Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Targeting calpains in myocardial ischemia/reperfusion injury
Time:2020-07-03
Number:13766
Author Affiliations

Conditioning Medicine 2020. 3(3):160-170.
Abstract
Loss of intracellular Ca2+ homeostasis occurring during ischemia/reperfusion (IR) induces dysregulated overactivation of calpains, Ca2+-dependent cysteine proteases. In this review we describe the mechanisms of calpain activation and analyze the evidence for calpain contribution to myocardial reperfusion injury. Exaggerated calpain activation occurs during reperfusion after intracellular pH normalization, and results in altered contractility and cell death through the cleavage of numerous protein substrates involved in sarcolemmal structure, cellular contractility, mitochondrial function, and cellular signalling. Finally, we provide an overview of the calpain inhibitors that have been evaluated in experimental models of IR and the advances in the design of new potent and selective compounds that may serve as candidates for testing the efficacy of calpain inhibition in patients with acute myocardial infarction.
Abstract
Loss of intracellular Ca2+ homeostasis occurring during ischemia/reperfusion (IR) induces dysregulated overactivation of calpains, Ca2+-dependent cysteine proteases. In this review we describe the mechanisms of calpain activation and analyze the evidence for calpain contribution to myocardial reperfusion injury. Exaggerated calpain activation occurs during reperfusion after intracellular pH normalization, and results in altered contractility and cell death through the cleavage of numerous protein substrates involved in sarcolemmal structure, cellular contractility, mitochondrial function, and cellular signalling. Finally, we provide an overview of the calpain inhibitors that have been evaluated in experimental models of IR and the advances in the design of new potent and selective compounds that may serve as candidates for testing the efficacy of calpain inhibition in patients with acute myocardial infarction.
1. Introduction
The pathophysiology of reperfusion injury is a complex process that causes cardiomyocyte death via multiple mechanisms and involves the alteration and interplay of a variety of molecules and proteins (Davidson et al., 2019). A pronounced consequence of reperfusion is the generation of intracellular Ca2+ overload and a burst of reactive oxygen species (ROS), which have the potential to induce an anomalous increase in the activity of several enzymes. One of the families of proteins directly modulated by changes in the intracellular Ca2+ concentration are calpains, Ca2+-dependent, non-lysosomal cysteine proteases (Goll et al., 2003). Calpains are essential for many physiological processes regulated by Ca2+ including cell spreading and migration (Dewitt and Hallett, 2020), cell cycle (Santella, 1998), membrane repair (Mellgren et al., 2007), embryonic development (Arthur et al., 2000), and platelet function (Azam et al., 2001). In contrast to other major proteolytic systems, calpains do not induce protein digestion but act in a regulatory way by performing limited proteolysis of their substrates. Its activity is tightly controlled by their specific endogenous inhibitor calpastatin and the energy-dependent regulation of Ca2+ homeostasis (Goll et al., 2003). However, under pathological conditions associated with the loss of intracellular Ca2+ control, as occurs during ischemia/reperfusion (IR), calpains are overactivated resulting in the proteolysis of a wide variety of proteins (Inserte et al., 2012). The use of transgenic models and calpain inhibitors demonstrate that calpain deregulation contributes to myocardial reperfusion injury through different mechanisms, which include increased sarcolemmal fragility and defects in Ca2+ handling, altered myofibrillar contractility, and mitochondrial dysfunction. Calpains are sensitive to cellular acidosis and they appear to activate only during reperfusion, once intracellular pH has been normalized (Hernando et al., 2010). This feature is critical because it provides a window of opportunity for preventing their activation. Yet, confirmation of the potential cardioprotective value of strategies designed to prevent calpain activation in clinically relevant experimental models is scarce, mainly due to the lack of appropriate compounds. Development of potent and selective calpain inhibitors is challenging, but ongoing research in the pharmaceutical industry and academia continuously generates potential candidates based on new structures or in the optimization of current compounds (Ono et al., 2016). As a result of these efforts, some of these compounds have entered clinical trials to test their safety and pharmacodynamics (Lon et al., 2019) (jRTC2021190013). Finally, recent data demonstrating the involvement of calpains in the development of adverse postinfarction myocardial remodelling has re-awakened interest for calpains as potential treatment target in patients with acute myocardial infarction.
2. Regulation of the calpain system
At present, 15 calpain isoforms have been identified in humans. Some of these isoforms have tissue-specific distribution whereas others, including the most abundant and best studied forms, calpain-1 and calpain-2, are ubiquitously expressed. Both calpains are heterodimers composed of a large (80kDa) catalytic subunit (Capn1 and Capn2, respectively), and a small (30kDa) common regulatory subunit (Capn4) needed to maintain stability and activity of the large subunit (Goll et al., 2003). These two isoforms differ in the concentration of Ca2+ required for their activation in vitro (Ono et al., 1998), which is substantially greater than the cytosolic concentrations achieved in normal beating cardiomyocytes. Additional mechanisms have been therefore proposed to contribute to lower the Ca2+ threshold of calpain activation reported in vivo. The most accepted one includes the translocation of calpain in its inactive form from the cytosol to the membrane where it binds to phospholipids reducing its Ca2+ requirements (Chakrabarti et al., 1996) or localizes in microdomains with high local Ca2+ concentrations (Kifor et al., 2003). However, although membranes are generally considered the preferential site of calpain activity, different studies suggest that under conditions of severe Ca2+ overload as those present during ischemia, translocation is not a critical step for calpain activation (Liu et al., 2001; Hernando et al., 2010). Calpastatin is the only specific endogenous inhibitor of calpain-1 and calpain-2. However, calpastatin has been shown to be largely downregulated during reperfusion due to the proteolytic activity of calpains, reducing its inhibitory function (Sorimachi et al., 1997; Hernando et al., 2010).
Several studies suggest that the oxidative/nitrosative stress generated during the first minutes of reperfusion may modify the activity of calpains. However, the mechanisms involved in the redox-regulation of calpain activity and their overall relevance in the context of acute reperfusion injury are still not fully understood (Randriamboavonjy et al., 2019). On the one hand, ROS can activate calpains indirectly via changes in Ca2+ levels. It has been described that ROS can elevate cytosolic Ca2+ by favouring calcium release from the sarcoplasmic reticulum and decreasing Ca2+ removal from the cell due to the oxidative damage of Ca2+ transporters (Itoh et al., 1999; Menshikova and Salama, 2000; Zima and Blatter, 2006). On the other hand, different studies suggest that calpain activity is inhibited by direct oxidation and S-nitrosylation of the protease (Michetti et al., 1995; Guttmann and Johnson, 1998). The resulting inactivation of calpains could partially explain the proposed cardioprotective effects of nitrosative signalling against reperfusion injury (Penna et al., 2014; Totzeck et al., 2017). Other post-translational modifications that have been proposed to modulate calpain activity include phosphorylation by protein kinase A (PKA) and extracellular signal-regulated kinase (ERK) (Shiraha et al., 2002; Glading et al., 2004), and carbonylation (Norberg et al., 2010). Although PKA and ERK have been implicated in the mechanisms of IR injury and cardioprotection (Hausenloy and Yellon, 2006), and IR has been shown to increase protein nitrosylation (Hamilton et al., 2004) and carbonylation (Khaliulin et al., 2006), whether or not these protein modifications are relevant modulatory mechanisms of calpain activation during IR compared to Ca2+ overload has not yet been determined.
Highlights: calpain activity is tightly regulated by variations in Ca2+ concentration and calpastatin. Although post-translational modifications of calpains have been reported, their pathophysiological relevance remains to be determined.
3. Calpain contribution to acute myocardial reperfusion injury
Experimental evidence obtained using several independent approaches, including the cleavage of known calpain substrates, the measurement of cellular calpain activity, and the use of calpain inhibitors and transgenic animals with altered calpain system in a variety of experimental models of IR, unambiguously demonstrate that calpains are important contributors to IR injury in the heart (Inserte et al., 2012; Neuhof, 2014) (see Table 1). This is not surprising, as IR is associated with a significant and sustained alteration of Ca2+ homeostasis in cardiomyocytes (Garcia-Dorado et al., 2012). Importantly, intracellular pH (pHi) determines the time-course of calpain activation secondary to transient ischemia. Our group has reported that ischemia-induced intracellular acidosis inhibits calpain activation despite Ca2+ overload, while the rapid pHi normalization during reperfusion allows its overactivation (Hernando et al., 2010). In agreement with that, experimental interventions applied in Langendorff-perfused hearts specifically addressed to delay pHi recovery (i.e. acidic perfusion) or those interfering with pHi normalization during the first minutes of reperfusion (i.e. ischemic postconditioning), attenuated calpain activation and reduced reperfusion injury (Inserte et al., 2009). The causative role of calpains on reperfusion-induced cell death has been established by experiments in which the administration of calpain inhibitors exclusively during reperfusion reduced infarct size in in vivo and in situ models of IR (Neuhof et al., 2008; Hernando et al., 2010). Several mechanisms, summarized in Figure 1, have been proposed to explain the contribution of calpains to reperfusion injury.
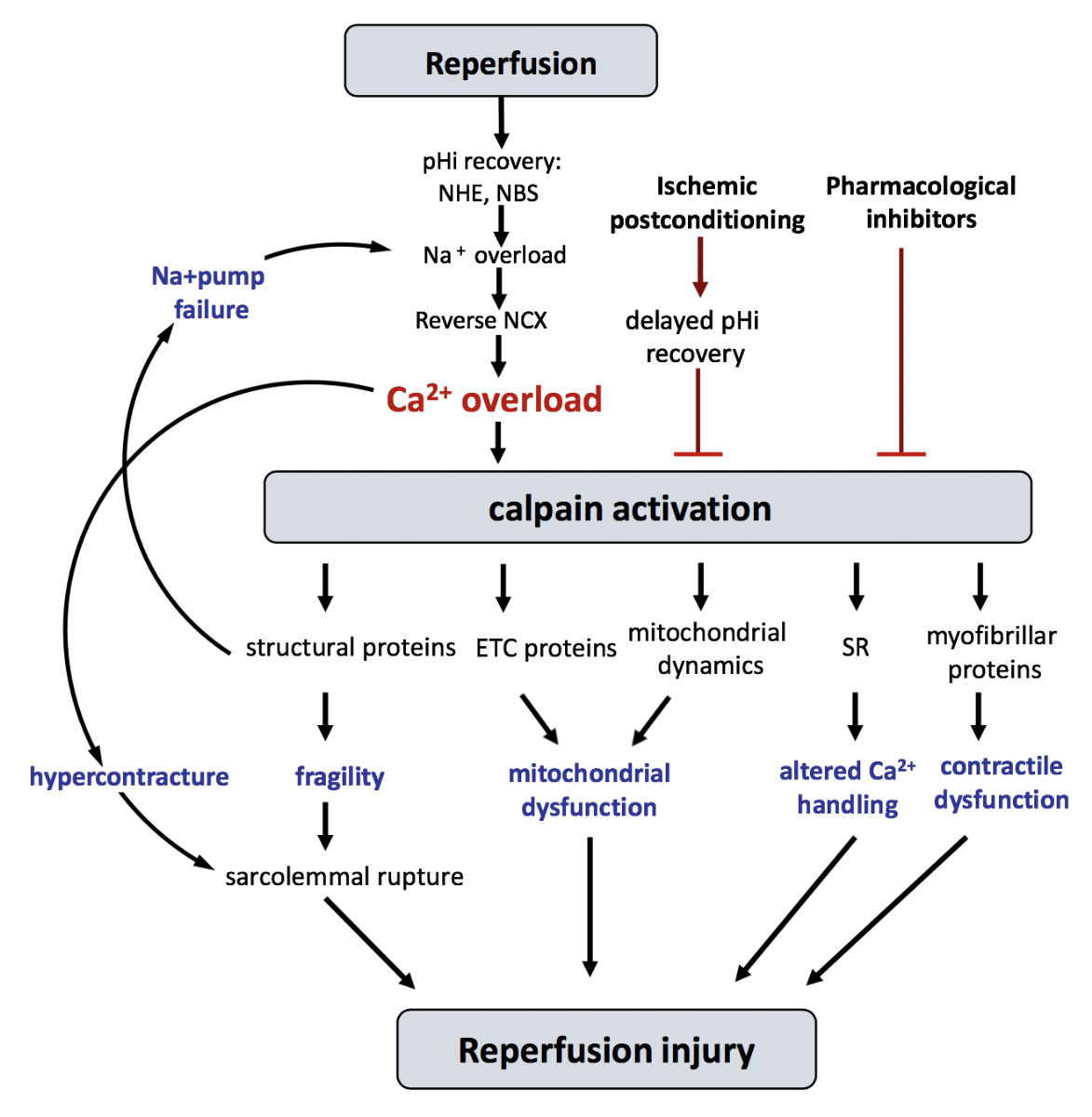
In a new window | Download PPT
Figure 1: Schematic diagram showing the main proposed mechanisms discussed in the text by which calpains participate in reperfusion injury. ETC, electron transport chain; NCX, Na+/Ca2+ exchanger; NBC, Na+/HCO3- cotransporter; NHE, Na+/H+ exchanger; pHi, intracellular pH; SR, sarcoplasmic reticulum.
3.1 Sarcolemmal fragility
Calpain activation during IR is implicated in the degradation of various sarcolemmal and cytoskeletal proteins (Neuhof, 2014). Calpain-dependent cleavage of α-fodrin, which form the backbone of the membrane cytoskeleton (Bennett and Gilligan, 1993), has been associated with increased membrane fragility (Armstrong et al., 2001). Upon conditions of mechanical stress, as those imposed by myocardial reperfusion, α-fodrin cleavage reduces the tolerance of the sarcolemma to the mechanical stress induced by cell contraction and swelling and increases the probability of sarcolemmal rupture and cardiomyocyte necrosis (Inserte et al., 2004). Other calpain substrates that may contribute to increase sarcolemmal fragility include dystrophin, talin, paxilin, vinculin, filamin, tau, α-tubulin, and vimentin (Davies et al., 1978; Nelson and Traub, 1983; Yoshida et al., 1992; Franco et al., 2004; Cortesio et al., 2011).
3.2 Impairment of Ca2+homeostasis
Calpain degradation of α-fodrin not only increases sarcolemmal fragility but in concert with the cleavage of ankyrin causes the detachment of several membrane receptors and channels including the α-subunit of Na+/K+ ATPase and the Na+/Ca2+ exchanger from their anchorage to the ankyrin-fodrin complex, which is essential for their proper membrane localization and correct function (Li et al., 1993; Mohler et al., 2003). As a consequence, the elevated cytosolic Na+ concentration present in ischemic cells cannot be normalized upon reperfusion, resulting in further aggravation of Ca2+ overload through reduced Ca2+ extrusion, following a vicious circle that aggravates reperfusion injury (Inserte et al., 2005; Müller et al., 2013a). In addition, it has been suggested that calpains may impair sarcoplasmic reticulum control of cytosolic Ca2+ by cleaving sarcoplasmic reticulum Ca2+ ATPase (SERCA2) and the ryanodine receptor (Singh et al., 2004b; French et al., 2006; Pedrozo et al., 2010).
3.3 Myofibrillar derangement
Calpains have been largely involved in post-ischemic myocardial dysfunction through the proteolysis of several proteins involved in cell contraction. The calpain dependent cleavage of titin, desmin, cardiac troponin T (cTnT), cardiac troponin I (cTnI) and α-actinin is described in different experimental models of IR (Kositprapa et al., 2000; Maekawa et al., 2003; Zhang et al., 2006; Blunt et al., 2007; Feng et al., 2008; Li et al., 2009) and has been reported in human myocardial samples (Barta et al., 2005). Overexpression of calpastatin prevents the degradation of cTnI and attenuates contractile dysfunction in rat hearts subjected to IR (Maekawa et al., 2003). The regulation of its degradation by calpain and the identification of the cleavage sites is being exhaustively investigated (Wijnker et al., 2015; Li et al., 2017; Martin-Garrido et al., 2018; Mahmud et al., 2019). Overexpression of heat shock protein-27 (HSP27) has been proposed to improve post-ischemic contractility by preventing the proteolytic cleavage of cTnI, cTnT, and desmin via blockade of calpain interaction with these proteins (Blunt et al., 2007; Lu et al., 2008). Cardiac myosin binding protein-C (cMyBP-C) is hydrolyzed by calpain during IR generating a N-terminal fragment (Sadayappan et al., 2008) that it is also observed in infarcted human hearts (Barefield et al., 2019). Recently, it has been reported that the cardiac-specific ablation of the calpain-target site of CMyBP-C reduces myocardial infarction in an in situ IR model (Barefield et al., 2019). Junctophilin-2, an essential protein for efficient excitation-contraction coupling in adult cardiomyocytes, is reduced after transient ischemia leading to cardiac contractile dysfunction and heart failure due to the calpain-dependent cleavage of its N-terminal domain (Murphy et al., 2012; Guo et al., 2015). More recently, it has been proposed that the resulting junctophilin-2 N-terminal fragment translocates to the nucleus where it acts as a stress-adaptive transcription regulator (Guo et al., 2018).
3.4 Mitochondrial dysfunction
Although traditionally considered cytoplasmic proteases, different studies have reported that calpains 1, 2, 4, and 10 are also present in the mitochondria (Arrington et al., 2006; Chen et al., 2011; Shintani-Ishida and Yoshida, 2015). Mitochondrial calpain-1 (mCPN1) is detected in the inter-membrane space (Ozaki et al., 2007) and matrix (Chen and Lesnefsky, 2015), and mitochondrial calpain-2 (mCPN2) in the matrix of mouse heart mitochondria (Shintani-Ishida and Yoshida, 2015). Studies performed in isolated liver mitochondria suggest that the activity of mCPN1 and mCPN2 is regulated through their association to the molecular chaperones endoplasmic reticulum resident protein 57 (ERp57) and glucose regulated protein 75 (Grp75), respectively (Ozaki et al., 2008, 2009). Calpain activation during IR were initially involved in the induction of the mitochondrial-dependent apoptotic programme by activating the pro-apoptotic factors BH3 interacting-domain death agonist (BID) (Chen et al., 2001; Luo et al., 2015) and apoptosis-inducing factor (AIF) (Chen et al., 2011). However, the relevance of apoptotic cardiomyocyte death in the context of acute reperfusion injury has been questioned due to the repression of the canonical caspase pathway in post-mitotic cardiomyocytes (Sanchis et al., 2008; Inserte et al., 2016). By contrast, several recent studies have proposed that mitochondrial calpains alter the electron transporter chain function by targeting the NADH dehydrogenase [ubiquinone] iron-sulfur protein 7 (NDUFS7) (Chen et al., 2019) and NADH-ubiquinone oxidoreductase chain 6 protein (ND6) (Shintani-Ishida and Yoshida, 2015) subunits of complex I. Pharmacological inhibition of calpains in rats subjected to ex vivo and in situ transient ischemia prevented mitochondrial calpain activation, complex I cleavage and inactivation, and reduced the occurrence of mitochondria permeability transition pore (mPTP) (Shintani-Ishida and Yoshida, 2015; Thompson et al., 2016; Chen et al., 2019). Mitochondrial calpains have also been involved in the disruption of the mitochondrial FoF1 ATP synthase through the proteolysis of its ATP5A1 subunit (Ni et al., 2016). Recently, by using transgenic mice with upregulation of calpain-1 restricted to cardiomyocyte mitochondria, the same group has proposed that calpain-mediated cleavage of ATP5A1 is causally associated with ROS generation, mPTP opening, and cell death (Cao et al., 2019).
Finally, mitochondrial calpains have been suggested to modulate mitochondrial dynamics. Recent findings suggest that impairment of mitochondrial dynamics may contribute to myocardial damage caused by IR (Ong et al., 2010; Forte et al., 2020). Cardiac-specific downregulation of OPA1 has been associated with mitochondrial fusion, mitophagy inhibition, and enhanced reperfusion injury (Zhang et al., 2019). In a recent study, calpastatin overexpression in mice subjected to in vivo transient ischemia prevented OPA1 degradation and mitochondrial fission, and improved mitochondrial fusion and mitophagy (Guan et al., 2019). Furthermore, calpain inhibition attenuated beclin-1 cleavage, a key component of the autophagy pathway required to form autophagosomes, and improved mitophagy in ex vivo hearts subjected to IR (Chen et al., 2019), suggesting that calpains negatively modulate mitophagy by acting at multiple levels.
3.5 Modulation of signaling pathways
Other known calpain substrates are protein kinases, protein phosphatases, and transcription factors (Neuhof, 2014). The calpain-dependent cleavage of these regulatory proteins may alter the function of several signaling pathways at different levels and may indirectly affect reperfusion injury. Proteolytic processing of protein kinase C (PKC) by calpain-1 generates a C-terminal fragment with unregulated kinase properties that contributes to reperfusion injury (Kang et al., 2010). Calpain activity also induces the activation of glycogen synthase kinase 3-beta (GSK-3β) (a well-known regulator of mPTP opening) through the cleavage of its inhibitory domain, while its inhibition upregulates the GSK-3β downstream signaling pathways insulin/phosphoinositide-3-kinase (PI3K) and WNT/β-catenin (Potz et al., 2017). Different groups have demonstrated that calpain activation during reperfusion induces the cleavage of the nuclear factor kappa B (NFκB) inhibitor IκBα, inducing the translocation of NFκB to the nucleus, where it promotes the expression of proteins involved in the development of adverse ventricular remodeling (Hamid et al., 2011; Ma et al., 2012; Poncelas et al., 2017). In addition, it has been proposed that calpains elicit calcium-triggered cell injury by activating calcineurin in a calmodulin-independent form through the cleavage of its 60 kDa subunit (Lakshmikuttyamma et al., 2003) and the proteolysis of the calcineurin inhibitor cain/cabin1 (Kim et al., 2002). Finally, in vitro experiments show that purified m-calpain promotes partial degradation of G protein-coupled receptor kinase 2 (GRK2) and inhibits its activity (Lombardi et al., 2002). GRK2 is a central regulator of β-adrenergic receptors and many other G protein-coupled receptors (GPCR) involved in cardiovascular pathophysiology, but also a key regulatory node in non-GPCR pathways (Mayor et al., 2018). Recently we have reported that GRK2 levels are transiently reduced at the onset of reperfusion, at least in part due to the action of calpains, which reduced endogenous cardioprotection through impaired overall protein kinase B (Akt) functionality (Penela et al., 2019).
Highlights: A broad range of experimental studies demonstrates that calpain activation contributes to IR injury by different mechanisms including increased sarcolemmal fragility, impairment of Ca2+ regulation, myofibrillar derangement, mitochondrial dysfunction, and modulation of signaling pathways.
4. Pharmacological inhibition of calpains
Despite the compelling experimental evidence supporting the contribution of calpains to myocardial reperfusion injury, no clinical trials and only a few pre-clinical studies in large animal models have explored the use of pharmacological calpain inhibitors as a therapeutic strategy to attenuate myocardial infarction (see Table 1). This is in part due to the limitations of most of the available calpain inhibitors, which include poor selectivity (Ali et al., 2012), metabolic instability, limited membrane permeability, and poor water-solubility. The association of other non-cardiac pathological conditions, including myopathies, neurodegenerative disorders, ophthalmic diseases, or cancer with disturbances in the calpain system and the demonstration that transgenic models with deficient calpain expression or overexpression of calpastatin attenuate the symptoms of these pathologies (Zatz and Starling, 2005), make inhibition of calpains an attractive target for the industry, promoting active research for development of novel calpain inhibitors (examined in detail in previous reviews (Ono et al., 2016; Dókus et al., 2020)).
Table 1. Selected studies of calpain inhibition in ischemia/reperfusion models.
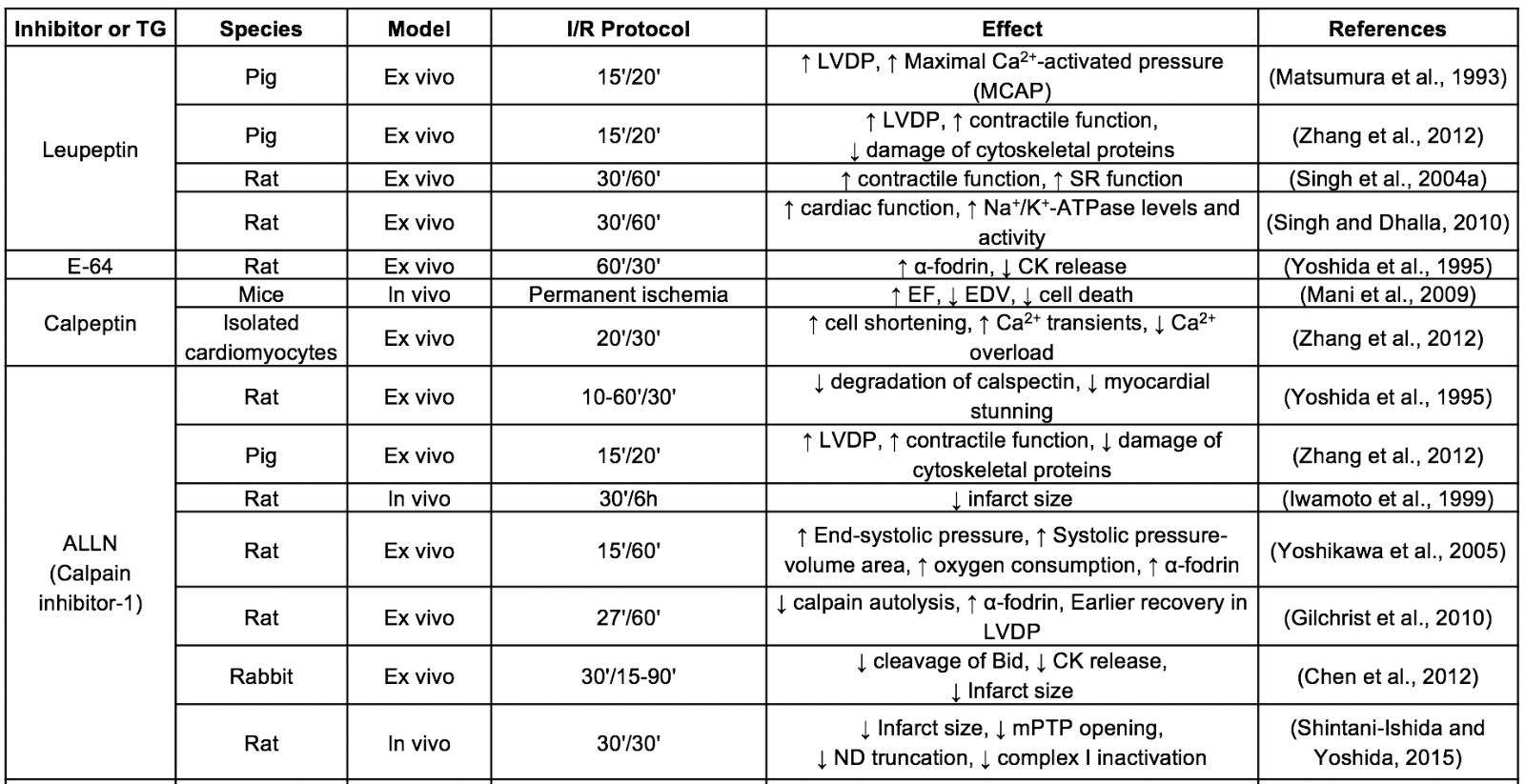
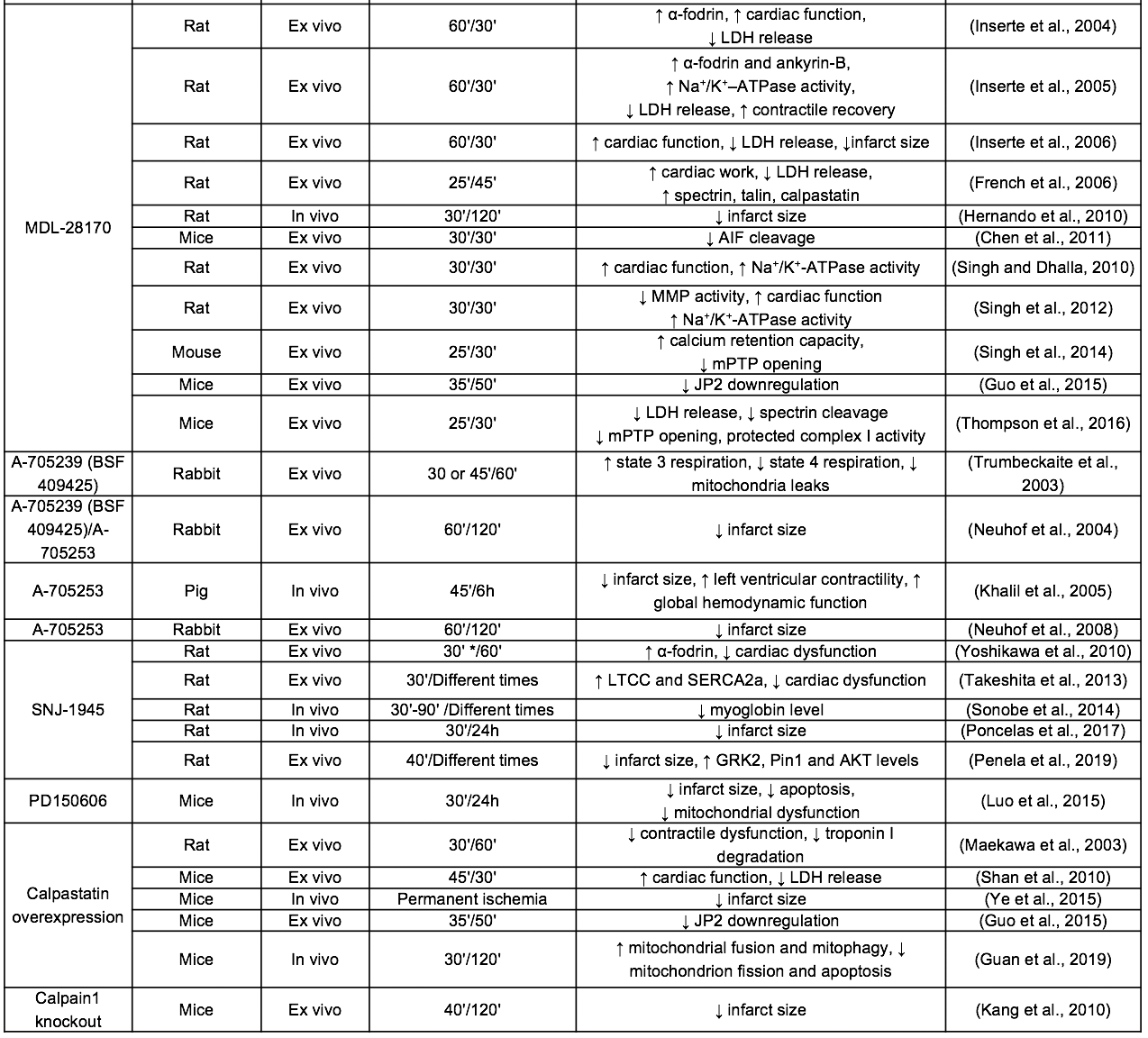
CK: creatine kinase; EDV: end-diastolic ventricular volume; EF: ejection fraction; LDH: lactate dehydrogenase; LTCC: L-type calcium channel; LVDP:left ventricular developed pressure; MMP: metalloproteinase; ND: NADH-ubiquinone oxidoreductase chain 6 protein; SR: sarcoplasmic reticulum; TG: transgenic model
Leupeptin and E-64, both naturally isolated from microorganisms, constitute the first-generation of calpain inhibitors. These peptide aldehydes have an electrophilic center for a covalent interaction with the catalytic cysteine residue of calpain. Leupeptin was the first inhibitor tested in the context of IR injury, improving functional recovery in isolated guinea pig hearts subjected to IR (Matsumura et al., 1993). However, although these inhibitors have been extensively used for many years in in vitro and in vivo studies, they exhibit little specificity for calpains and low membrane permeability (Mehdi, 1991). The physicochemical properties of leupeptin were improved by substituting the terminal amino acid for a hydrophobic cap group (Donkor, 2011). The resulting synthetic leupeptin derivatives calpeptin, ALLN (calpain inhibitor I) and MDL-28170 (calpain inhibitor III) have demonstrated an improved efficacy to protect against reperfusion injury in isolated hearts and in vivo models of transient coronary occlusion (Inserte et al., 2004; Mani et al., 2009; Gilchrist et al., 2010; Hernando et al., 2010). Nevertheless, the high chemical reactivity of aldehydes and poor aqueous solubility limits the stability and bioavailability of these compounds, therefore precluding their progression into the clinic. Efforts to solve these problems have led to the development of peptidomimetic inhibitors derived from the MDL-28170 structure with improved water solubility, cell permeability, and metabolic stability over previous inhibitors (Lubisch et al., 2003). Among them, the benzoylalanine-derived ketoamides A-705239 and A-790253 developed by AbbVie reduced infarct size and mitochondrial dysfunction in isolated rabbit hearts (Neuhof et al., 2003, 2004) and in an in vivo pig model of IR (Khalil et al., 2005). An improved derivative of A-705239 namely Alicapistat (ABT-957), with high calpain selectivity versus cathepsins (Jantos et al., 2019), has been recently tested in a phase I clinical study analysing its safety and pharmacological properties for the treatment of Alzheimer’s disease (Lon et al., 2019). However, in this study, Alicapistat failed to induce a measurable pharmacodynamic effect, which can be a consequence of selecting inadequate concentrations, suggesting that this inhibitor has indeed a moderate potency. Another ketoamide derived inhibitor with an encouraging pharmacokinetic profile is SNJ-1945, produced by Senju Pharmaceutical (Shirasaki et al., 2006; Yoshikawa et al., 2010). Intraperitoneal administration of SNJ-1945 attenuated ventricular dysfunction induced by IR (Takeshita et al., 2013) and reduced infarct size when given immediately before reperfusion in in vivo rat and mouse models of transient coronary occlusion (Poncelas et al., 2017; Penela et al., 2019). Currently, a phase II clinical trial designed to test the efficacy and safety of oral administration of SNJ-1945 in central retinal artery occlusion is in progress (jRTC2021190013). The limited specificity for calpains over other cysteine proteases present in many calpain inhibitors has been attributed to the highly conserved active site among cysteine proteases. Therefore, a novel approach to develop new inhibitors with increased specificity for calpains is based on the use of compounds that induce allosteric inhibition of the enzyme by binding to other position than the catalytic site. One of these allosteric inhibitors, PD150606, which is supposed to bind to the Ca2+ binding site of calpain, has been demonstrated to be effective in attenuating myocardial infarction in mice subjected to in vivo IR (Luo et al., 2015). Importantly, a derivative of PD150606 (PD151746) has been shown to be more selective for calpain-1 than for calpain-2. Because the two isoforms may display some differences in their substrate specificity (Shinkai-Ouchi et al., 2016) and biological function (Santos et al., 2012; Wang et al., 2016), this type of compounds opens the door to the development of new isoform-selective inhibitors. In addition to calpain isoforms, differences also seem to exist between cytosolic and mitochondrial calpains. It has been proposed that mitochondrial but not cytosolic calpains bind to chaperons (ERp57 for calpain-1 and Grp75 for calpain-2) (Ozaki et al., 2011). Based on these observations, Ozaki et al. (2012) developed a novel peptide that blocks the binding site between calpain-1 and ERp57 and inhibits the mitochondrial activity of calpain-1 in a specific manner. Considering the central role of mitochondria as a trigger of cell death during IR injury (Davidson et al., 2020), and that mitochondrial calpain-1 activity is increased in mice subjected to IR (Chen et al., 2011), the potential cardioprotective effects of these types of inhibitors deserve further investigation.
Highlights: Translation of the use of calpain inhibitors into clinical trials has been hampered due to the lack of inhibitors with appropriate pharmacological properties for use in patients. The development of new calpain inhibitors is an increasing research trend.
5. Perspectives
Calcium overload occurring during IR invariably results in aberrant activation of calpains and the concomitant cleavage of a wide variety of proteins. Calpain activation contributes to the aggravation of reperfusion injury by reducing the likelihood of a cell to survive. Several experimental studies have demonstrated in different models of IR that calpains are an attractive therapeutic target and that pharmacological inhibitors applied at the onset of reperfusion are capable of preventing cell death and limiting myocardial infarction. However, translation of the use of calpain inhibitors into clinical context has been frustrating, in part due to the lack of specific compounds with an appropriate pharmacologic profile to make them candidates for use in patients, but also due to the negative results obtained when using some promising cardioprotective interventions, like ischemic postconditioning (Engstrøm et al., 2017), which has been shown to prevent calpain activity in experimental studies (Inserte et al., 2009), in patients with acute coronary syndrome.
Fortunately, due to the involvement of calpain in other non-cardiac pathologies, there is a high interest for the development of novel and specific calpain inhibitors, and plenty of molecules are being described in the literature and registered in patent offices. The safety and pharmacokinetics of some of the drugs that have demonstrated efficacy in animal models of IR are currently being tested for safety in clinical trials (jRTC2021190013; (Lon et al., 2019)). The 3D structure of the calpastatin-calpain complex has already been solved (Hanna et al., 2008; Moldoveanu et al., 2008) and it is being used to find new chemical structures that can be candidates for the design of new calpain inhibitors or for the optimization of the current compounds. One of the historical limitations of many calpain inhibitors is their poor selectivity over the cysteine proteases cathepsins (Mehdi, 1991). However, given the evidence for the involvement of the altered activity of both cathepsins and calpains in different pathophysiological conditions including myocardial infarction (Müller et al., 2013b), it has been questioned whether the design of pure specific inhibitors is essential for their clinical efficacy (Siklos et al., 2015).
In addition to their role in reperfusion injury, different studies using transgenic models have demonstrated that calpains participate in the progression of adverse post-infarction remodeling (Ma et al., 2012; Kudo-Sakamoto et al., 2014; Ye et al., 2015). In two recent studies, the chronic administration of SNJ1945 and MDL-28170 during reperfusion was effective in attenuating ventricular remodeling and cardiac dysfunction in a mouse model of transient coronary occlusion with no signs of toxic effects (Poncelas et al., 2017; Wang et al., 2018). Overall, these promising experimental results demonstrate that calpain inhibition is feasible and safe and may be an effective therapeutic intervention to attenuate both reperfusion injury and the development of adverse postinfarction remodeling and heart failure in patients with acute myocardial infarction.
Highlights: Recent studies suggest that calpains participate in the progression of post-infarction remodeling. However, translational research involving calpains is still at the development stage.
Conflicts of interest:
The authors declare that they have no conflicts of interest.
Funding
This study was supported by Instituto de Salud Carlos III, Spain (PI-16/00232); CIBERCV-Instituto de Salud Carlos III, Spain (CB16/11/00479), co-funded with European Regional Development Fund-FEDER contribution.
References
David Aluja1
1Cardiovascular Diseases Research Group, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Vall d’Hebron Barcelona Hospital Campus, Passeig Vall d'Hebron 119-129, 08035 Barcelona, Spain.
Andrea Rodriguez-Lopez1
1Cardiovascular Diseases Research Group, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Vall d’Hebron Barcelona Hospital Campus, Passeig Vall d'Hebron 119-129, 08035 Barcelona, Spain.
Marisol Ruiz-Meana1,2,3
1Cardiovascular Diseases Research Group, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Vall d’Hebron Barcelona Hospital Campus, Passeig Vall d'Hebron 119-129, 08035 Barcelona, Spain. 2Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain. 3CIBER de Enfermedades Cardiovasculares (CIBERCV), Instituto de Salud Carlos III, Avenida de Monforte de Lemos 3-5, 28029 Madrid, Spain.
Javier Inserte1,2,3
1Cardiovascular Diseases Research Group, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Vall d’Hebron Barcelona Hospital Campus, Passeig Vall d'Hebron 119-129, 08035 Barcelona, Spain. 2Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain. 3CIBER de Enfermedades Cardiovasculares (CIBERCV), Instituto de Salud Carlos III, Avenida de Monforte de Lemos 3-5, 28029 Madrid, Spain.
Corresponding author:
Javier Inserte
Email: javier.inserte@vhir.org
In a new window | Download PPT
Figure 1: Schematic diagram showing the main proposed mechanisms discussed in the text by which calpains participate in reperfusion injury. ETC, electron transport chain; NCX, Na+/Ca2+ exchanger; NBC, Na+/HCO3- cotransporter; NHE, Na+/H+ exchanger; pHi, intracellular pH; SR, sarcoplasmic reticulum.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13766 | 51 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA