Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
New therapeutic targets to prevent diastolic dysfunction in heart failure with preserved ejection fraction
Time:2020-07-03
Number:15535
Author Affiliations

Conditioning Medicine 2020. 3(3):171-183.
Abstract
Heart failure with preserved ejection fraction (HFpEF) is poised to be the leading cause of heart failure in the near future. Currently, there are no specific therapies for improving morbidity and mortality associated with HFpEF and this has been attributed, in part, to its diverse etiology. One common feature, which defines HFpEF is diastolic dysfunction, a condition in which impaired left ventricular (LV) relaxation results in increased end-diastolic pressures and impaired blood filling. This manifests with signs and symptoms of congestive heart failure, despite LV systolic function being relatively preserved. Studies that have investigated the mechanisms underlying diastolic dysfunction have linked it to impaired cardiomyocyte relaxation and extracellular matrix (ECM)-related stiffening of the heart. Current treatment strategies for heart failure that target the sympathetic nervous system and the renin–angiotensin–aldosterone system have failed to improve prognosis in HFpEF. As such, there is an unmet need to identify new therapies that can directly ameliorate diastolic function and improve clinical outcomes in HFpEF. In order to achieve this, a comprehensive understanding of the multi-level processes that lead to diastolic dysfunction is required, which entail abnormal calcium handling, myofilament and cytoskeleton dysfunction, and abnormal ECM depositions. In this review article, we focus on the molecular mechanisms underpinning diastolic dysfunction, and also discuss potential therapeutic strategies for alleviating the impaired relaxation associated with HFpEF and improving clinical outcomes.
Keywords: Diastolic dysfunction, heart failure with preserved ejection fraction, myocardial stiffness, impaired relaxation, calcium handling, myofilament
Abstract
Heart failure with preserved ejection fraction (HFpEF) is poised to be the leading cause of heart failure in the near future. Currently, there are no specific therapies for improving morbidity and mortality associated with HFpEF and this has been attributed, in part, to its diverse etiology. One common feature, which defines HFpEF is diastolic dysfunction, a condition in which impaired left ventricular (LV) relaxation results in increased end-diastolic pressures and impaired blood filling. This manifests with signs and symptoms of congestive heart failure, despite LV systolic function being relatively preserved. Studies that have investigated the mechanisms underlying diastolic dysfunction have linked it to impaired cardiomyocyte relaxation and extracellular matrix (ECM)-related stiffening of the heart. Current treatment strategies for heart failure that target the sympathetic nervous system and the renin–angiotensin–aldosterone system have failed to improve prognosis in HFpEF. As such, there is an unmet need to identify new therapies that can directly ameliorate diastolic function and improve clinical outcomes in HFpEF. In order to achieve this, a comprehensive understanding of the multi-level processes that lead to diastolic dysfunction is required, which entail abnormal calcium handling, myofilament and cytoskeleton dysfunction, and abnormal ECM depositions. In this review article, we focus on the molecular mechanisms underpinning diastolic dysfunction, and also discuss potential therapeutic strategies for alleviating the impaired relaxation associated with HFpEF and improving clinical outcomes.
Keywords: Diastolic dysfunction, heart failure with preserved ejection fraction, myocardial stiffness, impaired relaxation, calcium handling, myofilament
Introduction
Cardiovascular disease (CVD) is the leading cause of death in the world and it has been estimated that more than 22.2 million people will die from CVD by 2030 (Virani et al., 2020). Among all the risk factors, heart failure (HF) has been the fastest-growing cause of CVD (Ziaeian et al., 2016) and accounted for 1 in 8 deaths in the U.S.A. in 2017 (https://www.cdc.gov/, 2019). Normal heart contractile function is critically dependent on a compliant left ventricle (LV) filling during diastole, and contraction of the LV in systole. HF occurs when the normal activity of the heart fails to pump enough blood to meet the demands of the body for oxygen and nutrient supply. Because of their mutual dependency, the impairment of either diastolic or systolic function can result in congestive HF. Based on the LV ejection fraction, HF is broadly divided into HF with reduced ejection fraction (HFrEF, when LVEF is < 40%), HF with mid-range ejection fraction (when LVEF is 40-50%), and HF with preserved ejection fraction (HFpEF, when LVEF is > 50%)(Ponikowski et al., 2016; Yancy et al., 2017). Epidemiology studies have shown that HFpEF has morbidity and mortality that rivals HFrEF (Yancy et al., 2017). A variety of factors and comorbidities are associated with the development of HFpEF including advanced age, female sex, obesity, systemic arterial hypertension, diabetes mellitus, renal dysfunction, sleep disorders, and chronic obstructive pulmonary disease, conditions that result in systemic inflammation and endothelial dysfunction that may trigger adverse LV remodeling and result in HFpEF (Butler et al., 2014; Shah et al., 2016). Unlike HFrEF, for which there is a growing armamentarium of pharmacological agents with prognostic benefit, there are no specific therapies for improving clinical outcomes in patients with HFpEF. This likely relates to the latter’s multi-factorial etiology, and an incomplete understanding of the pathophysiology underlying HFpEF. A key feature defining HFpEF is diastolic dysfunction, which is characterized by increased myocardial stiffness, impaired LV relaxation, increased LV end-diastolic pressures, and impaired LV filling, the clinical manifestations of which are congestive HF.
HFpEF was previously known as “diastolic HF (DHF)” a term referring to any clinical syndrome where congestive HF occurs in the presence of relatively normal LV systolic function. However, the term “diastolic HF” does not always equate to HFpEF, and this often generates confusion. Firstly, many HFrEF patients develop diastolic dysfunction, suggesting that diastolic dysfunction is not an exclusive feature of HFpEF (Aziz et al., 2013). Also, maintaining a normal ejection fraction does not necessarily mean that systolic function of HFpEF patients is normal. Other systolic parameters such as global longitudinal strain (GLS) have been shown to be compromised in HFpEF (Lekavich et al., 2015; Morris et al., 2014). Moreover, although diastolic function measurements remain the most heavily-weighted parameters according to the latest Heart Failure Association-Pretest Assessment (P), Diagnostic workup with echocardiogram and natriuretic peptide score (E), Advanced workup with functional testing in case of uncertainty (F), and Final etiological workup (F) (HFA–PEFF) diagnostic algorithm, an absence of diastolic dysfunction does not necessarily exclude the diagnosis of HFpEF (Pieske et al., 2019). Thus, the term “DHF” is much less commonly used nowadays since it does not describe the real scope of this syndrome. Nonetheless, the overall majority of HFpEF patients do have diastolic dysfunction despite the foregoing controversies, and there is no evidence supporting other more predominant contributors.
In order to improve HFpEF-related outcomes, a comprehensive understanding of the multi-level processes that lead to diastolic dysfunction is required. In this article, we review the molecular mechanisms (such as abnormal calcium handling, myofilament and cytoskeleton dysfunction, and abnormal extracellular matrix [ECM] depositions) underpinning diastolic dysfunction, and also discuss potential therapeutic strategies for alleviating the impaired relaxation associated with HFpEF for improved clinical outcomes.
Myocardial relaxation and passive LV stiffness as determinants of diastolic function
At the beginning of diastole, the ventricles relax and increase their volume with a rapid drop in intraventricular pressure. When the pressure is lower than atrial pressure, the atrioventricular valves open, allowing the majority of the blood to transit into the ventricles from the vena cavae and pulmonary veins (Fig. 1). Thus, the ventricles work as a "suction pump" during the early phase of blood filling (Robinson et al., 1986). After that, the subsequent atrial contraction pumps a relatively smaller volume of blood into the ventricles, which concludes diastole. Interestingly, since the heart does not have extensors unlike skeletal muscle, the driving force of behind ventricular relaxation in early diastole does not come from the work of contractile units. Instead, it is the recoiling movement powered from the elastic elements of the heart, such as elastin and collagen of the extracellular matrix (ECM), titin of cardiomyocytes, and even pericardium. During systole when the chambers contract against arterial load, it simultaneously compresses these elements, storing the elastic energy, much like a compressed spring. When electrical excitation is over, the contractile units of the cardiomyocytes stop generating force, the stored energy releases and powers the whole chamber to recoil back to the original position.

In a new window | Download PPT
Figure 1: Wiggers diagram and cardiac cycle. (1) Isovolumetric contraction: closure of atrioventricular (AV) valves with rapid rise of intraventricular pressure without volume change; (2) Ejection: intraventricular pressure surpasses aortic pressure and opens aortic valves. Rapid blood outflow from the ventricle to aorta followed by slowing down of outflow because of the turning-off of contractile units; (3) Isovolumetric relaxation: intraventricular pressure becomes lower than aortic pressure. AV valves close with rapid decrease in intraventricular pressure without volume change; (4) Atrial filling: start of blood filling into atrial with AV valves closed; (5) Intraventricular pressure becomes lower than atrial pressure. AV valves open and blood filling into the ventricle (E or early wave); (6) Ending of diastole with atrial contraction to eject smaller amount of blood into the ventricle (A or atrial wave).
Two key determinants of myocardial relaxation are cardiomyocyte contractile mechanisms and passive stiffness of the LV. Cardiomyocyte relaxation is an energy-consuming process, mediated by the calcium (Ca2+) handling system and the basic relaxation properties of the myofibrils. The Ca2+ handling system controls how fast intracellular Ca2+ is removed from the cytoplasm and is mainly governed by the sarcoplasmic reticulum Ca2+ ATPase pump (SERCA) (Maruyama et al., 1989). Also, the relaxation of myofibrils requires the binding of ATP to proceed (Poggesi et al., 2005; Weber et al., 1973). Passive stiffness of the myocardium relates to LV compliance and is strongly influenced by changes in the composition of ECM (e.g. interstitial fibrosis), and modulation of myofibrillar proteins (e.g. Titin) and cytoskeleton components. Fig. 2 summarizes the complexity of the regulatory processes that impact on diastolic function. This complexity possibly explains why HFpEF is such a heterogeneous syndrome as any molecular and cellular alterations on different biological levels could compromise diastolic function. In the following sections, we will focus on how these alterations impact on myocardial relaxation and passive stiffness, and highlight potential new targets for ameliorating diastolic dysfunction in HFpEF.
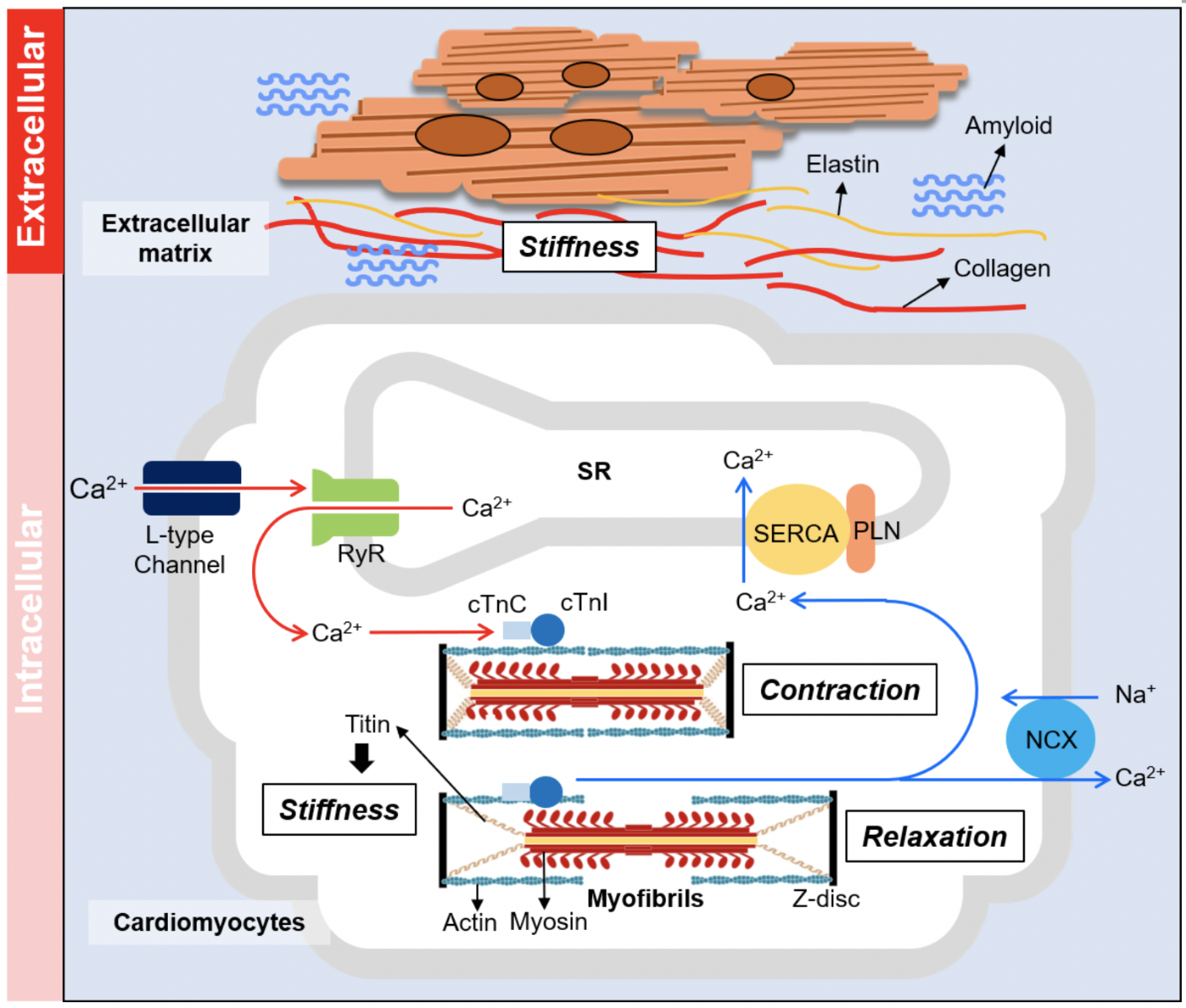
In a new window | Download PPT
Figure 2: Determinants of myocardium relaxation. Diastolic function of the heart is governed by the stiffness of the myocardium as well as the relaxation action of the contractile unit. Extracellular regulation (top panel): the composition of extracellular matrix mediates the stiffness of the non-contractile elements of the myocardium. Amyloidosis is another extracellular cause of myocardial stiffening. Intracellular regulation (lower panel): muscle relaxation property is mediated by the calcium handling system of the cardiomyocyte or the basic contractile/relaxation properties of the myofibrils. Stiffness of cardiomyocyte is mainly mediated by giant protein titin.
Calcium dysregulation and impaired cardiomyocyte relaxation
Calcium (Ca2+) handling plays a central role as a mediator of contraction and relaxation of cardiomyocytes. The sarcoplasmic reticulum (SR) is a subcellular membrane network that stores the majority of Ca2+ in cardiomyocytes. Contraction of cardiomyocytes relies on a process termed calcium-induced calcium release (CICR) in which the action potential stimulates Ca2+ influx from L-type Ca2+ channel, which opens the ryanodine receptor (RyR), followed by Ca2+ release from SR through the RyR to activate myofibrils (Bers 2002; Fabiato et al., 1979). After electrical stimulation, the relaxation of cardiomyocytes requires removal of Ca2+ from the cytosol to deactivate the myofibrils and this is mainly mediated via SERCA. The cardiac-specific isoform SERCA2a is an integral SR membrane protein that has a high affinity for Ca2+ on the cytosolic side, and transports Ca2+ to the lumen side of the SR with the consumption of ATP, essentially functioning as a Ca2+ pump (Fig. 2). The activity of SERCA2a is regulated by two smaller integral SR membrane proteins: phospholamban (PLN) and sarcolipin (SLN). Phospholamban serves as a SERCA2a inhibitor by interacting with SERCA2a and decreasing its affinity for Ca2+ (Akin et al., 2013; MacLennan et al., 2003). The activity of PLN is regulated by its phosphorylation at Ser16 and/or Thr17, which promotes the oligomerization of PLN and releases its inhibition on SERCA2a (Ablorh et al., 2015; Tada et al., 1996). SLN is structurally similar to PLN and also inhibits SERCA2a function (Bhupathy et al., 2007), but acts more like an uncoupler between Ca2+ transport and ATP hydrolyzation (Shaikh et al., 2016).
Even though SERCA2a function has been linked to myocardial relaxation, most studies have investigated the role of impaired SERCA2a function in systolic HF (Hasenfuss et al., 1993; Houser et al., 2000; Morgan 1991). This is not surprising since SERCA2a not only plays a central role in Ca2+ removal for cardiomyocytes to relax, but also the availability of Ca2+ to activate myofibrils. Impaired SERCA2a activity can be either transcriptional or post-translational. Gene expression of SERCA2a has been shown to be down-regulated in end-stage failing hearts (Arai et al., 1993; Eisner et al., 2013). Also, the chronic sympathetic nervous system in HF can desensitize β-adrenergic signaling, the downstream effect of which is reduced PLN phosphorylation that inhibits SERCA2a (Marks 2013; Schmidt et al., 1999). SERCA2a is also a direct target of oxidative modification by increased reactive oxygen species (ROS) (Balderas-Villalobos et al., 2013; Viner et al., 1999; Zima et al., 2006), which commonly occurs in acute myocardial ischemia-reperfusion injury or diabetic HF with impaired mitochondrial function (Zarain-Herzberg et al., 2014).
Overexpression of SERCA2a has been shown to be beneficial in HFrEF (Baker et al., 1998; Del Monte et al., 1999), and it has been demonstrated that diastolic function can also be improved by increasing SERCA2a function (Baker et al., 1998; Iwanaga et al., 2004). Several groups are now focusing on the role of SERCA2a in HFpEF. Groban et al. (2012) first reported that diastolic function was improved along with reduced PLN/SERCA2a ratio when aging rats were treated with angiotensin converting enzyme inhibitor (ACE-I). This finding is consistent with later reports showing reduced SERCA2a activity in hypertensive animal models of diastolic dysfunction with preserved ejection fraction (Rouhana et al., 2019; Tanaka et al., 2014). Furthermore, anti-oxidative agents have been shown to benefit diastolic dysfunction with a potential link to restored SERCA2a function (Scotcher et al., 2016; Wilder et al., 2015), but the mechanisms are unclear.
The sodium-calcium exchanger (NCX) is another mediator of Ca2+ removal during cardiomyocyte relaxation, but only makes a small contribution (Bassani et al., 1994; Pieske et al., 1999). NCX is a plasma membrane integral protein that uses the electrochemical gradient of sodium (Na+) to drive one Ca2+ ion out of the cell in exchange for 3 Na+ ions (Bers 2002). The expression of NCX is usually increased in animal models of HFrEF (Bers et al., 2010), although the findings in human HFrEF are more inconsistent (Hasenfuss et al., 2002). The overall contribution of NCX for Ca2+ removal is increased because of reduced SERCA activity (Piacentino III et al., 2003).NCX appears to act differently in HFpEF. In the hypertensive animal, Rouhana et al. (2019) established that NCX activity is decreased along with SERCA2a, suggesting it might be another contributor to elevated diastolic [Ca2+], which potentially leads to relaxation impairment. In some end-stage HFrEF hearts, the activity of NCX is limited because extra Na+ efflux results in reversed Ca2+ extrusion, which does not happen in HFpEF (Nuss et al., 1992).
Therapeutic targeting of SERCA2a in HFpEF
Given the beneficial roles of SERCA2a in both systolic and diastolic function in animal models, clinical trials have tested the upregulation of SERCA2a as a therapeutic strategy for HF. The “Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID)” trial demonstrated that adeno-associated virus serotype 1-delivered SERCA2a gene (AAV1/SERCA2a) therapy in HFrEF patients reduced the rate of re-hospitalization for HF (Hajjar et al., 2008; Jaski et al., 2009). Despite these promising results, the follow-up larger phase 2b CUPID2 trial failed to show any benefit (Fernández-Ruiz 2016; Greenberg 2017). The failure of SERCA2a gene therapy has been attributed to the potential effects of B-type natriuretic peptide (BNP) on SERCA2a expression (Zhai et al., 2018), and it has been suggested that SERCA2a gene therapy may be beneficial in HFpEF where BNP levels tend to be lower than in HFrEF. The funny (If) channel inhibitor, ivabradine, has been reported in a small clinical trial of 61 patients to improve exercise capacity, LV filling pressures, and diastolic function of HFpEF patients (Kosmala et al., 2013). Although the main action of ivabradine is to reduce heart rate, it is also known to be a SERCA2a activator (Reil et al., 2013; Xie et al., 2020), supporting a beneficial role of SERCA2a in HFpEF. However, the larger “Preserved Left Ventricular Ejection Fraction Chronic Heart Failure With Ivabradine Study (EDIFY)” trial failed to demonstrate improved diastolic function with ivabradine in HFpEF patients (Komajda et al., 2017). Istaroxime is another activator of SERCA2a that acts by blocking the SERCA2a–PLN interaction (Huang 2013). It has been tested in the Hemodynamic Effects of Istaroxime in Patients with Worsening HF and Reduced LV Systolic Function (HORIZON-HF) trial in HFrEF patients (Shah et al., 2009), and was shown to lower pulmonary capillary wedge pressure (PCWP) and improve diastolic function, effects that might benefit HFpEF. A clinical trial with this agent is currently ongoing in HFpEF patients (clinicaltrials.gov, NCT02772068).
Myofilament dysfunction and impaired cardiomyocyte relaxation
Myofibrils are the most abundant organelle and are the basic contractile unit of the cardiomyocytes (Gerdes 2012). Contraction and relaxation are mediated by alternating sliding actions between the thin and thick filaments toward or away from the M-band (Huxley 1957). The thin filament is composed of filamentous sarcomeric α-actin with regulatory proteins troponin complex (Tn) and tropomyosin (Tm). The thick filament consists of myosin heavy chains (MHC), regulatory myosin light chains (RLC), essential myosin light chain (ELC), and myosin binding protein C (MyBP-C). The protruding globular region of MHC, termed cross-bridge, can interact with actin using ATP to generate force to pull the thin filament towards the center of the sarcomere (Weber et al., 1973). Myofibril function is tightly coupled with intracellular Ca2+ signaling. During contraction, elevated [Ca2+] from SR propels Ca2+ to bind to TnC. This results in conformational changes in the troponin complex and releases the allosteric inhibition of Tm, allowing the cross-bridges to interact with actin and generate force (Fig. 2). During relaxation, Ca2+ is removed by SERCA2a and NCX, the interaction of cross-bridges to actin arrests, thereby allowing the sarcomere to slide back into the relaxed state (Ebashi et al., 1968; Gordon et al., 2000; Lin et al., 2019).
At the myofibril level, cardiomyocyte relaxation can be modulated via two distinct pathways, Ca2+ sensitivity and relaxation properties of the myofibril itself. Ca2+ sensitivity refers to how readily the myofibrils are activated by Ca2+. During cardiomyocyte relaxation, Ca2+ sensitivity of the myofibrils determines the rate of relaxation of the myofibrils. As for myofibril relaxation, it is important to appreciate the distinction between “relaxation of cardiomyocytes” and “relaxation of myofibrils.” The inactivation of myofibrils begins when dissociation of Ca2+ turns off cross-bridges. The pure relaxation kinetics of the myofibrils after Ca2+ removal has a unique “bi-phasic” pattern – linear phase and exponential phase of relaxation (Stehle et al., 2002; Tesi et al., 2002), a finding which is not observed in cardiomyocyte relaxation. The reason for this is that cell relaxation is the combination of Ca2+ handling and myofibril relaxation processes, and the fall of cytosolic [Ca2+] is a much more gradual process than myofibrils (Backx et al., 1995; Blinks et al., 1978). In many cases, alterations in general myofibril relaxation kinetics can translate to the whole-cell level as several recent studies have demonstrated (Coppini et al., 2017; Jeong et al., 2018; Lin et al., 2020). However, the different phases of relaxation from the myofibril study can provide further insights into the potential molecular basis of these abnormalities. For example, the investigation of the impaired linear phase relaxation should focus on how the thin filament is inactivated by the action of the Tn complex, whereas the impaired exponential phase of relaxation is more relevant to the cross-bridge cycling between the thin and thick filaments and the elastic properties of the myofibrils provided by titin (Lin et al., 2019; Stehle et al., 2009). Titin is the third major structure of the myofibrils. In contrast to the thin and thick filaments, titin does not have a contractile function but is the main player that mediates passive stiffness of the myofibrils (Granzier et al., 2002). Alterations of sarcomere proteins can directly affect stiffness and relaxation of the cardiomyocytes and potentially result in HFpEF.
Therapeutic targeting of titin in HFpEF
Titin is the largest human protein (35,000 amino acids in length), as evidenced by the length of one single titin molecule spanning from the Z disc to the central M line. Titin provides structural support and elasticity of the myofibrils, a property that can directly translate to passive stiffness at the whole heart level (Chung et al., 2011). Because of its extreme length, titin comprises many different structural elements (LeWinter et al., 2010), and goes through a series of alternative splicing during development (Guo et al., 2010). The shorter and stiffer N2B isoform is the predominant isoform in the mature human heart in contrast to the longer and more compliant N2BA isoform (Warren et al., 2004). Another determinant of titin stiffness are post-translational modifications (PTMs) of amino acids located in its key structural elements, many of which have been characterized: (1) Decreased stiffness by phosphorylation at Ser4010 and/or Ser4099 of the N2B unique sequence (N2Bus) by protein kinase A and G (PKA and PKG) (Borbély et al., 2009; Krüger et al., 2009); (2) Increased stiffness by phosphorylation at Ser11878 and/or Ser12022 of the PEVK (Pro-Glu-Val-Lys) domain by protein kinase C alpha (PKCα) (Hidalgo et al., 2009); (3) Increased stiffness by disulfide bond formation at 6 cysteine residues in N2Bus (Grützner et al., 2009); and (4) Decreased stiffness by S-glutathionylation at the cryptic cysteines located in immunoglobulin (Ig) domains (Alegre-Cebollada et al., 2014).
The ratio of N2B/N2BA isoforms has long been shown to be altered in pathological conditions in response to stress (Neagoe et al., 2002; Wu et al., 2002). In 2006, van Heerebeek et al. (2006) compared the stiffness and N2B/N2BA ratio between heart tissues from HFrEF and HFpEF patients and found HFpEF patients had higher N2B expression than HFrEF patients, but it was later found that N2B/N2BA ratio is unchanged when HFpEF hearts are compared to non-failing hearts (Hopf et al., 2018; Zile et al., 2015). Instead, Hamdani et al. (2013) used a mouse HFpEF model stimulated by a combination of metabolic dysfunction and high fat diet, and found the N2Bus region of titin to be hypophosphorylated, which was accompanied by diastolic dysfunction and increased titin stiffness, and this could be ameliorated by activated PKG signaling (Hamdani et al., 2014). These results were confirmed by Zile et al. (2015) using heart tissues from HFpEF patients. The nitric oxide (NO)/cyclic GMP/PKG pathway is known to be impaired with increased oxidative stress (Kasner et al., 2011; Park et al., 2018), likely explaining why increased stiffness is the signature feature of HFpEF observed in diabetic cardiomyopathy (van Heerebeek et al., 2009). In contrast, reports from phosphorylation in the PEVK region of titin in HFpEF hearts have been inconsistent (Hamdani et al., 2013; Hopf et al., 2018).
Other strategies that have been tested in the experimental setting to reduce titin stiffness include: (1) exercise, which was shown to reduces titin stiffness and improve diastolic function in a mouse model of HFpEF (Slater et al., 2017), although the mechanism was not addressed; (2) Metformin, a commonly used diabetic drug, was shown to reduce titin stiffness and improve diastolic function via PKA phosphorylation in the N2Bus region of titin (Slater et al., 2019). This result is consistent with an earlier study using another antidiabetic drug empagliflozin (SGLT2 inhibitor) on ventricular trabeculae from HF patients although no explanation for the underlying mechanism was provided (Pabel et al., 2018). However, one can speculate that amelioration of metabolic dysfunction is beneficial to titin stiffness-regulated diastolic function; and (3) RNA binding motif 20 (RBM20), the known regulator that switches titin isoforms by alternative splicing to the stiffer N2B isoform has been investigated as a therapeutic target for improving diastolic function (Guo et al., 2012). In animal studies, cardiac-specific RBM20 knockout mice were shown to have much more compliant myofibrils, ventricle chamber, and improved diastolic function in response to HFpEF (Methawasin et al., 2016). Based on the promising experimental results showing that PKG phosphorylation of the titin N2Bus region could improve diastolic dysfunction, the RELAX trial was performed using the phosphodiesterase 5 inhibitor, sildenafil, which can enhance PKG activity, but six months treatment with sildenafil did not improve exercise capacity or clinical status (Redfield et al., 2013). The above new findings shed light on using titin as a potential therapeutic target to improve the compliancy of the HFpEF heart despite earlier failure of the RELAX trial.
Alterations in myofibril mechanics and impaired cardiomyocyte relaxation
The cardiac troponin complex (cTn) directly regulates the response of the thin filament to Ca2+ and impacts on myofibril relaxation (Gordon et al., 2000). Failure to efficiently turn off cTn leads to enhanced Ca2+ sensitivity and prolonged relaxation (Chung et al., 2016). Thus, it is not surprising that the inhibitory cardiac troponin I subunit (cTnI) was among the earliest proteins identified in inherited restrictive cardiomyopathies (RCM) (Gomes et al., 2005; Mogensen et al., 2003), the class of cardiomyopathy known to result from relaxation impairment without showing systolic dysfunction. cTnI mutations can be also seen in some hypertrophic cardiomyopathy (HCM) patients, whose pathological phenotypes sometimes show prolonged relaxation as well. In HCM and RCM heart tissues, Ca2+ sensitivity is generally elevated accompanied by prolonged myofibril relaxation kinetics (Cheng et al., 2015; Iorga et al., 2008; Kruger et al., 2005), with occasional exceptions (Dvornikov et al., 2016). cTnI can be phosphorylated by several protein kinases (Solaro et al., 2013), the most well-known of which is PKA. PKA-phosphorylated cTnI reduces Ca2+ sensitivity and is beneficial for relaxation (Cheng et al., 2015; Zhang et al., 1995). Recently, Lin et al. (2020) reported that acetylation-mimic mutations in the actin-binding region of cTnI leads to reduced Ca2+ sensitivity and faster relaxation at both the cellular and myofibril levels in a PKA-independent manner, but the effect on the whole heart was not addressed.
In addition to the regulatory system of the thin filament, the kinetics of force-generating myosin cross-bridges can also modulate Ca2+ sensitivity and myofibril relaxation (Robinson et al., 2002). Myosin RLC and ELC are a pair of myosin light chains located in the neck of the myosin head. In two RCM models, mutations in RLC and ELC increased Ca2+ sensitivity and diastolic dysfunction of the heart (Abraham et al., 2009; Yuan et al., 2017). Also, phosphorylation of RLC results in increased Ca2+ sensitivity and delayed relaxation (Colson et al., 2010; Sevrieva et al., 2020). Cardiac specific MyBP-C3 is a thick filament-associated protein that controls the interactions between the myosin S2 domain and α-actin. Ablation of MyBP-C3 has been shown to result in increased Ca2+ sensitivity and faster relaxation because of accelerated cross-bridge kinetics (Moss et al., 2015). Phosphorylation of MyBP-C3 also speeds up relaxation, which alleviates diastolic dysfunction in an aging mouse model (Rosas et al., 2015; Rosas et al., 2019). Despite these interesting results showing that the thick filament proteins could be closely linked to muscle relaxation and diastolic dysfunction, detailed myofibril mechanics of these alterations are still not fully understood and need further investigation.
Therapeutic targeting of the contractile apparatus to improve myofibril relaxation in HFpEF
A number of pharmacologic interventions for improving maladaptive contractile function of the myofibrils in HFrEF have been investigated (Bristow 2000; McMurray et al., 2014; Teerlink et al., 2016). There are several challenges associated with targeting myofibrils to improve diastolic function in HFpEF. The onset of HFrEF, which mainly results from reduced contractility, can be targeted by pharmacological agents to either improve contractility of the myofibrils or decrease the afterload of the heart. In contrast, HFpEF is more complex and is characterized by passive stiffening and impaired cardiomyocyte relaxation. Furthermore, the mechanisms underlying the relaxation impairment of the myofibrils is difficult to dissect from disturbed calcium handling at the level of cardiomyocyte.
With advances in cellular or ex vivo myofibril mechanics system, it is now possible to decipher the contribution of myofibril mechanics to the development of diastolic dysfunction in HFpEF. Jeong et al. (2018) have investigated myofibril mechanics of explanted LV tissue from two patients diagnosed as idiopathic RCM with symptoms of HFpEF. They found prolonged myofibril relaxation compared to control donors as seen in two small animal models of diastolic dysfunction and preserved EF. Interestingly, the diastolic dysfunction as well as myofibril relaxation were improved by the pan-histone deacetylase (HDAC) inhibitor givinostat (Jeong et al., 2018). This was the first evidence showing that relaxation at the myofibril level could be improved by a pharmacological intervention. Recently, Wallner et al. (2020) treated a feline model of HFpEF with another pan-HDAC inhibitor, suberoylanilide hydroxamic acid, and observed similar results. With the advancement of techniques, it is now feasible to assess the mechanical properties of myofibrils from cardiomyocytes derived from human induced pluripotent stems cells (hiPSCs) (Pioner et al., 2020; Pioner et al., 2016) or living human heart tissue culture (Ou et al., 2019). Whether human myofibrils with relaxation impairment in HFpEF patients can be corrected by HDAC inhibition remains to be determined. A recent publication from Wu et al. (2019) assessed three different hiPSC-derived cardiomyocytes (CM) from inherited diastolic HF patients with myofibrillar protein mutations, and found enhanced Ca2+ sensitivity and slower relaxation in all of them. Despite no direct mechanism for their finding, this shows iPSC-CMs could be an ideal tool for drug screening in the near future.
The cytoskeleton and cardiomyocyte relaxation
Microtubule and intermediate filament desmin
A microtubule (MT) is composed of α/β tubulin heterodimers that polymerize to form a hollow tubes-shaped cytoskeleton about 25 nm in diameter (Stephens et al., 1976). The role of MTs in cardiomyocytes and how they mediate mechanical function has been overshadowed by the myofilament system for years. In 2016, it was unveiled by the Prosser Lab that MTs can mechanically couple to the sarcomere and provide resistance, acting like a spring that deforms into sinusoidal buckles during cardiomyocyte contraction (Robison et al., 2016). This “buckling” action requires the detyrosination (dTyr) of α-tubulin and the presence of intermediate filament desmin. Down-regulation of dTyr or desmin substantially reduced stiffness of the entire cardiomyocyte (Granzier et al., 1995). Follow-up studies from the same group demonstrated that detyrosination of MTs is increased in human cardiomyocytes isolated from HFpEF patients and can be relieved by adenoviral overexpression of tubulin tyrosine ligase (TTL) or the knock-down of a newly identified α-tubulin detyrosinase vasohibin-small vasohibin binding protein (Caporizzo et al., 2020; Chen et al., 2018; Chen et al., 2020). However, there are two issues that need to be addressed before MT-based stiffness correction can be considered a therapeutic option: (1) Despite promising data from human cardiomyocytes, it remains to be shown whether detyrosination of the MTs can be demonstrated in an in vivo system; (2) Pharmacological agents that can specifically target detyrosination at the level of MTs are needed. Nonetheless, the breakthrough finding of MT-regulated cardiomyocyte stiffness has offered a novel therapeutic target for the treatment of HFpEF.
Filamin C
Filamins are a family of large cytoskeletal proteins that organize filamentous actin (f-actin) into networks and also anchor various plasma membrane integral proteins to the actin cytoskeleton to provide a scaffold for a variety of cytoplasmic signaling pathways (Stossel et al., 2001). Filamin C is the muscle-specific filamin that modulates actin dynamics and plays an important role in myofibrillogenesis (Chiang et al., 2000). Filamin C deficiency in mice is lethal at birth due to underdeveloped skeletal muscles and respiratory system failure (Dalkilic et al., 2006). In matured cardiomyocytes, filamin C acts as a signaling hub to repair damaged myofibrils (Leber et al., 2016). Filamin C has high expression in the heart and is one of the major non-myofibrillar proteins that could result in cardiomyopathies. Recently, it was found that missense mutations of filamin C leads to RCM and HCM (Brodehl et al., 2016; Gómez et al., 2017), which tend to develop diastolic dysfunction and HFpEF (Seferović et al., 2019). The mechanism through which abnormal filamin C causes these phenotypes is unclear. However, histological staining of RCM heart tissue from filamin C mutation showed cytoplasmic aggregates formed by mutant filamin (Brodehl et al., 2016). The authors speculated that protein aggregates could increase cellular stiffness and relaxation properties (Plodinec et al., 2011), which may explain the restrictive phenotype. Further identification of the molecular causes underlying HCM/RCM is required to get a better understanding for gene-specific diagnosis and potential therapy.
Extracellular matrix and stiffening of the myocardium
Myocardial fibrosis in HFpEF
Fibrosis is defined as the abnormal deposition of ECM in tissues via a cascade of cellular responses to injuries. It commonly begins as an adaptive response but ultimately progresses into cellular dysfunction (Rockey et al., 2015). Myocardial fibrosis is the hallmark of detrimental adaptation of the heart that likely progresses to HF. It is a well-known feature of HFrEF after myocardial infarction and results from replacement of dead cardiomyocytes, which eventually contributes to systolic dysfunction because of impaired myocardial force transduction or increased workload of the remaining cardiomyocytes (Baicu et al., 2003; Heineke et al., 2006). HFpEF has been previously reported to have a similar collagen level as HFrEF (Borbély et al., 2005; van Heerebeek et al., 2006). Since one of the features of abnormal ECM deposition during fibrosis is increased stiffness of organs including the heart (Norton et al., 1997; Yamamoto et al., 2002), it was frequently speculated that a stiffened myocardium is the cause of diastolic dysfunction in HFpEF. The major cause of myocardial fibrosis of HFpEF is the inflammatory responses driven by comorbidities such as hypertension, coronary microvascular disease, or metabolic dysfunction (Friebel et al., 2019; Glezeva et al., 2014; Paulus et al., 2013). For example, chronic hypertension commonly leads to endothelial damage and vascular inflammation, thereby triggering the differentiation of circulating monocytes into macrophages, which secretes pro-fibrotic factors such as transforming growth factor-β (TGF-β), and eventually leads to fibrosis (Mouton et al., 2020). Some studies reported the elevated stiffness induced by fibrosis was directly linked to diastolic dysfunction in animal HFpEF models (Cieslik et al., 2011; Martos et al., 2007) and HFpEF patients (Rommel et al., 2016; Su et al., 2014). Recently, HFpEF has been shown to have more collagen volume than HFrEF (Dai et al., 2012; Echegaray et al., 2017), and a different spectrum of ECM composition that results in higher stiffness (Bielecka-Dabrowa et al., 2016; Kasner et al., 2011). This could explain why the myocardium of HFpEF typically has higher stiffness than HFrEF and worse diastolic function (Aziz et al., 2013; Røe et al., 2017).
Amyloidosis is a common disease in the elderly that occurs when insoluble misfolded proteins deposit in tissues and organs (Dubrey et al., 2011). Amyloidosis is different from fibrosis as the origin of amyloid is from proteins arranged into a fibril like structure instead of ECM. In the heart, amyloidosis results in stiffening of the heart chamber, which results in diastolic dysfunction (Bhupathi et al., 2011; Pislaru et al., 2019), and is frequently present (5-13%) in patients diagnosed with HFpEF (González-López et al., 2015). There are several types of amyloidosis derived from different proteins such as immunoglobulin light and heavy chain (AL and AH) or transthyretin (ATTR) triggered from totally different etiologies (Kholova et al., 2005). Hearts with amyloidosis might have higher fibrosis, which is likely linked to inflammatory responses (Buxbaum et al., 2012; Mohammed et al., 2014), but they do not seem to have a direct correlation.
Therapeutic targeting of fibrosis to improve myocardial relaxation
The common end point of myocardial fibrosis is the activation of cardiac fibroblasts when TGF-β binds to its receptor and initiates signaling pathways (Bujak et al., 2007). This leads to the nuclear import of Smad2/3 to prime pro-fibrotic genes (Khalil et al., 2017). The activation of fibroblasts plays pivotal roles in the secretion of major ECM materials such as collagen, fibrin, fibronectin, proteoglycans, and glycosaminoglycans (Tracy et al., 2016). The anti-fibrotic compound, pirfenidone, blocks the transport of Smad2/3 by inhibiting p38-γ (Dosanjh 2007) and has been shown to ameliorate myocardium fibrosis in two rodent models of diastolic dysfunction (Miric et al., 2001; Mirkovic et al., 2002). In HFpEF, the clinical trial “PIROUETTE (Efficacy and Safety of Pirfenidone in Patients With Heart Failure and Preserved Left Ventricular Ejection Fraction)” is ongoing to test the efficacy and safety of pirfenidone to treat HFpEF patients with evidence of myocardial fibrosis (clinicaltrials.gov, NCT02932566). Advanced glycation endproducts (AGE) are the products of glycated proteins or lipids that can crosslink and act as another major determinant of the stiffness of ECM (Hartog et al., 2007). Increased AGE crosslinking has been found in HFpEF patients (Kasner et al., 2011; Willemsen et al., 2012). Several studies in diabetic animal models have shown improvement of diastolic function when AGE crosslinking is blunted (Kranstuber et al., 2012; Ma et al., 2009). Earlier in 2005, a small, open-label trial also showed an AGE crosslinking breaker, ALT-711, improved diastolic function in aging patients (Little et al., 2005), suggesting a potential approach for treatment. In general, fibrosis exists in virtually all organs in the body with complex dynamics and large numbers of players, thus, it is challenging to focus on specific pathways to treat fibrosis. Recently, high throughput screening for synthetic or natural compounds has been applied to search for more potent therapeutics for cardiac fibrosis (Palano et al., 2020; Schimmel et al., 2020). However, there is still a void between basic science and clinical application in most studies to date.
Future perspective and conclusion
Among all HF patients, approximately 50% of them are diagnosed to have HFpEF (Dunlay et al., 2017). Furthermore, the prevalence of HFpEF relative to HFrEF is increasing with the majority being > 65 years old (Gerber et al., 2015; Steinberg et al., 2012). With the increasing trend of an aging population, HFpEF is projected to be the most common form of HF (Ambrosy et al., 2014). Unfortunately, despite abundant research efforts on HFpEF, its morbidity and mortality are still rising (Ponikowski et al., 2016; Yancy et al., 2017) without pharmacological therapy showing similar benefits as in HFrEF (Ponikowski et al., 2016; Roh et al., 2017). This is most likely due to the multifactorial pathophysiology that involves not only diastolic dysfunction but also perturbations in non-cardiac factors such as pulmonary and systemic vascular systems and renal function (Lam et al., 2018). This heterogeneity is not surprising given the definition of HFpEF comes from the mix of HF patients that do not have a single symptom (reduced EF). Thus, HFpEF can essentially be regarded as many different diseases based on this notion.
Despite the heterogeneity of HFpEF, it is possible to extrapolate common features of HFpEF and assess how they are initiated in response to the pathological stimulus. The most recent example is a “bona fide” HFpEF murine model developed by the Hill Lab (Schiattarella et al., 2019), in which they described a defective unfolded protein response (UPR), which involves reduced inositol-requiring enzyme 1 alpha-X-box binding protein signaling that is not seen in other HFpEF models that only represent a transitional state between normal condition and HFrEF. Interestingly, Schiattarella et al. (2019) demonstrated defective relaxation properties at the cardiomyocyte level without a mechanism described in the same report. Based on our perspective introduced in this review, these cardiomyocytes may have (1) impaired Ca2+ handling system, (2) increased cell stiffness, or (3) prolonged myofibril relaxation. Potentially, with further investigation of both Ca2+ signaling of cardiomyocytes and mechanics of isolated myofibrils, researchers could determine which mechanisms are involved and develop a therapeutic strategy for HFpEF. If no abnormalities are observed at the level of the cardiomyocyte or the myofibril, the most likely contributor of diastolic dysfunction could be alterations in ECM such as altered ECM deposition or amyloid infiltration. With the advancement of technologies it is now feasible to specifically assess detailed ECM protein expression (Barallobre-Barreiro et al., 2016; Cui et al., 2019) and detailed stiffness/elastic properties of heart tissues (Borin et al., 2018), with experimental systems allowing for the analysis of cellular and myofibril mechanics (Lin et al., 2019; Sala et al., 2018).
There are many clinical trials targeting the sympathetic nervous system (SNS) and renin–angiotensin–aldosterone system (RAAS) to indirectly ameliorate heart function in HFrEF (Brann et al., 2019; Pellicori et al., 2020), but new therapeutic targets are necessary to directly improve diastolic function in HFpEF patients. It is important to focus on the detailed mechanisms leading to the diastolic dysfunction of the heart, including stiffness and active relaxation of the myocardium, cardiomyocyte, and myofibril. Current therapeutic approaches that ameliorate the stiffness of the myocardium have focused on targeting fibrosis and titin. Emerging fields such as tubulin detyrosination and filamin C also provide promising targets for further investigation. In contrast, treatments to improve muscle relaxation either through calcium handling or sarcomere function are mostly still preliminary, but have great potential to unveil more targets for clinical application. Knowledge obtained from these studies could help in the identification of new targets and discovery of new therapeutic strategies to ameliorate diastolic function and improve clinical outcomes in patients with HFpEF.
Acknowledgments
Sang-Ging Ong is supported by National Institutes of Health grant R00 HL130416 and R01 HL148756. Chrishan Ramachandra is supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (NMRC/OFYIRG/0073/2018), the National Health Innovation Centre Singapore under its Innovation to Develop Grant (NHIC-I2S-1811007) and the SingHealth Duke-NUS Academic Medical Centre under its SingHealth Duke-NUS Academic Medicine Research Grant (AM/TP033/2020 [SRDUKAMR2033]). Derek Hausenloy is supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
References
Ying-Hsi Lin1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Shuo Cong1
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
Sang-Ging Ong3,4
3Department of Pharmacology, University of Illinois College of Medicine, Chicago, Illinois, United States of America. 4Division of Cardiology, Department of Medicine, University of Illinois College of Medicine, Chicago, Illinois, United States of America.
Chrishan J.A. Ramachandra1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Derek J. Hausenloy1,2,4-7
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore. 4Division of Cardiology, Department of Medicine, University of Illinois College of Medicine, Chicago, Illinois, United States of America. 5Yong Loo Lin School of Medicine, National University Singapore, Singapore. 6The Hatter Cardiovascular Institute, University College London, London, UK. 7Cardiovascular Research Center, College of Medical and Health Sciences, Asia University, Taiwan.
Corresponding author:
Professor Derek J. Hausenloy
Email: derek.hausenloy@duke-nus.edu.sg
In a new window | Download PPT
Figure 1: Wiggers diagram and cardiac cycle. (1) Isovolumetric contraction: closure of atrioventricular (AV) valves with rapid rise of intraventricular pressure without volume change; (2) Ejection: intraventricular pressure surpasses aortic pressure and opens aortic valves. Rapid blood outflow from the ventricle to aorta followed by slowing down of outflow because of the turning-off of contractile units; (3) Isovolumetric relaxation: intraventricular pressure becomes lower than aortic pressure. AV valves close with rapid decrease in intraventricular pressure without volume change; (4) Atrial filling: start of blood filling into atrial with AV valves closed; (5) Intraventricular pressure becomes lower than atrial pressure. AV valves open and blood filling into the ventricle (E or early wave); (6) Ending of diastole with atrial contraction to eject smaller amount of blood into the ventricle (A or atrial wave).
In a new window | Download PPT
Figure 2: Determinants of myocardium relaxation. Diastolic function of the heart is governed by the stiffness of the myocardium as well as the relaxation action of the contractile unit. Extracellular regulation (top panel): the composition of extracellular matrix mediates the stiffness of the non-contractile elements of the myocardium. Amyloidosis is another extracellular cause of myocardial stiffening. Intracellular regulation (lower panel): muscle relaxation property is mediated by the calcium handling system of the cardiomyocyte or the basic contractile/relaxation properties of the myofibrils. Stiffness of cardiomyocyte is mainly mediated by giant protein titin.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 15535 | 57 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA