Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
[Special Issue] Time to exercise: Circadian regulation of cardiac preconditioning
Time:2020-05-08
Number:10933
Author Affiliations

Conditioning Medicine 2020. 3(2):71-81.
Abstract
Myocardial ischemia reperfusion injury remains a significant health concern worldwide. The pursuit of therapeutic interventions to reduce the severity of ischemia reperfusion (IR) injury by preconditioning the heart has been ongoing for decades, and through preclinical studies the approach broadly includes three methodological categories; ischemic, pharmacological, and exercise preconditioning. These efforts have yielded many exciting experimental candidates, which have largely been unsuccessfully translated into clinical practice. One potential hurdle in clinical translation and efficacy of myocardial preconditioning may involve the circadian rhythm (i.e. – the ~24 hour recurring cycles in physiological processes), and the experimental or clinical timing of interventions designed to improve cardiac outcomes. Circadian rhythms are regulated by the direct influence of environmental input (light, food, activity), as well as the cell autonomous transcriptional mechanism known as the circadian clock. While circadian rhythms (and their disruption) are implicated in various cardiovascular disease states and IR-injury (with increased incidence and severity in the early morning), more recent attention has been directed toward the circadian regulation of myocardial preconditioning. Indeed, intact circadian rhythms, which are modulated by physical activity, are a cornerstone of health. As such, exercise may play an important role as a cardioprotective, and chronoprotective intervention. However, the circadian timing of interventions eliciting cardioprotection has not been explored. Thus, it is the purpose of this novel review to highlight cardioprotective mechanisms that are directly and indirectly controlled or influenced by the circadian rhythm, concluding with the perspective that preconditioning the heart may be optimally timed based on the circadian rhythm for maximal efficacy.
Keywords: cardioprotection; chronoprotection
Abstract
Myocardial ischemia reperfusion injury remains a significant health concern worldwide. The pursuit of therapeutic interventions to reduce the severity of ischemia reperfusion (IR) injury by preconditioning the heart has been ongoing for decades, and through preclinical studies the approach broadly includes three methodological categories; ischemic, pharmacological, and exercise preconditioning. These efforts have yielded many exciting experimental candidates, which have largely been unsuccessfully translated into clinical practice. One potential hurdle in clinical translation and efficacy of myocardial preconditioning may involve the circadian rhythm (i.e. – the ~24 hour recurring cycles in physiological processes), and the experimental or clinical timing of interventions designed to improve cardiac outcomes. Circadian rhythms are regulated by the direct influence of environmental input (light, food, activity), as well as the cell autonomous transcriptional mechanism known as the circadian clock. While circadian rhythms (and their disruption) are implicated in various cardiovascular disease states and IR-injury (with increased incidence and severity in the early morning), more recent attention has been directed toward the circadian regulation of myocardial preconditioning. Indeed, intact circadian rhythms, which are modulated by physical activity, are a cornerstone of health. As such, exercise may play an important role as a cardioprotective, and chronoprotective intervention. However, the circadian timing of interventions eliciting cardioprotection has not been explored. Thus, it is the purpose of this novel review to highlight cardioprotective mechanisms that are directly and indirectly controlled or influenced by the circadian rhythm, concluding with the perspective that preconditioning the heart may be optimally timed based on the circadian rhythm for maximal efficacy.
Keywords: cardioprotection; chronoprotection
Introduction
Cardiovascular disease (CVD) is, and has been, the uninterrupted leading cause of death in the United States since the middle of the 20th century (Center for Disease Control). Within the scope of CVD, ischemic heart disease (primarily manifesting as myocardial ischemia-reperfusion injury; IR-injury), represents a significant burden, comprising nearly fifty percent of CVD-related deaths. The extent of IR-injury occurs in a dose-dependent manner, where injury is greater following increasing duration of ischemia prior to reperfusion. Thus, prompt reperfusion is essential for preservation of myocardial function and survival. In the case that reperfusion therapies are successfully deployed (thrombolytic infusion, angioplasty, etc), the restoration of blood flow is accompanied by a secondary reperfusion injury, creating the cumulative insult of IR-injury. Myocardial IR-injury occurs in evolutionary fashion through three phases that accumulate in a time-dependent manner (as reviewed in Powers et al., 2007). At the onset of ischemia, the supply-demand mismatch rapidly produces a series of potentially lethal ventricular arrhythmias (0-5 minutes) (Miller et al., 2012). In the absence of immediate reperfusion, the pathological progression devolves to left ventricular pump dysfunction and myocardial stunning (5-20 minutes) (Bolli and Marban, 1999). Finally, unremitting ischemia eventually results in myocardial infarction (MI) (>20 minutes) through a combination of apoptotic and necrotic cell death processes (Powers et al., 2008; Quindry et al., 2010; Quindry et al., 2012; McGinnis et al., 2015; Miller et al., 2015).
From a cellular perspective, the biological underpinnings of ischemic tissue death are well described and the reader is directed to several well-articulated reviews on the topic (Kloner and Jennings, 2001a, 2001b; Bolli et al., 2004; McCully et al., 2004; Gross and Auchampach, 2007; Buja and Weerasinghe, 2010; Vetterlein et al., 2003). In brief, the initiating events central to IR-injury include, but are not limited to, free-radical production/oxidative stress, calcium overload, a drop in cellular pH and metabolic substrate shift towards anaerobic fuels, activation of apoptosis, and the opening of the mitochondrial permeability transition pore (mPTP). Relevant to the current review, the foundational mechanisms of IR-injury are also subject to circadian influence. With respect to the aforementioned mechanisms, links have been established for autophagy, apoptosis, and necrosis (Rabinovich-Nikitin et al., 2019).
Circadian rhythms in myocardial autophagy been appreciated since the 1980’s (Pfeifer and Strauss, 1981), where autophagic vacuoles appear to peak in the resting phase. In the years since, mechanistic connections between autophagic signaling (microtubule-associated proteins 1A/1B light chain 3B (LC3), p62, etc) and flux (measured in the presence of inhibitors) to the circadian clock have been made (Ma et al., 2011), which may contribute to rhythms in IR tolerance (Rotter and Rothermel, 2012). Several studies have established circadian regulation of cardiac mammalian target of rapamycin (mTOR)/adenosine monophosphate-activated protein kinase (AMPK) balance, which serve as the proverbial brake/gas pedal for autophagy induction (Khapre et al., 2014; McGinnis et al., 2017). Mechanistic studies in other metabolically active peripheral tissues have found that Rev-erbα is both necessary and sufficient for skeletal muscle autophagy (Woldt et al., 2013), while Period 2 (Per2) inhibits hepatic mTOR complex formation, and increases autophagy (Wu et al., 2019).
Evidence also exists for circadian regulation of apoptosis in the heart. A large scale screening of putative p53 activators identified the core clock component, brain and muscle arnt-like 1 (Bmal1) as a candidate that actively binds to transcriptional promotor sites regulating p53 (Mullenders et al., 2009), a key regulator of apoptosis signaling. Additionally, Bmal1 was recently found to control cardiomyocyte Bcl-2/adenovirus E1B 19-kD interacting protein (Bnip3), a key regulator of mitochondrial dynamics (Li et al., 2020). As such, deletion of Bmal1 was associated with reduced Bnip3 expression, leading to decreased mitochondrial fission and mitophagy, and increased apoptosis, Ca2+ dysregulation, and cardiac dysfunction (Li et al., 2020). This represents transcriptional circadian control of apoptosis.
Based on this collective understanding, there is a growing body of knowledge to indicate mediators of IR-mediated injury and death are under circadian control, and loss of circadian control is central to post-infarct clinical outcomes. Thus, the scientific process of uncovering novel countermeasures to IR-injury should be holistically mindful of proper circadian control. This is particularly applicable to the appropriate design of experiments taking circadian biology into consideration. For example, nocturnal rodents are active during the dark cycle, but acute exercise interventions are commonly performed during work hours (which, unless mice are housed on reverse light/dark cycles means exercising in the middle of the sleep phase). Savvy readers of the conditioning/preconditioning literature should be aware that, more often than not, scientific reporting of the chronobiological considerations in experimental design goes completely unreported. In these instances, the reader then remains uncertain of whether circadian influences are optimized or confounding to the key dependent outcomes being examined. Further considerations of circadian control are forthcoming.
In addition to considerations of circadian control, and in the context of exercise preconditioning research, even if exercise intervention is appropriately timed, it may not be carried out under dim red light conditions or in darkness, thus imposing a bright light stimulus in the middle of the dark phase (light is an extremely potent zeitgeber, which can shift circadian rhythms) (Morin and Studholme, 2014). This technical shortcoming is of course not restricted to the field of exercise, but also extends to pharmacology studies, as a recent study reported that 50 of the top 100 prescribed drugs in the US have a target that displays a circadian rhythm in at least one tissue (Zhang et al., 2014), yet the timing of prescription is rarely, if ever, considered. Within the experimental paradigm of exercise and the cardioprotected phenotype, there is a burgeoning body of work to suggest that the now established subfield of exercise cardiac preconditioning may be influenced by circadian biology in ways that have not been understood previously. For the purposes of this review, we will briefly overview the foundational concepts of cardiac preconditioning, cellular-level circadian control, circadian links to a cardioprotected phenotype, and chrono-exercise mechanisms.
Cardiac Preconditioning –
Protecting the heart against IR-injury holds great potential for alleviating the significant burden presented by IR-injury. Historic understanding of cardiac preconditioning has centered upon beneficial adaptations to cardiac anatomy and architecture such as optimizing myocardial vascularization, chamber size, and transmural wall thickness, which take place over prolonged periods of training. Important as these structural adaptations are, they don’t attend to a wealth of understanding, which clearly demonstrates that the biochemistry of the myocardium can be acutely modified by a variety of stressors to produce a robustly protected phenotype (Hoshida et al., 2002).
While indirect evidence of biochemical cardioprotection extends back to the 1970s (Paffenbarger et al., 1970), the first direct evidence stems from an investigation of what is now called ischemic preconditioning (IPC) (Murry et al., 1986). In this now classic publication, Murry and colleagues employed a surgical model of regional ischemia (left anterior descending artery (LAD) of anesthetized dogs) to demonstrate that the myocardium could be largely spared when an experimental infarction was preempted by a series of short duration (4 x 5 minutes ischemia, separated by 5 minutes reperfusion) sub-lethal ischemic bouts (Murry et al., 1986). Importantly, no damage to the heart is induced by these preemptive short bouts of preconditioning ischemia (Reimer et al., 1986). Because the surgery to induce the short duration ischemic bouts preceded the extended duration, and clinically relevant infarct challenge 40 minutes later, it was apparent that IPC was biochemical in nature. Indeed, in the years since this foundational investigation, it has been revealed that IPC evokes acute allosteric changes and up-regulation in a concert of mediators that prevent the collective pathology of IR insults (Yellon and Downey, 2003). This protection extends to all major forms of IR-injury including reactive oxygen species (ROS), calcium (Ca2+) overload, and cellular perturbations leading to cell death (Bolli, 2007; Sprick et al., 2019). Importantly, from Murry’s first observation of IPC in 1986, it took only a matter of years to demonstrate that the phenomenon encompasses comprehensive protection against the core mediators of ischemic injury. From this point the race was on to identify the mechanism(s) responsible for IPC in an attempt to reverse engineer a druggable solution to MI (Bolli et al., 2004).
Is circadian biology the missing link in the translation of cardiac preconditioning research? Unfortunately, these pioneering findings in the subfield of IPC have not resulted in successful clinical translation. Indeed, although preliminary results from preclinical trials have offered promise, confirmation of these findings into humans remains problematic. The barriers to progress include a host of independent factors. While delineation of these scientific sticking points is beyond the scope of the current manuscript, there is a rationale to believe that deleterious alterations to circadian biology may be among the mitigating factors that has not been fully considered to date (Heusch, 2017; Montaigne and Staels, 2018). In contrast to these dead ends in preconditioning research, there is a parallel rationale to believe that exercise, being cardioprotective in nature, may partially exert its effects through a restoration of, or optimization in, circadian control within the heart (i.e. – chronoprotection). In order to advance these parallel rationales, we will discuss exercise preconditioning as a prelude to a review of chronobiology related to cardioprotection against IR-injury.
Exercise preconditioning of the heart against an ischemic insult - In order to advance the foundational postulate of this review paper, that cardiac preconditioning via exercise includes the essential influence of circadian control mechanisms, it is important to reflect on the key descriptive and mechanistic observations of exercise-induced cardioprotection against an ischemic insult. Because these details are briefly addressed in this manuscript, the reader is directed to several authoritative reviews by several prominent groups (Starnes and Taylor, 2007; Quindry and Hamilton, 2013; Powers et al., 2014; Powers, 2017; Quindry and Franklin, 2018; Chowdhury et al., 2019).
The early descriptive observations indicate that exercise-induced cardioprotection, primarily examined using rodent models (rats and mice) of surgical ligation of the LAD, prevents and/or mitigates all major forms of ischemic injury. Specifically, exercise prevents the severity and frequency of several lethal forms of ventricular ectopy, during both ischemia and reperfusion (Hamilton et al., 2004; Quindry et al., 2010; Quindry et al., 2012; Frasier et al., 2013; Miller et al., 2015; Alleman et al., 2016). Just as important, ventricular pump dysfunction associated with an acute ischemic insult are also largely prevented by exercise training (note that some of these observations are derived from isolated perfused heart preparations using global ischemia, but are confirmed by LAD ligation models) (Bowles & Starnes, 1994; Taylor et al., 1999; Demirel et al., 2001; Quindry et al., 2005; French et al., 2006; Taylor et al., 2007; French et al., 2008). Finally, an exercise regimen prevents myocardial tissue death by both frank necrosis (Yamashita et al., 1999; Quindry et al., 2007; McGinnis et al., 2015; Miller et al., 2015) and apoptosis (Quindry et al., 2005; Quindry et al., 2007; Quindry et al., 2012; McGinnis et al., 2015).
Central to these observations of potent cardioprotection against all the major forms of ischemic pathology, the dose of exercise used to precondition the heart is just a few days. Indeed, between 1-3 days of moderate intensity treadmill exercise in rats and mice underpin the cardioprotective outcomes mentioned above (Demirel et al., 2001; McGinnis et al., 2015). Relative to the question of dose, cross-study comparisons indicate that longer duration exercise programs (weeks to months) are no more protective than a few days of exercise (Demirel et al., 1998; Demirel et al., 2001; Lennon et al., 2004a). Just as interestingly, high intensity exercise (~75% VO2max) is no more protective than moderate intensities (~50% VO2max) (Lennon et al., 2004b), though there are indications that high intensity exercise may have added benefits (Esposito et al., 2011).
Of importance to the current discussion, the exercise-protected phenotype appears to be heavily reliant upon acute modulation of biochemical factors local to the myocardium. Once introduced, the exercise stimulus appears to evoke a cardioprotected phenotype for at least 9 days (Lennon et al., 2004a). This important observation contrasts with the less robust time frame (3-4 days) associated with non-exercise approaches to cardioprotection (Kuzuya et al., 1993; Guo et al., 1998; Yellon and Downey, 2003). While still debated, observational differences between exercise and non-exercise forms of preconditioning may infer that the various methodologies protect the heart through divergent mechanisms (Frasier et al, 2011; Quindry and Hamilton, 2013). Moreover, that moderate and high intensity exercise is equally protective suggests that from the perspective of the exercised myocardium, the stimulus is threshold-independent. Indeed, within circadian biology, the phase-shifting capacity of exercise as a zeitgeber is similarly threshold-independent, and vigorous physical activity was no more effective than moderate intensity exercise (Buxton et al., 1997).
As a final consideration of exercise preconditioning, a number of mechanisms appear to be responsible for the exercised phenotype. Early observations confirmed that exercise improves Ca2+ control during ischemia and reperfusion (Bowles & Starnes, 1994; French et al., 2006; Starnes et al., 2007). Accordingly, and despite the supply-demand mismatch of an ischemic challenge, bioenergetic control is better supported in the exercised heart (Bowles & Starnes, 1994; Kavazis et al., 2014; Alleman et al., 2016). Related to the above mechanisms, the mitochondrial isoform of superoxide dismutase is upregulated in the exercised heart and appears to be essential to prevent ventricular dysrhythmias and necrotic tissue death in the exercised heart (Hamilton et al., 2004; French et al., 2008). How improvements in mitochondrial control are facilitated is not fully understood, but improvement in KATP channels on the sarcolemma and mitochondrial membranes are likely contributors (Brown et al., 2005; Chicco et al., 2007; Quindry et al., 2010; Quindry et al., 2012). Finally, several preliminary instances of receptor-mediated protection have been observed in the exercised hearts. To date, the delta opioid receptor and interleukin-6 (IL-6) mediated cardioprotection has been observed (Dickson et al., 2008; McGinnis et al., 2015; Miller et al., 2015). In summation, exercise evokes a robustly protective phenotype against ischemic injury and death. As will be discussed shortly, many of the mechanisms of exercise-induced cardioprotection also have chronobiological underpinnings. First, however, it is important to provide an overview of the foundational mechanisms of circadian rhythm as they pertain to both ischemic injury and cardioprotection.
Diurnal/Circadian Rhythms in physiology, circadian clock, and rhythms in the heart –
Numerous behavioral, cellular, and metabolic processes have evolved to maximize the integration of physiology with the recurring cycles of light and dark in our environment, and compose our circadian rhythm. These rhythms are driven by extrinsic factors (i.e. - exposure to light, intake of nutrients, and activity), as well as intrinsic mechanisms (i.e. – the molecular circadian clock), and are present in almost all forms of life on earth (Roenneberg and Merrow, 2005). The molecular circadian clock, which has been identified in essentially every tissue studied to date, is composed primarily of a core heterodimer of the transcription factors circadian locomotor output cycles kaput (Clock) and Bmal1, which bind to conserved E-box domains in the promotor regions of clock-controlled genes (CCG’s) (Hogenesch et al., 1998). While variability exists between tissues, it is estimated that between 10-15% of the cardiac transcriptome oscillates with a circadian rhythm (Storch et al., 2002; Martino et al., 2004), which is ameliorated by cardiomyocyte specific deletion of the Clock (Bray et al., 2008) or Bmal1 (Young et al., 2014). Among CCG’s are the negative regulators of the clock mechanism, Period (Per1/2/3), Cryptochrome (Cry1/2), and Rev-erbα, which feed back to either inhibit the heterodimerization of Clock/Bmal1 or inhibit the expression of Bmal1, respectively, in effect ‘turning off’ the clock. Circadian rhythms in the heart have been shown to display significant control of cardiac physiology (signaling, metabolism, and function) and is reciprocally linked to pathophysiology (i.e. – circadian rhythm disruption leads to CVD, and cardiometabolic diseases disrupt cardiac circadian rhythms) (Young et al., 2001; Young et al., 2002; Kung et al., 2007; Martino and Young, 2015; Crnko et al., 2019; Zhang et al., 2019; Rana et al., 2020).
It is now well appreciated that the time of day and circadian rhythms has a marked effect on the incidence and severity of MI (Muller et al., 1985; Mistry et al., 2017). Several large studies, old and new, have consistently shown that there is a morning peak in MI. This pattern (Muller et al., 1985) and a review of the precipitating factors (morning surge in blood pressure, coagulation, etc.) (Muller 1989; (Muller et al., 1989) was originally revealed by Muller et al (1989) in the 1980’s. In the 30 plus years following, numerous prospective and retrospective observations have supported these original claims (Suarez-Barrientos et al., 2011; Mogabgab et al., 2012; Bulluck et al., 2017; Mistry et al., 2017). While many factors are involved, one key mechanism is the diurnal rhythm in plasminogen activator inhibitor 1 (PAI-1) activity, which shows increased activity in the morning hours for healthy individuals and those with a history of MI (Angleton et al., 1989), indicating increased coagulability of blood in the morning. PAI-1 expression was subsequently shown to be under direct circadian control, as its expression is induced by the binding of Clock:Bmal1 to its transcriptional promoter sites (Schoenhard et al., 2003). Thus, the body is biologically primed for clotting in the morning, creating a temporal window when thrombi are more likely to form.
The contribution of the molecular circadian clock on cardiovascular function has been elucidated mechanistically via global and cardiomyocyte specific deletion of core clock genes including clock (Bray et al., 2008; Durgan et al., 2010; Durgan et al., 2011; Podobed et al., 2014; Alibhai et al., 2017) bmal1 (Lefta et al., 2012; Kohsaka et al., 2014; Young et al., 2014; Ingle et al., 2015; He et al., 2016; McGinnis et al., 2017), and per2 (Eckle et al., 2012; Bonney et al., 2013; Virag et al., 2013; Bartman et al., 2017). The subsequent section will discuss the interplay between circadian rhythms, the molecular clock components, and myocardial IR-injury, with specific reference to the potential for preconditioning with exercise or IPC (See Figure 1).
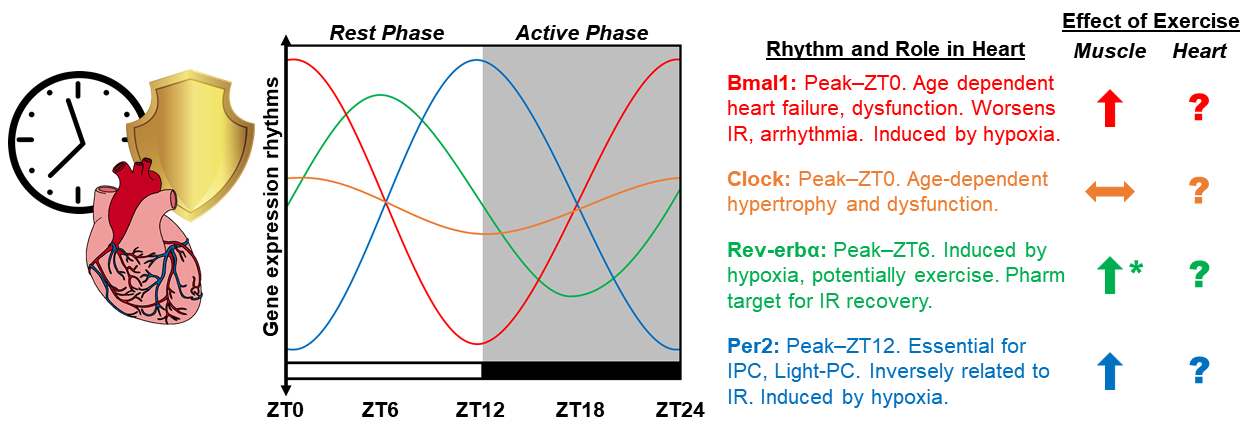
In a new window | Download PPT
Figure 1: Diagram depicting the proposed integration of chronobiology and exercise-induced cardioprotection. Graphic representation of circadian rhythms in the mRNA expression of core clock genes CLOCK, BMAL1, PER2, REV-ERBα, in the mouse heart over the 24-hour light dark cycle. Established roles for clock genes in the heart based on whole body and cardiomyocyte specific deletion models, as well as extrapolation from relevant studies in skeletal muscle. Additionally, the role of exercise as a means to induce the expression of clock genes is summarized in skeletal muscle and the heart. The amplitude of animated lines is based on published observations of clock gene expression. ∧ Induced by exercise, ㏒No change, ? Unknown influence of exercise, * effects of exercise only at ZT22.
Circadian clock and cardioprotection –
Clock: Clock is a core component of the transcriptional heterodimer in the positive arm of the molecular circadian clock, and binds with Bmal1 to induce transcription of CCG’s. The amplitude of clock gene expression is not as robust as other molecular components of the clock in wild-type (WT) mice, presenting less than 2 fold oscillations in gene expression (peak ~Zeitgeber Time (ZT)-0) (Young et al., 2002; Young et al., 2014; Alibhai et al., 2017). The role of Clock in the heart has been investigated mechanistically using Clock 19/∆19 mice, overexpressing a loss of function mutated clock gene (Durgan et al., 2010; Alibhai et al., 2017; Alibhai et al., 2018). These mice display cardiac hypertrophy and alterations in metabolism, transitioning to dilation and failure with age (Bray et al., 2008; Durgan et al., 2011; Alibhai et al., 2017). Using a closed-chest model of IR, it was found that IR-injury severity displays a robust rhythm in WT mice that was absent in Clock19/∆19 mice, which was similar at all times of the day, corresponding to ZT0 in WT hearts (Durgan et al., 2010). Others have shown that whole body Clock19/∆19 has worsened cardiac outcomes in response to aging, as well as permanent coronary artery ligation (Alibhai et al., 2017).
The role of Clock in the heart in the context of IPC and exercise preconditioning has not been investigated. It is worth noting that in a recent study examining the time-of-day dependent regulation of gene expression in skeletal muscle found that moderate intensity exercise had no effect on Clock mRNA levels, whether it was performed in the early or late portion of the dark phase (Ezagouri et al., 2019). Whether the same is true in the heart is currently not known.
Bmal1: Bmal1 is a core component of the positive arm of the circadian clock, which interacts with its binding partner, Clock, as a heterodimeric transcription factor initiating transcription of CCG’s. Bmal1 mRNA expression peaks at ZT0 (lights on) in mouse hearts, displaying a high amplitude rhythm (up to >15-fold) (Schroder et al., 2013; Rotter et al., 2014; Young et al., 2014; Zhang et al., 2015; Alibhai et al., 2017). Global germline deletion of Bmal1 results in an accelerated aging phenotype with reduced lifespan, associated with reduced activity levels and cardiovascular pathology (Kondratov et al., 2006; Kondratov et al., 2009; Lefta et al., 2012), though some effects are attributable to developmental, and not recapitulated in adult deletion (Yang et al., 2016). Cardiomyocyte specific deletion of Bmal1 similarly reduces lifespan and tolerance to hypertrophic stimuli and stress (Durgan et al., 2011; Young et al., 2014; Ingle et al., 2015; McGinnis et al., 2017) and increases cardiac arrhythmias (Schroder et al., 2013). At the cellular level, Bmal1 regulates the hypoxia-induced activation of hypoxia-inducible factor (HIF)-1α in C212 cells (Peek et al., 2017), suggesting its importance for adaptation to hypoxia/ischemia. Pathological aspects of Bmal1 loss have been rescued with antioxidants (Kondratov et al., 2009), and rapamycin supplementation (Khapre et al., 2014; McGinnis et al., 2017) (both of which are exercise mimetics), while the laudatory effects of exercise have not yet been investigated. Though exercise has been shown to have a modest impact on Bmal1 expression in skeletal muscle (Zambon et al., 2003), the effects appear to be time-of-day independent (Ezagouri et al., 2019), and the effects of exercise on the physiological and pathological cardiac-related outcomes have not been investigated in a bmal1-dependent manner.
Per2: The period proteins (Per1/2/3) are components of the negative arm of the circadian clock and feedback to prevent the heterodimerization of clock and bmal1, thereby reducing their ability to initiate transcription of clock genes. Per2 mRNA expression oscillates with a peak in expression at ZT12 (lights off) in mouse hearts, antiphase with bmal1 (Young et al., 2014). Several studies have shown that Per2 plays an important role in myocardial IR-injury and is necessary for IPC (Eckle et al., 2012; Bonney et al., 2013; Bartman et al., 2017; Oyama et al., 2018; Oyama et al., 2019). Per2-/- mice were shown to have larger infarct sizes compared to control littermates, suggesting Per2 is essential for myocardial adaptation to and recovery from ischemia. A series of elegant studies showed that in response to ischemia, myocardial adenosine signaling through the adenosine receptor 2b (Adora2b) induces a strong upregulation and stabilization of Per2 in the heart. Ischemic preconditioning activated strongly Adora2b signaling and Per2 expression, leading to a reduction of IR-injury, which was lost in Adora2b-/- and Per2-/- mice. Furthermore, treatment of mice with an Adora2b agonist (BAY 60-6583), reduced IR-injury in a Per2 dependent manner. Per2 dependent cardioprotection allowed for metabolic flexibility in the heart, increasing anaerobic glycolysis and lactate consumption during ischemia, when aerobic metabolism is limited (Eckle et al., 2012). Interestingly, consistent with Per2 being a core clock component with input from the sun/light, exposure to intense light (>10,000 lux, equivalent to sunlight) induced the expression of myocardial Per2 in a time-dependent manner (Eckle et al., 2012), which was absent in mice lacking the ability to perceive light (Oyama et al., 2019). The light-induced Per2 expression was associated with a strongly protected phenotype, which was absent in Per2-/- mice (Eckle et al., 2012). Additionally, intense light was shown to induce the expression of micro-RNA (miR)-21 in a Per2 dependent manner, inducing cardioprotected phenotype in mice that was abolished in miR-21-/- mice (Bartman et al., 2017). While numerous studies support the notion that myocardial Per2 is important for myocardial preconditioning, one contradictory study found that Per2-/- mice were protected against IR-injury, showing lower rates of cardiac apoptosis and infarction (Virag et al., 2013). This discrepancy could be the result of differing experimental models of IR or timing of surgery. Regardless of the exact combination of confounding factors, these uncertainties reinforce the underpinning premise that dynamic interplay between chronobiology to physiologic challenges have the potential to influence study outcomes in unpredictable ways. Collectively, these findings strongly implicate Per2 as a critical mediator of IPC and intense light-mediated preconditioning, with strong potential for pharmacological targeting.
While similar and divergent pathways exist between different mechanisms of preconditioning, the role of Per2 in exercise preconditioning has not been investigated. As previously described, acute exercise (1-3 days) elicits equal or greater protection against IR-injury compared to IPC and pharmacological methods. Exercise training has the capability to shift the circadian rhythm of Per2 expression in several peripheral tissues, including skeletal muscle (Sasaki et al., 2016), though the heart was not investigated. A recent investigation on the time-of-day dependent effects of acute exercise in skeletal muscle found that exercise increases the expression of Per2 to a similar extent (~1.5-2 fold) regardless of when it is performed, though the levels of Per2 are still strongly effected by time-of-day independent of exercise (>10 fold) (Ezagouri et al., 2019). A thorough investigation of the acute effects of exercise on myocardial Per2 expression is lacking, so the contribution of Per2 to exercise-induced preconditioning is currently unknown.
Rev-erbα: Rev-erbα (nr1d1) is a nuclear receptor in the negative arm of the circadian clock, which serves as a transcriptional repressor, specifically inhibiting Bmal1 expression. Rev-erbα mRNA expression peaks during the light phase (~ZT6) in mouse hearts (Young et al., 2014; Alibhai et al., 2017). Several studies have shown a role for Rev-erbα on myocardial IR-injury severity (Stujanna et al., 2017; Zhang et al., 2017; Reitz et al., 2019). These findings suggest that pharmacologically targeting Rev-erbα is sufficient to reduce inflammation, apoptosis, as well as dilation and heart failure following IR or transverse aortic constriction (TAC). However, these studies have all been conducted in the post-IR period. Thus, the role for Rev-erbα in cardiac preconditioning remains unknown.
Interestingly, recent clinical observations on the diurnal rhythm in subacute cardiovascular complications following surgery involving cross-clamping (inducing a predictable period of ischemia) found that morning operations were associated with greater risk/injury. Transcriptomic analysis revealed that Rev-erbα is higher in the morning (when risk was higher) vs evening samples, suggesting a negative role for Rev-erbα in the time-dependent susceptibility to ischemic stress. Mechanistic studies using ex vivo Langendorff perfused hearts confirmed that genetic (Rev-verbα-/- hearts) and pharmacologic (SR8278, a Rev-erbα antagonist) inhibition of Rev-erbα protected hearts from ischemic damage (Montaigne et al., 2018).
Several studies have also explored the role for Rev-erbα in skeletal muscle (which may be reflective of its role in the heart) (Woldt et al., 2013), as well as the immune system (Sato et al., 2014). Rev-erbα plays an important role in the regulation of skeletal muscle metabolism and autophagy (Woldt et al., 2013), as well as muscle mass (Mayeuf-Louchart et al., 2017), both of which are implicated in IR-injury and cardioprotection. Deletion of Rev-erbα in muscle leads to mitochondrial dysfunction and pronounced changes in muscle mass, morphology, reduced exercise capacity (Woldt et al., 2013; Mayeuf-Louchart et al., 2017), while overexpression or pharmacological agonism had the opposite effect (i.e. increasing exercise capacity and mitochondrial content) (Woldt et al., 2013), suggesting activation of Rev-erbα could be beneficial in the heart.
The ability for IPC or exercise to acutely induce the expression of Rev-erbα has not been conclusively shown, although a scientifically plausible rationale exists. Hypoxia (Adamovich et al., 2017) and exercise (Zambon et al., 2003; Ezagouri et al., 2019) were shown to modestly increase Rev-erbα expression in skeletal muscle, but neither has been tested in the heart, or in the context of preconditioning. Interestingly, Ezaguori et al. (2019) showed that exercise performed at ZT22 (late in the active period) increased Rev-erbα expression, while exercise at ZT14 (early in the active period) had no effect, suggesting exercise timing to induce Rev-erbα can be optimized. Pharmacological activation of Rev-erbα has been achieved with several ligands, of which SR9009 has received considerable attention. While several studies demonstrate that the benefits of SR9009 are dependent on Rev-erbα, it was recently shown that SR9009 also has Rev-erb-independent effects (Dierickx et al., 2019). Thus, the benefits of this compound may not be dependent on the circadian rhythm. Time-dependent administration of SR9009 (to mimic the in vivo rhythm of Rev-erbα expression), has important implications for chronopharmacological cardioprotection.
Chrono-Exercise and mechanisms of cardioprotection –
Time-of-day dependent regulation of exercise performance and adaptation is an area of increasing interest, as it holds great potential for human performance and athletics, as well as precision exercise prescription for health. While it is well appreciated that diurnal and nocturnal animals significantly consolidate their habitual activity to a single phase of their circadian rhythm, several recent excellent studies have highlighted that, within the respective active phases, there are distinct time-of-day dependent differences in both acute exercise responses and training induced adaptations (Dalbram et al., 2019; Ezagouri et al., 2019; Sato et al., 2019). A thorough appreciation of interaction between exercise physiology and the circadian rhythm may accelerate mechanistic underpinnings of the laudatory effect of exercise, and optimize exercise interventions to maximize their efficacy. This section will focus on the concept of ‘chrono-exercise,’ based on the tenet that exercise timing augments activation of protective pathways.
Reciprocal regulation exists between exercise and the circadian rhythm, as there are appreciable time-of-day dependent rhythms in exercise performance (peaking in the afternoon in humans), and exercise serves to synchronize or phase shift the circadian clock in peripheral tissues (Gabriel and Zierath, 2019; Wolff and Esser, 2019). Forced and voluntary exercise in the early night or late night can significantly phase shift the rhythms in cardiovascular function and clock gene expression in several peripheral tissue (Wolff and Esser, 2012; Sasaki et al., 2016), although subsequent investigation revealed that the effects in the heart were modest (Schroeder et al., 2012). These molecular insights have metabolically meaningful impact, as time-restricted exercise in the late active period prevented high fat diet induced obesity, while exercise in the early active period exercise did not (Dalbram et al., 2019).
While the topic of chrono-exercise has been relatively understudied in the cardiovascular system, two recent excellent studies sought to elucidate the mechanistic underpinnings of chrono-exercise in skeletal muscle using state-of-the-art omics approaches (Ezagouri et al., 2019; Sato et al., 2019). These studies employed a range of exercise intensities (low, moderate, high), performed in either the early-active or late active phases (Ezagouri et al., 2019), or in the early light phase and early dark phase (Sato et al., 2019). Exercise capacity was shown to be time-of-day dependent, with high intensity exercise performance being higher at the beginning of the active period (ZT14), and low-moderate intensity exercise performance being better at the end of the active period (ZT22) (time-of-day differences in performance were abolished in Per2-/- mice) (Ezagouri et al., 2019). Interestingly, among transcripts that were influenced by exercise, only 4% were common between the two different times of exercise tested, indicating strong time-of-day dependent influence of exercise (Sato et al., 2019). Robust time-of-day dependent signatures of genomic and metabolomic exercise responses were identified, profiling a critical role for critical cardioprotective pathways.
The AMPK pathway has been implicated in exercise and pharmacological approaches to cardiac preconditioning against IR-injury (Kristiansen et al., 2009; Barr et al., 2017), and is also important for exercise training-induced protection against myocardial fibrosis (Ma et al., 2015). Exercise-induced augmentation of glycolytic myocardial metabolism during reperfusion was associated with reduction in infarction, suggesting AMPK activation is critical for the metabolic response to IR (Kristiansen et al., 2009). Indeed, exercise in the late active period resulted in a comparably stronger activation of the AMPK pathway compared to exercise in the early active period, suggesting that exercise may be more protective at this time (Ezagouri et al., 2019). Interestingly, the regulation of this time-dependent response was potentially via 5-aminoimidazole-4-carboxamide ribonucleotide (ZMP). The adenosine analog and ZMP precursor 5-aminoimidazole-4carboxamide-1β-D-ribofuranoside (AICAR) also activates cardioprotection (Kristiansen et al., 2009), but these findings raise the chronopharmacological question as to if AICAR administration could be optimally timed.
Additionally, important to the chronobiological timing of exercise to induce robust cardioprotection, a significant time-of-day dependent regulation of exercise induced HIF-1α was identified (Sato et al., 2019). HIF-1α is strongly implicated in cardiac preconditioning (Ong and Hausenloy, 2012), and IR-injury is worse in HIF-1α knockout mice (Nanayakkara et al., 2015). Furthermore, there is an inextricable link between the circadian clock mechanism and hypoxia trough HIF that has been identified in multiple tissues (Adamovich et al., 2017; Peek et al., 2017; Wu et al., 2017). These recent findings suggest that exercise more strongly induces HIF-responsive glycolytic pathways in the early active period (ZT15) compared to early rest (ZT3) (Sato et al., 2019). These data agree with previous observations that exhaustive exercise induced expression of HIF-responsive genes in skeletal muscle in a time-of-day dependent manner (Peek et al., 2017). While almost all of the studies were performed in skeletal muscle, they highlight the potential for robust circadian control of exercise-induced adaptations in the heart, which may significantly impact cardioprotection.
Lastly, one integrating component in cardioprotective interventions (exercise, IPC, and remote IPC) is the contribution of muscle-derived cytokines, or myokines. Exercise elicits production and secretion of numerous myokines that have broad impact on exercise-induced adaptations in a variety of tissues, including the heart (Pedersen, 2013; Whitham and Febbraio, 2016). Previous research has shown that IL-6 is essential for both IPC (Dawn et al., 2004), and exercise-induced cardioprotection (McGinnis et al., 2015). Interestingly, it was recently found that the circadian clock regulates the production and secretion of a number of myokines in synchronized cultured human fibroblasts. Specifically, Perrin, et al. (2015) showed a circadian rhythm in IL-6 secretion into culture media, which was abolished by in vitro suppression of the clock. Furthermore, human studies have shown that exercise-induced IL-6 release is higher and more sustained following exercise performed in the evening (Abedelmalek et al., 2013; Kim et al., 2015). Together, these findings present a compelling rationale that time-of-day dependent exercise prescription may be a potential strategy to maximize the cardioprotective myokine response.
Conclusions and Future Directions –
Chronobiology is a burgeoning field of research with health and performance at its core. To this end, it is important to recognize the magnitude of circadian rhythm regulation of the molecular and physiological responses to exercise and ischemia. It is now critical at preclinical and clinical levels that we apply the principles of chronobiology to exercise and pharmacological interventions. Accordingly, there is reason to be hopeful that exercise induced preconditioning is intrinsically linked to circadian influences, yet the definitive investigations to support or refute this notion remained untested to date. Elucidation of time-dependent regulation of exercise-induced cardioprotection has the potential to maximize the efficacy of potent preconditioning against IR-injury and increase the likelihood of clinical translation, a goal that has been elusive for over 40 years.
References
Graham R. McGinnis1
1University of Nevada, Las Vegas. Las Vegas, NV. School of Integrated Health Sciences.
John C. Quindry2,3
2University of Montana. Missoula, MT. School of Integrative Physiology and Athletic Training. 3International Heart Institute, St. Patrick’s Hospital. Missoula, MT.
Corresponding author:
Graham R. McGinnis
Email: graham.mcginnis@unlv.edu
In a new window | Download PPT
Figure 1: Diagram depicting the proposed integration of chronobiology and exercise-induced cardioprotection. Graphic representation of circadian rhythms in the mRNA expression of core clock genes CLOCK, BMAL1, PER2, REV-ERBα, in the mouse heart over the 24-hour light dark cycle. Established roles for clock genes in the heart based on whole body and cardiomyocyte specific deletion models, as well as extrapolation from relevant studies in skeletal muscle. Additionally, the role of exercise as a means to induce the expression of clock genes is summarized in skeletal muscle and the heart. The amplitude of animated lines is based on published observations of clock gene expression. ∧ Induced by exercise, ㏒No change, ? Unknown influence of exercise, * effects of exercise only at ZT22.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 10933 | 33 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA