are impaired with aging (Cai et al., 2003). Most importantly, Boengler and colleagues reported that the lack of protection with ischemic postconditioning, observed in aged mice compared to younger animals, was associated with a lack of phosphorylation of STAT3 (Boengler et al., 2008). These data strongly suggest that reduced activation of the SAFE path with aging may contribute to the lack of protection with conditioning in aged mice.
Gender
Female and male hearts present a difference in susceptibility against ischemia-reperfusion injury with smaller infarcts observed in female compared to male when subjected to the same ischemia-reperfusion insult (Ostadal et al., 2009; Penna et al., 2009). Gender may also affect the effectiveness of conditioning. The benefit of ischemic postconditioning is larger in males compared to females, an effect which might be explained by the fact that the females were already better protected without the conditioning protocol (Penna et al., 2009). It is highly possible that some of these gender differences may be explained by a gender difference in sensitivity of the activation of the SAFE path. Indeed, gender differences in TNF signaling after ischemia/reperfusion have been reported, including a greater activation of TNFR2 in females compared to males (Wang et al., 2006; Wang et al., 2008). Similarly, male hearts subjected to an ischemia-reperfusion injury expressed a lower level of activated STAT3 compared to female hearts, an effect which was reversed in castrated males (Wang et al., 2009).
Diabetes
In both type 1 and type 2 diabetic animals, an increase in infarct size following an ischemia-reperfusion injury was observed, most likely as a consequence to an increase in hyperglycemia induced oxidative stress associated with the disease (see review (Lejay et al., 2016)). With no surprise, the cardioprotective effect of ischemic preconditioning and ischemic postconditioning was also compromised in diabetic animals (Przyklenk et al., 2011; Tyagi et al., 2019). The increased susceptibility of ischemia-reperfusion injury in the diabetic rats was associated with a decrease in STAT3 activation (Li et al., 2013). Similarly, STAT3 failed to be phosphorylated following a pharmacological or an ischemic conditioning stimulus in streptozotocin-induced diabetic rats (Li et al., 2013; Lei et al., 2019).
Hypertension
In spontaneously hypertensive rats, ischemic and pharmacological preconditioning confer benefits against ischemia-reperfusion injury (Boutros and Wang, 1995). Surprisingly, ischemic postconditioning failed to protect in spontaneously hypertensive rats, (Penna et al., 2010; Wagner et al., 2013), an effect that is attributed to the lack of phosphorylation of glycogen synthase kinase 3 (Gsk-3β), as Gsk-3β inhibitors restored the protection (Gonzalez Arbelaez et al., 2013). In contrast, postconditioning was successful in protecting rats with hypertensive dilated cardiomyopathy induced by administration of angiotensin II (Hernandez-Resendiz et al., 2013), an effect mediated by activation of Akt. Although remote ischemic and normobaric hypoxic conditioning can reduce blood pressure in hypertensive patients, there is no knowledge as to whether the SAFE pathway may be altered in the presence of hypertension (Lyamina et al., 2011; Madias, 2015).
In conclusion, the discovery of the SAFE path has brought a better understanding to the intrinsic prosurvival signaling cascade involved in conditioning-induced cardioprotection. Although the end targets of the SAFE pathway are still unknown, the discovery of this pathway has the potential to develop novel therapies/strategies to limit ischemia-reperfusion injuries by targeting some components of this pathway such as specific miRNAs, for example. It is important, however, to keep in mind that, if the activation of this pathway has been confirmed in small animals and pigs, its protective effect still needs to be confirmed in patients in which many co-morbidities and co-medications may affect its effectiveness.
Conflict of interest
None.
Acknowledgements
The research of SL is supported by the University of Cape Town and the National Research Foundation (111801), The Cancer Association of South Africa and Winetech. AI received a postdoctoral fellowship from the University of Cape Town and NH received a PhD fellowship from the Medical Research Council in South Africa.
References
Aqeela Imamdin1
1Hatter Institute for Cardiovascular Research in Africa, Department of Medicine, Faculty of Health Sciences, University of Cape Town, South Africa.
Nkanyiso Hadebe1,2
1Hatter Institute for Cardiovascular Research in Africa, Department of Medicine, Faculty of Health Sciences, University of Cape Town, South Africa. 2Department of Anaesthesia, Faculty of Health Sciences, University of Cape Town, South Africa.
Sandrine Lecour1
1Hatter Institute for Cardiovascular Research in Africa, Department of Medicine, Faculty of Health Sciences, University of Cape Town, South Africa.
Corresponding author:
Sandrine Lecour
Email: Sandrine.lecour@uct.ac.za
In a new window | Download PPT
Figure 1: Cardiovascular risk factors such as age, gender, diabetes, and hypertension may affect the benefit of cardioprotective strategies by modulating the activation of the SAFE pathway. TNF: tumor necrosis factor alpha; STAT3: signal transducer and activator of transcription 3.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 10115 | 25 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA