Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Pharmacological modulators of mitochondrial dynamics as novel therapeutics for cardiovascular and neurological diseases
Time:2020-07-03
Number:11729
Author Affiliations

Conditioning Medicine 2020. 3(3):144-159.
Abstract
Mitochondrial dysfunction lies at the heart of a diverse variety of cardiovascular and neurological diseases, many of which are major causes of patient morbidity and mortality. As such, therapeutic strategies that are capable of preventing mitochondrial dysfunction may provide new treatments to improve clinical outcomes in patients suffering from these conditions. In this regard, pharmacological targeting of mitochondrial fusion and fission proteins, which regulate mitochondrial morphology and influence mitochondrial function, have the therapeutic potential to prevent mitochondrial dysfunction in these debilitating conditions. In this article, we provide an overview of common and new pharmacological agents, which have been shown to target mitochondrial fusion and fission proteins, and focus on their use as novel therapeutics for potentially treating and ameliorating clinical outcomes in a variety of cardiovascular conditions (including acute myocardial infarction, various cardiomyopathies, and pulmonary arterial hypertension, and neurological conditions (including stroke, acute cerebral injury, Parkinson’s Disease, Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis).
Abstract
Mitochondrial dysfunction lies at the heart of a diverse variety of cardiovascular and neurological diseases, many of which are major causes of patient morbidity and mortality. As such, therapeutic strategies that are capable of preventing mitochondrial dysfunction may provide new treatments to improve clinical outcomes in patients suffering from these conditions. In this regard, pharmacological targeting of mitochondrial fusion and fission proteins, which regulate mitochondrial morphology and influence mitochondrial function, have the therapeutic potential to prevent mitochondrial dysfunction in these debilitating conditions. In this article, we provide an overview of common and new pharmacological agents, which have been shown to target mitochondrial fusion and fission proteins, and focus on their use as novel therapeutics for potentially treating and ameliorating clinical outcomes in a variety of cardiovascular conditions (including acute myocardial infarction, various cardiomyopathies, and pulmonary arterial hypertension, and neurological conditions (including stroke, acute cerebral injury, Parkinson’s Disease, Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis).
1. Introduction
Cardiovascular and neurological diseases are the leading causes of death and disability worldwide. Of these, acute myocardial infarction (AMI), heart failure (HF), and neurodegenerative diseases are key contributors of morbidity and mortality. Therefore, novel therapies capable of protecting the heart and brain are needed to treat such medical conditions, and improve clinical outcomes in patients with cardiovascular and neurological diseases. The central pathophysiology underlying these diverse medical conditions is mitochondrial dysfunction, which has been established to be a critical determinant of cell death in these settings. It has been shown that pharmacological modulation of mitochondrial morphology by targeting mitochondrial fusion and fission proteins, may influence mitochondrial function, and provide a therapeutic strategy for preventing mitochondrial dysfunction in these debilitating conditions. In this review article, we provide an overview of pharmacological agents, which have been shown to modulate mitochondrial morphology by targeting mitochondrial dynamics proteins, and focus on their novel therapeutic potential for treating and improving clinical outcomes in a variety of cardiovascular conditions (including AMI, various cardiomyopathies, and pulmonary arterial hypertension [PAH]) and neurological conditions (including stroke, acute cerebral injury, Parkinson’s Disease [PD], Alzheimer’s disease [AD], Huntington’s disease [HD], and amyotrophic lateral sclerosis [ALS]).
2. Mitochondrial morphology in the heart and brain
Mitochondria are dynamic organelles that continually change their shape by undergoing the processes of fission and fusion to generate fragmented or elongated mitochondria, respectively (Ong et al., 2015; Ong et al., 2010a; Ong et al., 2013; Hernandez-Resendiz et al., 2020). Mitochondrial fission is required for cell division, apoptosis, and selective removal of damaged mitochondria by the process of mitophagy, which is regulated by the cytosolic fission protein, dynamin-related protein 1 (Drp1) (Frank et al., 2001) that binds to its outer mitochondrial membrane (OMM) receptor, human fission protein 1 (hFis1) (James et al., 2003), mitochondrial fission factor (MFF) (Gandre-Babbe et al., 2008), and the mitochondrial dynamics proteins of 49 kDa (MiD49) and 51 kDa (MiD51) (Palmer et al., 2011; Samangouei et al., 2018) to execute scission of a single mitochondrion into two mitochondria. In contrast, mitochondrial fusion allows the replacement of damaged DNA, and helps to maintain normal mitochondrial respiratory function, and is controlled by the fusion proteins, mitofusin 1 (Mfn1) (Santel et al., 2003) and mitofusin 2 (Mfn2) (Santel et al., 2001), which mediate fusion of the OMM between two adjacent mitochondria, and optic atrophy protein 1 (OPA1) (Cipolat et al., 2004; Burke et al., 2015) that carries out inner mitochondrial membrane (IMM) fusion.
The relevance of mitochondrial dynamics to the adult heart has been questioned given the fragmented morphology present in cardiac mitochondria, and the fact that they are tightly packed into three discrete intracellular locations within the cardiomyocyte – between myofibrils, beneath the sarcolemmal membrane, and around the nucleus (Ong et al., 2015; Ong et al., 2010a; Ong et al., 2013; Hernandez-Resendiz et al., 2020). In this respect, genetic ablation of the mitochondrial fission proteins Drp1 (Ikeda et al., 2015) and MFF (Chen et al., 2015), or the mitochondrial fusion proteins Mfn2 (Papanicolaou et al., 2011) and OPA1 (Chen et al., 2012), have been shown to modify the mitochondrial shape and impair mitochondrial respiratory function. The disruption of balance between mitochondrial fission and fusion in the heart have been proven to contribute to the pathophysiology of a number of cardiovascular diseases including AMI, HF, and PAH, highlighting the importance of mitochondrial dynamics proteins in maintaining normal cardiac function, thereby showing their importance as therapeutic targets to treat and improve outcomes in these medical conditions (Ong et al., 2015; Ong et al., 2010a; Ong et al., 2013; Hernandez-Resendiz et al., 2020).
In the brain, mitochondrial dynamics (both movement and changes in morphology) play a key role in the functioning of neurons and brain development, supporting energy production, calcium buffering at synapses, mitochondrial trafficking to synapses, and mitochondrial quality control (Flippo et al., 2017). Genetic ablation of either Drp1 or Mfn2 in cerebellar neurons has been shown to result in mitochondrial dysfunction and neuronal degeneration, leading to an age-related loss of motor coordination (Chen et al., 2007; Kageyama et al., 2012), emphasizing the importance of mitochondrial dynamics for normal neuronal function. In these cells, mitochondrial dynamics allow the trafficking of mitochondria from the cell body where biogenesis occurs to synaptic sites and distal parts of the axons via anterograde transport (Sheng et al., 2012). Damaged mitochondria need to be retrogradely trafficked back to the cell body to be removed by mitophagy (Sheng et al., 2012). A disturbance in this delicate process with an imbalance of mitochondrial fission and fusion can, therefore, compromise normal neuronal function. Excessive mitochondrial fission has been shown to contribute to the pathophysiology of several neurological diseases including stroke, acute cerebral injury, AD, PD, HD, and ALS. Hence pharmacological agents, which can target and inhibit the mitochondrial fission machinery have the therapeutic potential to improve clinical outcomes in patients with these debilitating conditions.
3. Pharmacological modulators of mitochondrial fission
A number of pharmacological modulators of the mitochondrial fission machinery have been described, some of which target the mitochondrial fission proteins directly, or inhibit the protein-to-protein interaction between different fission proteins (Table 1A, Figure 1). These pharmacological agents include newly designed specific peptides, small molecules, and drugs already in clinical use, and have been either newly synthesized or identified from screening chemical libraries for their effects on inhibiting mitochondrial fission.
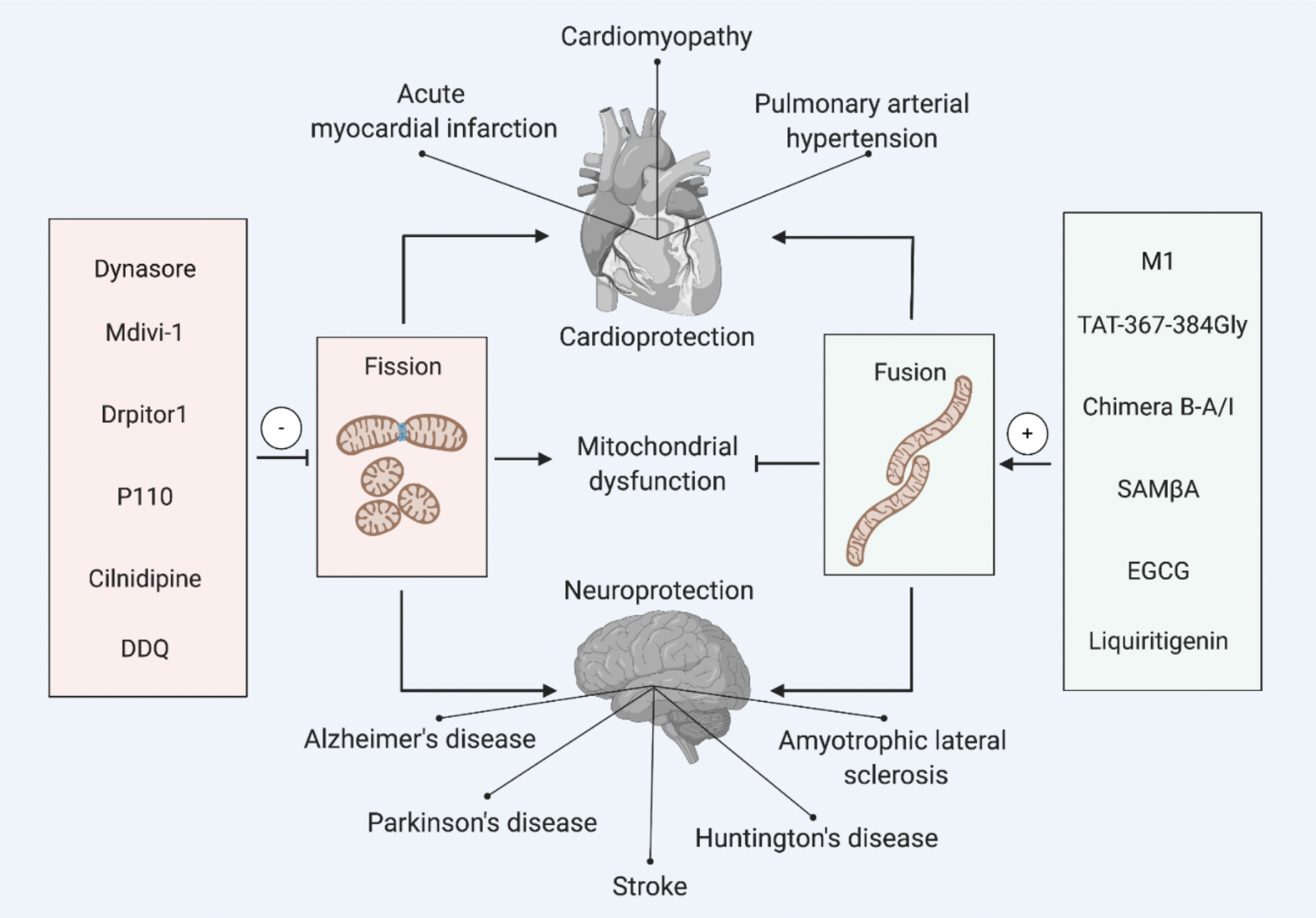
In a new window | Download PPT
Figure 1: Pharmacological modulators of mitochondrial dynamics for cardioprotection and neuroprotection. This scheme depicts the main pharmacological agents for inhibiting mitochondrial fission or activating mitochondrial fusion to prevent mitochondrial dysfunction, and confer cardioprotection (in acute myocardial infarction, cardiomyopathy, pulmonary arterial hypertension), and neuroprotection (in stroke, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis).
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone, DDQ; Selective antagonist of mitofusin 1-β2PKC association, SAMßA; Epigallocatechin gallate, EGCG.
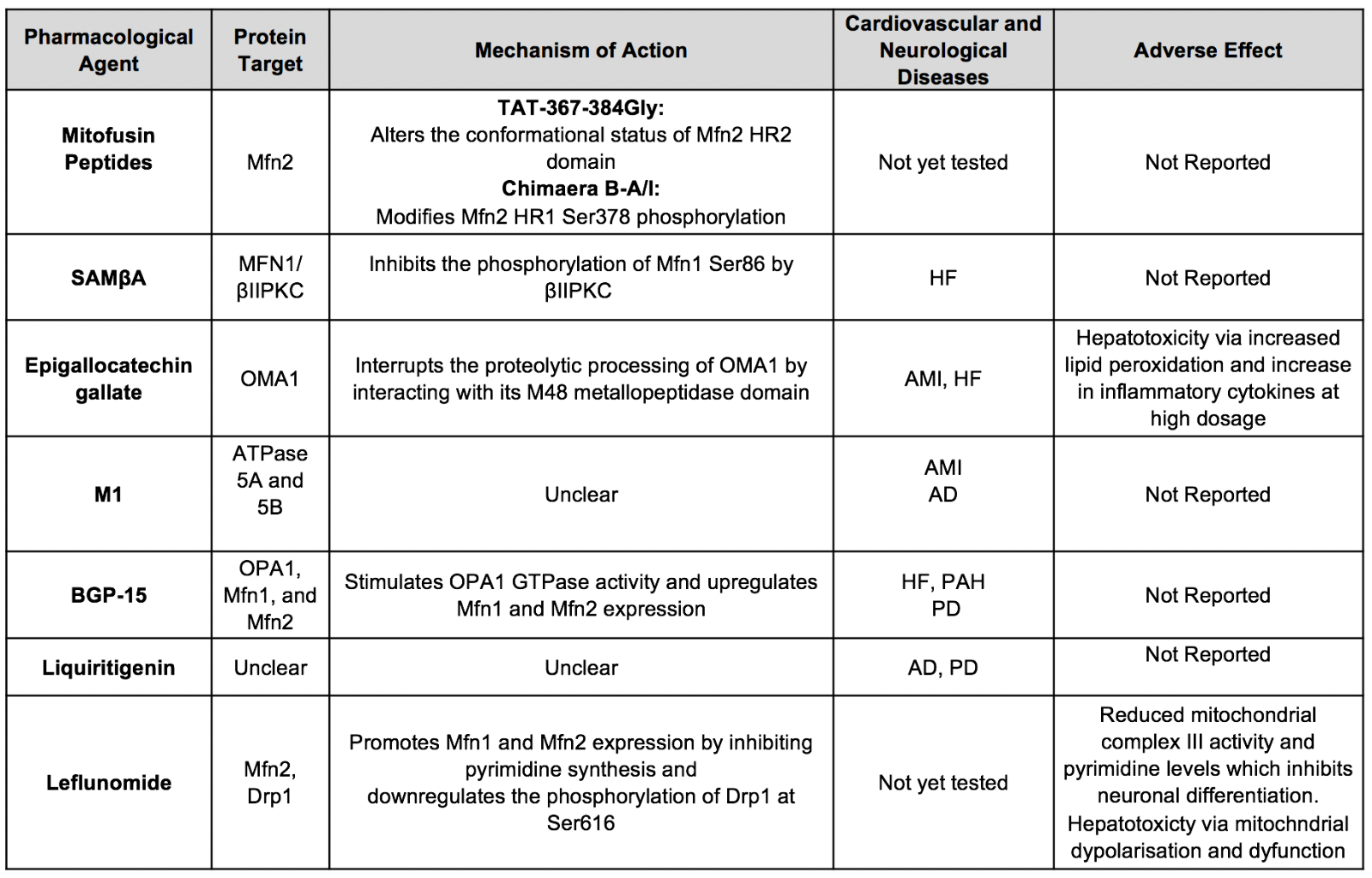
Table 1A. Summary of major pharmacological agents that prevent mitochondrial fission for cardioprotection and neuroprotection.
Dynasore and Dyngo4a – non-specific small molecule inhibitors of Drp1
In 2006, Macia et al. (2006) first discovered dynasore (identified from a small molecule screen) to be a novel inhibitor of dynamin GTPase activity (IC50 ~ 15 μM in vitro) of dynamin-1 (Dnm1), dynamin-2 and Drp1. It was shown to act by binding to the GTPase domain in a non-competitive manner, and to interfere with endocytic pathways and membrane trafficking, although the effects on Drp1-mediated mitochondrial fission were not investigated in this study. McCluskey et al. (2013) have described new analogues of dynasore, called Dyngo™ compounds (of which Dyngo4a was the most potent), which are more potent in terms of GTPase activity inhibition and that have reduced cytotoxicity compared to dynasore. However, both dynasore and Dyngo4a have been shown to have off-target effects as evidenced by their inhibition of endocytic pathways in mouse embryonic fibroblasts (MEFs) lacking dynamins 1, 2 and 3 (Park et al., 2013) and interference with cell signalling pathways such as vascular endothelial growth factor (VEGF) and extracellular signal-regulated kinase 1/2 (Erk1/2) (Basagiannis et al., 2017). As a non-specific inhibitor of Drp1 GTPase activity, dynasore has already been investigated as a pharmacological strategy to inhibit mitochondrial fission in AMI (Gao et al., 2013) and spinal cord injury (see later sections) (Li et al., 2017a).
Mitochondrial division inhibitor (mdivi-1) – a small molecule inhibitor of Drp1
In 2008, Cassidy-Stone et al. (2008) identified a selective inhibitor of Drp1-mediated mitochondrial fission from a small molecule screen on mitochondrial morphology in yeast cells, which they named mitochondrial division inhibitor (mdivi-1). It was shown in yeast cells that mdivi-1 (a quinazolinone derivative) selectively inhibited Dnm1 GTPase activity by blocking the self-assembly of Dnm1 (IC50 of approximately 1-10 µM), and induced mitochondrial elongation. However, this morphology change was not observed in yeast cells deficient in Dnm1, and did not affect either the actin cytoskeleton or the endoplasmic reticulum (ER), suggesting that the effect of mdivi-1 was dependent on the fission protein. In mammalian (COS-7) cells, mdivi-1 was shown to induce mitochondrial elongation by inhibiting self-assembly and translocation of Drp1 to the mitochondria, and also to inhibit apoptosis by blocking Bax/Bak-dependent mitochondrial outer membrane permeabilization (MOMP), but it failed to inhibit GTPase activity of human recombinant Drp1 (Cassidy-Stone et al., 2008). Since its discovery, a large number of experimental studies have used mdivi-1 to inhibit mitochondrial fission as a potential therapy for cardiovascular and neurological diseases (see later sections).
Recent studies have questioned the selectivity of mdivi-1 for Drp1, and have described several off-target effects of this molecule. Bordt et al. (2017) have reported that mdivi-1 weakly and reversibly inhibited mitochondrial respiratory complex I and attenuated reverse electron transfer-mediated mitochondrial reactive oxygen species (ROS) production in primary cortical neurons and COS-7 cells at concentrations (50 µM) used to inhibit mitochondrial fission. Interestingly, this unexpected effect of mdivi-1 was not recapitulated by genetic deletion of Drp1 and was observed in Drp1-deficient MEFs. Furthermore, mdivi-1 did not induce mitochondrial lengthening and was a weak inhibitor of Drp1-GTPase activity (Ki >1.2mM). Therefore, the authors concluded that the effect of mdivi-1 to reversibly inhibit complex I and modulate mitochondrial ROS production was independent of Drp1, and may explain, in part, the beneficial cardioprotective and neuroprotective effects observed in experimental models (Bordt et al., 2017). Similar findings have been observed in isolated cardiac mitochondria and adult cardiomyocytes from mice treated with mdivi-1, namely inhibition of mitochondrial respiration, prevention of transient mitochondrial permeability transition pore (MPTP) opening, and reduction in mitochondrial ROS production without having a marked effect on mitochondrial morphology (Zhang et al., 2017). However, in this study, genetic ablation of Drp1 had similar mitochondrial effects, and it is consequently not clear from this study whether the observed effect of mdivi-1 on mitochondrial respiratory function was dependent on Drp1 or not (Zhang et al., 2017). So et al. (2012) have shown that mdivi-1 (10-30 µM) treatment of HL-1 cardiomyocytes had off-target effects on ion channels and action potential (AP), with lengthening of AP duration and increased firing of APs, effects that were attributed to inhibition of rapidly activating K+ currents in heart cells.
In contrast to these studies, Manczak et al. (2019) have demonstrated in mouse neuroblastoma (N2a) cells that mdivi-1 inhibited Drp1 GTPase activity (25 µM or 75 µM), reduced Drp1 and Fis1 mRNA expression, and promoted mitochondrial elongation. In addition, Wu et al. (2020) have found that mdivi-1 (25µM) inhibited Drp1 GTPase activity, promoted mitochondrial elongation without affecting mitochondrial complex I activity in the human non-small cell lung cancer A549 cell line. The controversial results about the effect of mdivi-1 on mitochondrial function may be explained by cell-specificity, the concentration used, and duration of treatment tested. Either way more specific and potent inhibitors of Drp1 are needed to translate inhibition of mitochondrial fission as a cardioprotective and neuroprotective strategy. In this regard, Numadate et al. (2014) have chemically modified the structure of mdivi-1 to generate new inhibitors which are more potent and specific for Drp1, when compared to mdivi-1, but the functional effects of these new Drp1 inhibitors remain to be tested in disease models.
Drpitor1 – a small selective molecule inhibitor of Drp1
To overcome the off-target effects of mdivi-1, Wu et al. (2020) have identified from a virtual in silico screen of 4000 proprietary chemical compounds against the crystal structure of Drp1, identified two new Drp1 GTPase inhibitors, Drpitor1 and Drpitor1a, which are both orally bioavailable and non-toxic. The new Drp1 inhibitors were shown to be more potent than mdivi-1 at inhibiting Drp1 GTPase activity and suppressing mitochondrial fission in A549 cells. These agents did not inhibit dynamin-1 GTPase activity or inhibit mitochondrial complex I activity, suggesting specific Drp1 inhibition. These new Drp1 inhibitors were demonstrated to reduce right ventricular ischemia/reperfusion injury (IRI) in an ex-vivo perfused rat heart and to reduce mitochondrial ROS production, proposing a cardioprotective effect with these agents (Wu et al., 2020). Further studies are needed to assess whether they confer cardioprotective effects in vivo and whether they are also neuroprotective.
P110 – a peptide inhibitor of Drp1-hFis1 interaction
Given that Drp1 does not have a mitochondria-localizing sequence it needs to bind to an OMM receptor to mediate mitochondrial fission. One of these OMM Drp1 receptors is the mitochondrial fission protein, hFis1, and the protein interaction between Drp1 and hFis1 represents a potential target for inhibiting mitochondrial fission. In this regard, Qi et al. (2013) designed a novel 7 amino acid peptide conjugated to the cell-permeant TAT peptide, called P110, which specifically inhibited the interaction between Drp1 and hFis1, without affecting the binding of Drp1 with the other mitochondrial fission proteins, MFF and MiD51. In cultured human neuroblastoma SH-SY5Y cells, P110 was demonstrated to inhibit Drp1 GTPase activity, prevent mitochondrial translocation of Drp1, and antagonize the Drp1/hFis1 interaction in vitro (Qi et al., 2013). Crucially, P110 was demonstrated to have no effect on mitochondrial morphology and function under baseline conditions (Qi et al., 2013) and had no adverse effects when delivered in vivo for 5 months, suggesting that P110 was able to inhibit pathological mitochondrial fission (Joshi et al., 2018b). P110 has been shown in subsequent experimental studies to be cardioprotective in AMI (Disatnik et al., 2013) and neuroprotective in several different neurodegenerative conditions including PD, HD, and ALS, highlighting the Drp1/hFis1 interaction to be a critical therapeutic target (see later sections).
Interestingly, the same team has also designed a novel peptide, called P259, that specifically inhibits the interaction between Drp1 and MFF, and this was shown to induce mitochondrial elongation under baseline conditions (Kornfeld et al., 2018). However, in contrast to P110, this new peptide produced detrimental effects in both neuroblastoma SH-SY5Y cells and primary cortical neurons, with mitochondrial dysfunction and worsening of activity in wild-type mice and HD symptoms in a mouse model, suggesting that the Drp1-MFF interaction is required to maintain mitochondrial quality control, normal mitochondrial motility and function under baseline conditions. Other potential therapeutic strategies for inhibiting the mitochondrial fission machinery could target protein-to-protein interactions between Drp1 and MiD49/51 given that the binding sites between these proteins have been described (Kalia et al., 2018; Samangouei et al., 2018; Ma et al., 2019).
Cilnidipine - repurposing drugs to inhibit mitochondrial fission
It is possible that drugs already in clinical use can be repurposed and tested for their ability to inhibit mitochondrial fission, thereby providing a clinically available therapeutic option for treating medical conditions in which excessive mitochondrial fission is a major feature. Mitochondrial fission is known to be initiated by pre-constriction sites in mitochondria where they come into contact with the actin cytoskeleton, a process required to facilitate Drp1 oligomerization and subsequent scission of mitochondria (Korobova et al., 2013). Nishimura et al. (2018) screened several anti-hypertensive drugs for their ability to inhibit hypoxia-induced mitochondrial fission in cardiac fibroblasts and successfully identified cilnidipine, a voltage-dependent calcium channel blocker, to be a specific inhibitor of mitochondrial fission, in both cardiac fibroblasts and neonatal cardiomyocytes. This effect of cilnidipine was not reproduced by other calcium channel blockers and appeared to inhibit the interaction between Drp1 and the actin cytoskeleton protein, filamin A, and through this action, it was shown to reduce cell death and ROS production following hypoxia. The same authors also showed that cilnidipine could protect against mitochondrial fission and HF, following either AMI (Nishimura et al., 2018), or cardiotoxicity induced by the environmental pollutant methylmercury (Nishimura et al., 2019). In terms of neuroprotection, it has been demonstrated that cilnidipine has therapeutic potential as a treatment for AD, but its neuroprotective mechanism needs to be explored and has not been linked to the modulation of mitochondrial dynamics (Bachmeier et al., 2011).
Other pharmacological inhibitors of mitochondrial fission
In AD, it has been shown that Aβ-induced synaptic dysfunction may result from mitochondrial dysfunction with pathological interaction between Aβ and Drp1. Using a direct molecular docking screen, Kuruva et al. (2017) identified the oxidant 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to reduce Drp1 gene expression and inhibit Drp1 GTPase activity. However, DDQ was shown to have off-target effects on Mfn2 and was shown to downgrade hFis1 levels. Finally, a recent study has identified a family of 1H-pyrrole-2-carboxamide compounds to be selective inhibitors of Drp1 GTPase activity, and these agents were shown to preserve mtDNA copy numbers in a Mfn1-deficient cell line characterized by mitochondrial fission and low mtDNA copy numbers (Mallat et al., 2018).
4. Pharmacological modulators of mitochondrial fusion
Until recently, there were no effective and specific pharmacological activators of mitochondrial fusion. However, high-throughput screening has led to the discovery of several small molecules or peptides that can promote mitochondrial fusion by targeting the fusion proteins Mfn1, Mfn2, or OPA1, either directly or indirectly (Table 1B, Figure 1). Pharmacological manipulation of the mitochondrial fusion proteins can be challenging given the numerous pleiotropic non-fusion roles described for both Mfn2 (as a tethering protein between ER and mitochondria, and playing critical roles in mitophagy and autophagy), and OPA1 (mitochondrial cristae remodelling to prevent cytochrome C release and optimizing the respiratory function of mitochondrial supercomplexes) (Beikoghli et al., 2017; Ong et al., 2015; Ong et al., 2010a; Ong et al., 2013; Hernandez-Resendiz et al., 2020). Therefore, interpreting the effects of pharmacological modulators of mitochondrial fusion proteins as treatments of cardiovascular and neurological diseases must take into account these pleiotropic effects.
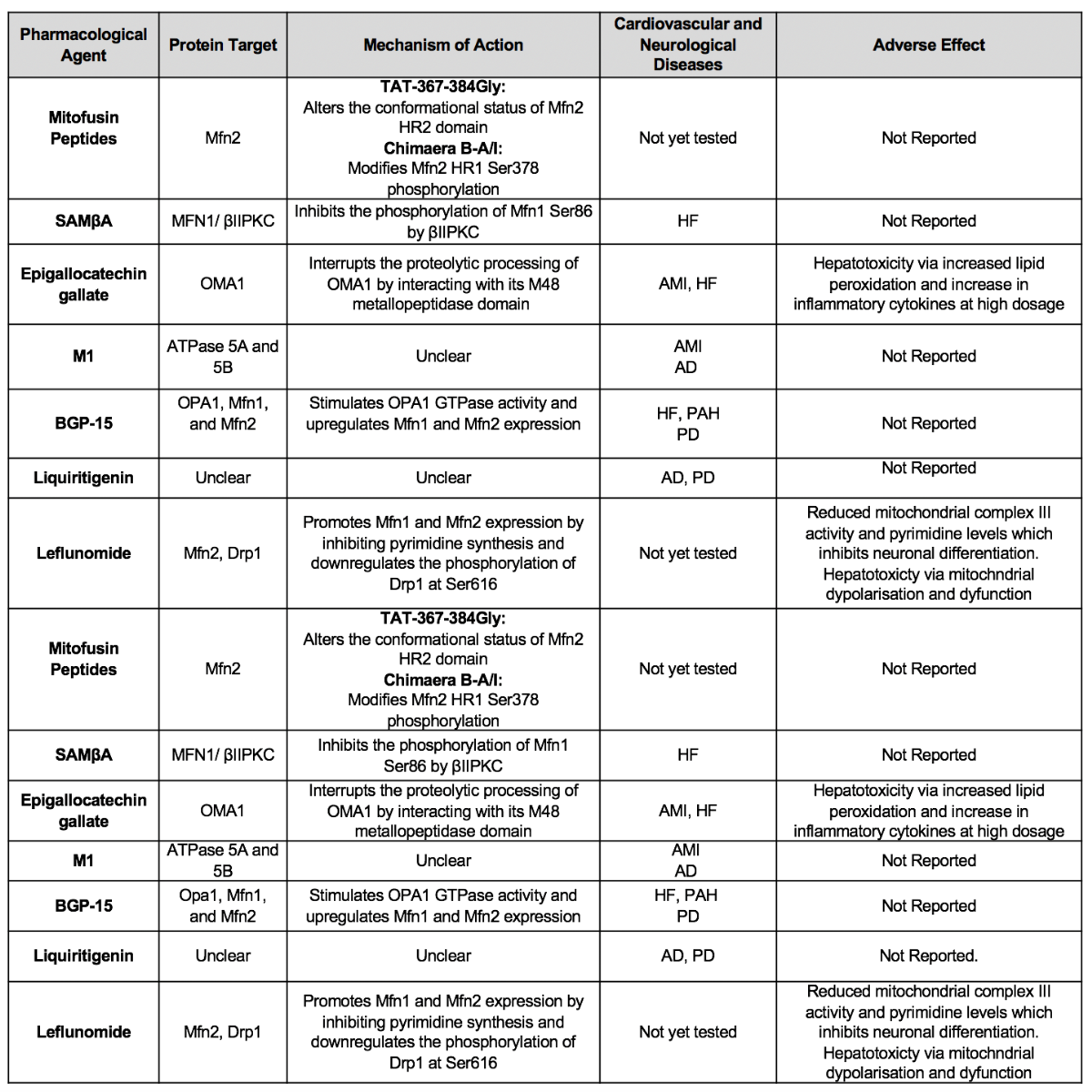
Table 1B. Summary of major pharmacological agents that promote/preserve mitochondrial fusion for cardioprotection and neuroprotection.
M1 – a small molecule activator of mitochondrial fusion
In 2012, Wang et al. (2012a) discovered a pharmacological activator of mitochondrial fusion called M1 (a hydrazone) from a screen of 75,000 small molecules for their ability to rescue mitochondrial fragmentation in Mfn1 knock-out MEFs. M1 was shown to promote mitochondrial elongation in MEFs devoid of either Mfn1 or Mfn2, but was unable to induce mitochondrial fusion in Mfn1/Mfn2 double knock-out and OPA1 knock-out cells. In addition, M1 was demonstrated to not influence the morphology of ER or peroxisomes, thereby making its effects specific to mitochondria (Wang et al., 2012a). Interestingly, M1 was found to promote mitochondrial fusion by increasing the expression levels of ATPase 5A and 5B, components of mitochondrial complex V, suggesting that the effect of M1 was not directly on the mitochondrial fusion proteins. M1 has been used as a tool to chemically induce mitochondrial fusion to demonstrate beneficial effects of mitochondrial DNA replenishment (Yang et al., 2015) and calcium signaling (Kowaltowski et al., 2019). Genetic or pharmacological (using M1) induction of mitochondrial fusion was shown to increase mitochondrial calcium uptake and retention, reduce basal cytosolic calcium levels, and to lower ER calcium stores (Kowaltowski et al., 2019). In addition, M1 has been used to promote mitochondrial fusion and differentiation of human-induced pluripotent stem cells (iPSCs) into cardiomyocytes (Lees et al., 2019). Lastly, M1 did not promote mitochondrial fusion in wild-type MEFs, suggesting that its action may be selective for fragmented mitochondria (Wang et al., 2012a), positioning M1 as a potential therapeutic for cardiovascular and neurological diseases characterized by excessive mitochondrial fission. In this regard, M1 has been investigated in experimental studies as a possible treatment for AMI, HD, and PD (see later sections).
Mitofusin peptides – specific modulators of Mfn2
The fusion of the OMMs of two individual mitochondria is mediated by the mitofusins, Mfn1 and Mfn2, which are able to form homodimers (Mfn1-Mfn1 or Mfn2-Mfn2) and heterodimers (Mfn1-Mfn2), inducing the ‘unfolding’ or ‘folding’ of these proteins, and promoting or inhibiting mitochondrial fusion, respectively. Franco et al. (2016) first designed novel TAT-conjugated peptides capable of promoting the fusion ‘unfolded’ (TAT-367-384Gly) or non-fusion ‘folded’ (TAT-398-418Gly) conformation of Mfn2, providing for the first time a pharmacological approach to modulate Mfn2-mediated mitochondrial fusion. These cell-permeant mini-peptides were able to correct the mitochondrial fragmentation in MEFs containing the Mfn2 mutation responsible for Charcot-Marie-Tooth Disease 2A (CMT2A), but not in MEFs deficient in both Mfn1 and Mfn2 (Franco et al., 2016). In addition, the beneficial effects were observed in cultured hippocampal and cortical neurons harvested from mice with Mfn2 mutations of CMT2A (Franco et al., 2016). In a subsequent study, the same authors have developed a novel small molecule (Chimera B-A/I), which promoted the unfolded conformational change of Mfn2 to induce mitochondrial fusion, correcting mitochondrial fragmentation and improving axonal mitochondrial trafficking in mice with a Mfn2 mutation of CMT2A (Rocha et al., 2018). These Mfn2-specific peptides and small molecules provide novel therapeutics for treating cardiovascular and neurological diseases, characterized by excessive mitochondrial fission or deficient mitochondrial fusion, and their role in these conditions remains to be tested.
SAMβA – a specific peptide modulator of Mfn1
The mitochondrial fusion activity of Mfn1 is regulated by Erk1/2 phosphorylation at its HR1 threonine-562 (Pyakurel et al., 2015). Conversely, the serine-threonine beta II protein kinase C (βIIPKC) is known to phosphorylate Mfn1 at the serine-86 site of its GTPase domain to inhibit the fusion activity of Mfn1. Ferreira et al. (2019a) have designed a peptide, called SAMβA, which blocks the effect of βIIPKC on Mfn1, thereby maintaining the fusion activity of Mfn1. This peptide has been shown to inhibit mitochondrial fragmentation and preserve mitochondrial function in both neonatal and adult murine cardiomyocytes exposed to oxidative stress, and has been reported to protect against post-AMI HF (see later section) (Ferreira et al., 2019a).
Epigallocatechin gallate – OMA1 inhibition to augment OPA1 levels
The regulation of OPA1 activity within the cell is complex and comprises 8 splice variants (in humans). Proteolytic processing by mitochondrial proteases, YME1L and OMA1, generates IMM-anchored long forms of OPA1 (termed L-OPA1), which mediate IMM fusion, and soluble short forms of OPA1 within the mitochondrial matrix (termed S-OPA1), which contribute to mitochondrial bioenergetics and cristae remodeling (reviewed in Burke et al., 2015). This provides the opportunity to pharmacologically inhibit OMA1, as a cardioprotective strategy. In this regard, Nan et al. (2019) has identified, epigallocatechin gallate (EGCG, a flavonoid found in green tea), from an in silico molecular docking screen of 2295 plant-derived compounds, for its ability to inhibit OMA1, and EGCG was shown to inhibit mitochondrial fragmentation induced by hydrogen-peroxide in MEFs, thereby giving a therapeutic approach for promoting mitochondrial fusion in cardiovascular and neurological diseases. However, EGCG has also been shown to inhibit mitochondrial fragmentation by modulating other fission and fusion proteins, suggesting the effects of this drug may not be fully specific for OMA1 (Chen et al., 2018b). Furthermore, EGCG has been reported to have multiple cytoprotective effects in a number of organs and tissues including the heart and brain, but the mechanisms underlying this effect have been mainly attributed to an anti-oxidant effect (Chen et al., 2018b).
Non-specific activators of mitochondrial fusion
BGP-15 is an insulin sensitizer that is known to mediate cytoprotection through several different mechanisms including through mitoprotective effects (Sumegi et al., 2017). A recent study has shown that it can induce mitochondrial fusion through Mfn1, Mfn2, OPA1, and Akt, effects which were associated with decreasing ROS production by targeting complex I and III of the mitochondrial respiratory chain, and preserving mitochondrial integrity (Szabo et al., 2018). Through its effects on mitochondrial fusion, BGP-15 has been shown to have beneficial properties in PAH (Szabo et al., 2018). On the other hand, by using a high-throughput screen of 1,120 FDA-approved compounds for their ability to induce the expression of Mfn2 in HeLa cells, Miret-Casals et al. (2018) identified the anti-rheumatic drug, leflunomide, which turned out to be a strong activator of Mfn2. In addition, it was also shown that leflunomide induced activation of Mfn1, reduced expression of Drp1, and blocked pyrimidine biosynthesis by inhibiting dihydroorotate dehydrogenase, a mitochondrial protein functionally connected with mitochondrial respiratory complex III. These findings suggest that the effect of leflunomide on inducing mitochondrial fusion is not specific for the mitofusins. Leflunomide has been shown to have potential benefit in inducing mitochondrial fusion in pancreatic cancer (Yu et al., 2019), and through its effects on pyrimidine biosynthesis, it may have benefit in AD (Pesini et al., 2019). Finally, in a phytochemical screen, Jo et al. (2016) identified liquiritigenin, a flavonoid with cytoprotective effects, to be an inducer of mitochondrial fusion in a neuroblastoma cell line, which was able to rescue mitochondrial fragmentation observed in cells deficient in Mfn1, Mfn2, and OPA1. Importantly, liquiritigenin was shown to prevent mitochondrial fragmentation and cytotoxicity induced by amyloid-β, providing a potential therapeutic strategy for AD (Jo et al., 2016).
5. Therapeutic targeting of mitochondrial morphology in cardiovascular diseases
Pharmacological modulation of mitochondrial fusion and fission proteins using the agents outlined in sections 3 and 4, have been shown to have beneficial effects in a number of cardiovascular diseases characterized by either excessive fission or deficient fusion including AMI, various cardiomyopathies, and PAH (Figure 1). Only an overview is given here, and the reader is referred to several detailed reviews on these topics (Ong et al., 2015; Ong et al., 2010a; Ong et al., 2013; Samangouei et al., 2018; Hernandez-Resendiz et al., 2020).
Acute myocardial infarction
A number of experimental studies have demonstrated that cardiac mitochondria undergo fission in AMI, primarily in response to the metabolic and biochemical disturbances induced by acute myocardial IRI, leading to mitochondrial dysfunction and cardiomyocyte death (Ong et al., 2010b). Genetic inhibition of Drp1 has been shown to inhibit IRI-induced mitochondrial fission, preserve mitochondrial function, and attenuate cardiomyocyte death (Ong et al., 2010b; Zepeda et al., 2014), positioning excessive mitochondrial fission as a therapeutic target for cardioprotection. Acute pharmacological inhibition of IRI-induced mitochondrial fission using a number of agents such as mdivi-1 (Ong et al., 2010b; Ishikita et al., 2016), dynasore (Gao et al., 2013), or P110 (Disatnik et al., 2013) has been reported to reduced myocardial infarct (MI) size in rodent models of acute myocardial IRI when administered either before ischemia or at the onset of reperfusion. The recently described more specific and potent Drp1 inhibitors, Drpitor1 and Drpitor1a, have also been shown to confer cardioprotection in the right ventricle in an isolated perfused rodent heart model (Wu et al., 2020). A key step in the translational pathway was to show cardioprotection in the clinically relevant large animal pig model of acute myocardial IRI, and in this regard, a small underpowered pilot study failed to show a reduction in MI size with mdivi-1 administered at reperfusion. Although acute inhibition of mitochondrial fission has been shown to be cardioprotective, chronic inhibition of mitochondrial fission is detrimental, with long-term genetic deletion of cardiac Drp1 (Ikeda et al., 2015) increasing susceptibility to acute myocardial IRI and susceptibility to cardiomyopathy, most likely due to suppression of mitophagy, and the accumulation of damaged mitochondria.
Targeting Mfn1 and Mfn2 to reduce MI size in the adult rodent heart has produced unexpected results with ablation of cardiac mitofusins protecting mitochondria and reducing MI size (Papanicolaou et al., 2011; Papanicolaou et al., 2012; Hall et al., 2016), suggesting the pleiotropic non-fusion effects of Mfn2, such as tethering ER to mitochondria, may dominate over any changes in mitochondrial shape (de Brito et al., 2008). This proposes that targeting mitofusins with cell-permeant peptides that inhibit Mfn2 (Franco et al., 2016) at the onset of reperfusion may limit MI size by dissociating mitochondria from sarcoplasmic reticulum (SR), thereby preventing mitochondrial overload and inhibiting MPTP opening, may be a therapeutic strategy for cardioprotection. In contrast to Mfn1 and Mfn2, genetic ablation of OPA1 has been demonstrated to increase MI size, suggesting a cardioprotective role for OPA1 (Chen et al., 2009). In this regard, OPA1 overexpression was shown to be cardioprotective with preservation of mitochondria cristae, prevention of apoptosis, and improved respiratory function. Upregulation of myocardial OPA1 levels by genetic ablation of its protease, OMA1, has also been shown to be cardioprotective (Xiao et al., 2014), providing the opportunity to pharmacologically inhibit OMA1, using newly discovered OMA1 inhibitors (such as EGCG) (Nan et al., 2019) as a future cardioprotective strategy.
Cardiomyopathy
Excessive mitochondrial fission has been shown to contribute to the pathophysiology of several cardiomyopathies including post-infarction HF, cardiotoxicity, and left ventricular hypertrophy (LVH) (Ong et al., 2015; Ong et al., 2010a; Ong et al., 2013; Hernandez-Resendiz et al., 2020). Long-lasting cardioprotection has been observed with the single application of P110 at the onset of reperfusion following AMI, in terms of reducing acute MI size, preserving mitochondrial function and preventing adverse LV remodeling three weeks later in a mouse model of acute myocardial IRI (Disatnik et al., 2013). Daily treatment with cilnidipine (the pharmacological inhibitor of Drp1-filamin A1) has been shown to reduce Drp1 GTPase activity in the peri-infarct zone and prevent adverse post-AMI remodeling (as evidenced by less LVH and preserved LV function at 1 month) when started either 1 or 7 days following AMI, potentially repurposing chronic therapy with cilnidipine as a therapeutic agent for preventing post-AMI HF (Nishimura et al., 2018).
Chronic treatment with mdivi-1 was also shown in a mouse total aortic constriction (TAC) model of LVH, to attenuate Drp1 activation and translocation, prevent LVH and preserve cardiac function (Chang et al., 2013). In another study, it was shown that LVH induced by chronic β1-adrenergic receptor (β1-AR) agonist is due to activation of Ca2+/calmodulin-dependent kinase II, phosphorylation of Drp1 at Ser616, mitochondrial translocation of Drp1, and mitochondrial fission, which in turn induces MPTP opening, resulting in mitochondrial and cardiac dysfunction. Genetic ablation of Drp1 or treatment with mdivi-1 was shown to attenuate mitochondrial fragmentation and prevent LVH in this mouse model of chronic β1-AR stimulation. Finally, Qi et al. (2018) reported that Angiotensin II (AgII)-induced cardiomyocyte apoptosis was mediated through a sirtuin 1-p53-Drp1 mediated pathway involving mitochondrial fission. In this study, chronic treatment of spontaneously hypertensive rats with mdivi-1 to inhibit Drp1 was shown to prevent AgII cardiomyocyte apoptosis, LVH, and to preserve cardiac function (Qi et al., 2018). These three studies provide evidence for Drp1 as a therapeutic target for HF associated with LVH.
It has been reported that cardiotoxicity due to the chemotherapy agent, doxorubicin, was associated with Drp1-mediated mitochondrial fission, mitochondrial dysfunction, and cardiotoxicity by alleviating cardiac performance and reducing MI size following acute IRI. (Gharanei et al., 2013) These effects were attributed to the activation of phosphorylated pro-survival kinases, including Akt and Erk1/2, and a reduction in phosphorylated Drp1 (Gharanei et al., 2013). Cardiomyopathy due to sepsis has also been shown to be due in part to mitochondrial fragmentation and dysfunction leading to impaired cardiac function. Haileselassie et al. (2019) have shown that treatment of H9C2 cardiomyocytes with lipopolysaccharide (LPS) impaired mitochondrial respiration, increased mitochondrial oxidative stress, and collapsed the mitochondrial membrane potential, effects which were abrogated by P110 administration. In mice treated with LPS to induce sepsis-related cardiomyopathy, treatment with P110 prevented mitochondrial fragmentation, preserved cardiac function, and reduced mortality (Haileselassie et al., 2019), highlighting inhibition of mitochondrial fission as a therapeutic strategy for preventing cardiomyopathy due to sepsis.
A limited number of studies have investigated the pharmacological modulation of mitochondrial fusion to prevent cardiomyopathy. One of them proved that SAMβA, which blocks the Mfn1-βIIPKC interaction, had beneficial effects on preventing mitochondrial fragmentation, rescuing mitochondrial respiration, and preventing post-infarct adverse LV remodeling (Ferreira et al., 2019b). Finally, given that HF has been associated with a reduction in myocardial OPA1 expression (Chen et al., 2009; Chen et al., 2012), pharmacological strategies aimed at inhibiting OMA1 to augment myocardial OPA1 levels, such as EGCG (Nan et al., 2019), may provide a novel agent therapeutic for preventing HF following AMI.
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a condition in which progressive obstruction of the pulmonary vasculature by vascular smooth muscle cell (VSMC) proliferation results in right ventricular HF. This clinical condition currently represents a significant cause of morbidity and mortality. Experimental studies have shown that Drp1-mediated mitochondrial fission is required for VSMC proliferation, positioning fission as a therapeutic target for preventing PAH (Chalmers et al., 2012; Marsboom et al., 2012; Parra et al., 2017). In three different experimental rat PAH models, phosphorylation of Drp1 at Ser616 together with mitochondrial fission was observed, and chronic treatment with the Drp1 inhibitor, mdivi-1, prevented Drp1-mediated fission, reduced VSMC proliferation, prevented PAH, and protected against right ventricular hypertrophy (Marsboom et al., 2012). Inhibiting Drp1 using mdivi-1 or preventing the Drp1-hFis1 interaction using P110, also attenuated mitochondrial fission and protected the right ventricle against acute IRI in a rodent PAH model (Tian et al., 2017). Interestingly, in lung tissue samples from patients with PAH, HIF-1α activation was observed in conjunction with cyclin B1/CDK1–dependent phosphorylation of DRP1 at Ser616 showing the relevance of Drp1 as a therapeutic target in humans (Marsboom et al., 2012).
Recently, it has been demonstrated that the mitochondrial fission proteins, MiD49 and MiD51, may also contribute to the VSMC proliferation observed in PAH. Chen et al. (2018a) found that MiD expression was increased in VSMC, and this was associated with Drp1-mediated mitochondrial fission, VSMC proliferation, and less apoptosis. Genetic ablation of MiD proteins induced mitochondrial fusion through the ERK1/2 and cyclin-dependent kinase 4 (CDK4)-dependent pathways. Conversely, genetic overexpression of MiD proteins induced mitochondrial fission, decreased miR-34a-3p, and reproduced the PAH phenotype (Chen et al., 2018a), positioning the MiD proteins as an additional therapeutic target for preventing PAH.
Finally, it has been shown that reduced levels of Mfn2 in VSMCs was present in animal PAH and human PAH lung tissue, and this was associated with mitochondrial fission, increased VSMC proliferation, and the development of PAH. Genetic upregulation of Mfn2 was shown to rescue mitochondrial fission, inhibit VSMC proliferation, and prevent PAH (Ryan et al., 2013), implicating Mfn2 as a therapeutic target for PAH treatment, and opening up the possibility of using cell-permeant Mfn2 profusion peptides (Franco et al., 2016) or Chimera B-A/I (Rocha et al., 2018) as therapeutics. The non-specific pharmacological inducer of mitochondrial fusion, BGP-15, has been reported in a rodent model of PAH to inhibit mitochondrial fission in type II pneumocytes (Szabo et al., 2018).
The proliferation of right ventricular (RV) fibroblasts is known to contribute to RV dysfunction and failure observed in PAH. Interestingly, Tian et al. (2018) found that Drp1 expression, mitochondrial fission, proliferation rates, and collagen production were increased in RV fibroblasts from PAH animals, and chronic treatment with mdivi-1 inhibited RV fibroblast proliferation, reduced RV fibrosis, and preserved RV function, positioning Drp1 as a therapeutic target for preventing RV dysfunction in PAH.
6. Therapeutic targeting of mitochondrial dynamics in neurological diseases
Pharmacological modulation of mitochondrial fusion and fission proteins using the agents outlined in sections 3 and 4, have been shown to have beneficial effects in a variety of neurological conditions (primarily neurodegenerative diseases) characterized by either excessive fission or deficient fusion (Figure 1). Only an overview is given here, and the reader is referred to several detailed articles on this topic (Oliver et al., 2019; Whitley et al., 2019).
Neuroprotection against acute cerebral injury
Ischemic stroke is one of the leading causes of death and long-lasting disability worldwide, and despite timely reperfusion using either thrombolytic therapy or thrombectomy, significant morbidity and mortality occurs, requiring the need for novel neuroprotective therapies to improve clinical outcomes following stroke. In common with AMI, acute cerebral artery occlusion and reperfusion in stroke also induces mitochondrial fission and dysfunction in neurons following acute IRI, making the fission machinery a therapeutic target for reducing cerebral infarct size and preserving motor and cognitive function in stroke patients. It has been shown in vitro that mitochondrial fragmentation and dysfunction occurs in hippocampal HT-22 cell-lines and primary cortical neurons subjected to simulated acute IRI, and the inhibition Drp1, using mdivi-1, was able to rescue mitochondrial morphology and reduce cell death (Grohm et al., 2012; Wang et al., 2014; Zhao et al., 2014). Furthermore, in mouse and rat middle cerebral artery occlusion (MCAO) stroke models, mitochondrial fragmentation and dysfunction occurs in neurons, and the administration of a single mdivi-1 bolus prior to MCAO was shown to reduce cerebral infarct size and preserve motor function (Grohm et al., 2012; Zhang et al., 2013; Zhao et al., 2014). Moreover, in rodent models of stroke, mdivi-1 administered at the clinically relevant time-point of reperfusion (Ma et al., 2016) also reduced cerebral infarct size. Another study using chronic treatment for 10 days provided sustained benefits in terms of neuroprotection (Cui et al., 2016). Inhibiting mitochondrial fission using mdivi-1 (or dynasore) has also exhibited neuroprotection in several other experimental models of acute cerebral and spine injury including subarachnoid hemorrhage (Fan et al., 2017), brain trauma (Wu et al., 2016; Wu et al., 2018; Omelchenko et al., 2019), chemical-induced epilepsy (Xie et al., 2013; Kim et al., 2016), anesthetic-induced cytotoxicity (Xu et al., 2016; Gao et al., 2018); post-cardiac arrest (Li et al., 2015; Wang et al., 2018c), and spinal cord acute IRI (Liu et al., 2015; Li et al., 2017b), highlighting the range of conditions in which excessive mitochondrial fission plays a pathophysiological role.
Alzheimer’s Disease
The leading cause of neurodegeneration worldwide is AD, a condition which results in a progressive decline in cognitive function. It is characterized by the extracellular accumulation of amyloid-β peptides, and the intracellular aggregation of hyperphosphorylated Tau protein, that induce synaptic dysfunction, neuroinflammation, and neuronal loss (Flannery et al., 2019). However, the etiological role of amyloid-β accumulation in the pathophysiology of AD has been questioned, given the failure of clinical trials aimed at reducing amyloid-β formation (Mehta et al., 2017). As such, alternative mechanisms have been proposed including mitochondrial dysfunction, highlighting the contribution of mitochondrial dynamics to the pathogenesis of AD. Electron microscopy studies of brain tissue from AD patients have demonstrated increased mitochondrial fragmentation, increased expression of Drp1 and hFis1, and lower expression of Mfn1, Mfn2, and OPA1 (Hirai et al., 2001; Baloyannis 2006; Manczak et al., 2011). Experimental studies have shown that amyloid-β and Tau induce Drp1-mediated mitochondrial fission, perturbed anterograde mitochondrial trafficking (hampered movement of newly synthesized mitochondria from the cell body to the axon), impaired retrograde mitochondrial trafficking (interference in mitophagic removal of damaged mitochondria), and mitochondrial dysfunction, resulting in the accumulation of toxic protein aggregates, thereby inducing synaptic dysfunction and neuronal loss in AD (Wang et al., 2008; Calkins et al., 2011; Tammineni et al., 2017) Amyloid-β has been shown to induce mitochondrial fission through the S-nitrosylation of Drp1 (Cho et al., 2009), and Tau has been reported to impair mitochondrial motility (Spires-Jones et al., 2014).
In support of a pathophysiological role for Drp1-induced mitochondrial fission in AD, pharmacological inhibition of Drp1 has been shown in several studies to be beneficial in experimental animal models of AD. In vitro studies demonstrated that treatment of neuroblastoma cells and primary hippocampal and cortical neurons with pharmacological inhibitors of mitochondrial fragmentation (mdivi-1 [Reddy et al., 2017; Baek et al., 2017; Reddy et al., 2018] or DDQ [Kuruva et al., 2017]) or inducers of fusion (M1 [Hung et al., 2018] or liquiritigenin [Jo et al., 2016]) protected against amyloid β-induced mitochondrial fragmentation, mitochondrial dysfunction, and synaptic toxicities. In amyloid precursor protein/presenilin 1 (APP/PS1) (Baek et al., 2017) and CRND8 APP (Wang et al., 2017) mouse models of AD, mitochondrial fragmentation, perturbed mitochondrial distribution, and mitochondrial dysfunction have been observed in the brain with changes occurring before the accumulation of amyloid. Importantly, chronic inhibition of mitochondrial fragmentation using mdivi-1 reversed these mitochondrial abnormalities, decreased extracellular amyloid deposition, improved synaptic function, and prevented the development of cognitive deficits in APP/PS1 (Baek et al., 2017) and CRND8 APP (Wang et al., 2017) mice. Joshi et al. (2018a) have reported that the interaction between Drp1 and hFis1, and mitochondrial fragmentation were increased in fibroblasts derived from AD patients, and chronic treatment with P110 reduced Aβ accumulation, preserved mitochondrial function, and reduced cognitive deficits in the brain of the 5X5AD mouse model of AD.
Parkinson’s Disease
Parkinson’s Disease (PD) is the second most common neurodegenerative disorder after AD, and is characterized clinically by a pill-rolling tremor, bradykinesia, muscle rigidity, and a slow shuffling gait (Feng et al., 2020). In most cases, it is due to the toxic accumulation of α-synuclein (termed Lewy bodies), which results in the progressive loss of dopaminergic neurons in the substantia nigra pars compacta. Most cases of PD are sporadic with 5-10% of PD cases being familial. Mutations in several different genes (such as α-synuclein, leucine-rich repeat kinase 2 [LRRK2], PTEN-induced kinase-1 [PINK-1], Parkin, DJ-1, and VPS35) have been associated with mitochondrial dysfunction and fragmentation in dopaminergic neurons (Feng et al., 2020).
Experimental studies have shown that mutations in PINK1 and Parkin induce perturbations in mitochondrial dynamics and quality control (such as mitophagy) and contribute to mitochondrial dysfunction characteristic of PD (Deng et al., 2008; Poole et al., 2008). However, the effects of the PINK1/Parkin pathway on mitochondrial dynamics have been mixed, playing roles in both mitochondrial fission and mitophagy (Deng et al., 2008; Yu et al., 2011; Yang et al., 2008; Yu et al., 2011; Liu et al., 2011), and mitochondrial fusion (Exner et al., 2007; Dagda et al., 2009; Lutz et al., 2009; Sandebring et al., 2009), depending on the species and cell-type. Treatment with mdivi-1 was shown to inhibit mitochondrial fission and preserve mitochondrial function in PINK1L347P and PINK1W437X mutant mice (Cui et al., 2010).
Mutations in α-synuclein has been shown to underlie many cases of sporadic and autosomal dominant PD. The toxic accumulation of mutant α-synuclein in dopaminergic neurons (Lewy bodies) has been demonstrated to induce mitochondrial fission and inhibit mitochondrial fusion (Kamp et al., 2010; Gui et al., 2012), resulting in mitochondrial dysfunction. The mechanisms through which mutant α-synuclein mediate changes in mitochondrial morphology are unclear with mixed results over whether this is via mitochondrial fusion and fission proteins, an indirect effect on other signalling proteins such as Erk1/2, or are a direct effect on the OMM (Kamp et al., 2010; Gui et al., 2012). Bido et al. (2017) have shown that in vivo treatment with the Drp1 inhibitor, mdivi1, rescued excessive mitochondrial fission, reduced neurodegeneration, decreased α-synuclein aggregates, and normalized motor function in a rat PD model over-expressing mutant hA53T-α-synuclein in the nigrostriatal pathway.
LRRK2 mutations are the most common cause of autosomal-dominant PD. LRRK2 is responsible for neuronal growth, synaptic trafficking, autophagy, and regulation of the neuronal cytoskeleton. Over-expression of LRRK2 in the human dopaminergic neuroblastoma SH-SY5Y cell line and rat E18 primary cortical neurons has been proven to increase mitochondrial localization of Drp1, induce mitochondrial fragmentation and dysfunction, and slow mitochondrial fusion events (Wang et al., 2012c). These effects of LRRK2 were rescued by transfection with either dominant-negative mutant of Drp1, and LRRK2 was shown to interact with Drp1 at the OMM (Wang et al., 2012c). The LRRK2 G2019S mutation is the most common genetic cause of PD. In both LRRK2 G2019S-expressing cells and PD patient fibroblasts carrying this specific mutant, Su et al. (2013) showed that treatment with P110 prevented mitochondrial fragmentation and dysfunction, and repressed excessive autophagy. Interestingly, LRRK2 G2019S was demonstrated to directly bind to Drp1 and increase Drp1 activity by phosphorylating the Threonine595 site instead of the typical Ser616 site, effects which were prevented by P110 (Su et al., 2013).
Mutations in the vacuolar protein sorting 35 (VPS35) gene, the third leading cause of autosomal-dominant PD, have also been shown to induce mitochondrial fragmentation and cell death in cultured neurons in vitro, in mouse substantia nigra neurons in vivo, and in human fibroblasts from a PD patient with the VPS35 D620N mutation, and these effects were prevented by treatment with the Drp1 inhibitor, mdivi-1 (Wang et al., 2016). Mutant VPS35 was shown to directly interact with Drp1, and its turnover via mitochondria-derived vesicles-dependent trafficking to lysosomes for degradation (Wang et al., 2016).
Mutations in the mitochondrial DJ-1 protein are known to induce mitochondrial dysfunction and cause an autosomal recessive form of PD (Bonifati et al., 2003). It has been shown that DJ-1 deficiency causes severe mitochondrial fragmentation (Irrcher et al., 2010; Krebiehl et al., 2010; Thomas et al., 2011). Over-expressing DJ-1 in M17 human neuroblastoma cells and primary hippocampal neurons reduced expression of Drp1 and induced elongation of mitochondria, whereas over-expressing PD-associated DJ-1 mutants (R98Q, D149A and L166P) induced mitochondrial fragmentation and dysfunction. Importantly, genetic ablation of Drp1 rescued these mitochondrial effects (Wang et al., 2012b).
Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant late-onset progressive neurodegenerative disease characterized by movement disorders and cognitive impairment, and is caused by the toxic accumulation of defective huntingtin (HTT) protein inside neurons, which results in transcriptional and protein defects, impaired axonal mitochondrial transport and synaptic function, mitochondrial dysfunction, and neuronal loss (Jimenez-Sanchez et al., 2017). In HD patients, decreased levels of mitochondrial biogenesis proteins (such as transcription factor A, mitochondria [TFAM] and peroxisome proliferator-activated receptor gamma coactivator 1 alpha [PGC1-α]) and mitochondrial fusion proteins (Mfn1, Mfn2, and OPA1), together with increased levels of mitochondrial fission proteins (Drp1 and hFis1) have been observed in brain tissue (Kim et al., 2010; Shirendeb et al., 2011). Over-expression of mutant HTT protein in HeLa cells has been shown to induce mitochondrial fragmentation and dysfunction, effects which were rescued with an expression of a dominant-negative mutant Drp1 or Mfn2 (Wang et al., 2009). A subsequent study by Song et al. (2011) showed that mutant HTT triggers mitochondrial fission and dysfunction in neurons and fibroblasts of HD patients in vitro, and HD mice in vivo before neurological deficits and HTT aggregates appeared, effects which were rescued by the dominant-negative mutant Drp1, suggesting for the first time a pathophysiological role for Drp1 in HD. Interestingly, the mutant HTT was found to bind to Drp1 and stimulate its GTPase activity, an effect which appears to be mediated by phosphorylation of Drp1 at Ser616 via mitogen-activated protein kinase 1 (Roe et al., 2018) and S-nitrosylation (Haun et al., 2013). Recently, mutant HTT-induced mitochondrial fission has also been demonstrated to disrupt ER-mitochondria contact points (essential for calcium signalling between these two organelles) resulting in perturbed mitochondrial calcium signalling and ROS production, and crucially, this effect was abrogated by the Drp1 inhibitor, mdivi-1 (Cherubini et al., 2020).
Experimental studies have reported beneficial effects with pharmacological inhibition of mitochondrial fission in HD. Manczak and Reddy (2015) found that treatment with the Drp1 inhibitor, mdivi-1, rescued mitochondrial fragmentation, reduced expression of mitochondrial fission proteins, increased expression of mitochondrial fusion and synaptic proteins, in both wild-type and mutant HTT striatal progenitor neurons. Additionally, it has been reported that P110 prevented HTT-induced mitochondrial fragmentation and dysfunction, prevented lysosomal dysfunction, and increased cell viability in HD cell culture models and in iPSC-derived neurons from HD patients (Guo et al., 2013; Joshi et al., 2019). Crucially, in vivo treatment of transgenic HD R6/2 mice with P110 prevented mitochondrial dysfunction, motor deficits, neuropathology, and mortality (Guo et al., 2013). In that study, p53, was shown to bind to Drp1 and induce mitochondrial fission and neuronal damage (Guo et al., 2013). Similar beneficial effects were seen in the R6/2 mouse model of chronic HD (Zhao et al., 2018).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neuromuscular degenerative disease characterized by motor neuron death and skeletal muscle wasting. For most patients, ALS occurs sporadically, with mutations in superoxide dismutase 1 (SOD1) accounting for up to 20% of familial ALS (Cabral-Costa et al., 2020). Mitochondrial fragmentation has been observed in spinal cord and skeletal muscle tissue samples from mutant SOD1 mice (including the SOD1G93A ALS mouse model) (Vinsant et al., 2013a; Vinsant et al., 2013b) and ALS patients (De Vos et al., 2007; Luo et al., 2013; Song et al., 2013; Tafuri et al., 2015). The Drp1 inhibitor, mdivi-1, was shown to prevent the mitochondrial fragmentation and dysfunction observed in skeletal muscle in the SOD1G93A ALS mouse model (Luo et al., 2013), confirming Drp1 to be a potential therapeutic target for ALS. Similarly, treatment with P110 has been shown to rescue mitochondrial fragmentation in ALS patient fibroblasts and in cultured motor neurons expressing SOD1 mutant (Joshi et al., 2018b). Crucially, chronic treatment of mice expressing the SOD1G93A mutation with P110, produced an improvement in motor performance and survival (Joshi et al., 2018b). Interestingly, it has recently reported that osteocytes in the SOD1G93A ALS mouse model have mitochondrial fragmentation and dysfunction, increased expression of Drp1, decreased expression of OPA1, changes which may in part contribute to bone abnormalities present in ALS. Importantly, these defects were corrected by the Drp1 inhibitor, mdivi-1 (Wang et al., 2018a). Finally, Wang et al. (2018b) have found reduced levels of Mfn2 in spinal cord motor neurons in the SOD1G93A ALS mouse model, and genetic expression of Mfn2 was able to prevent mitochondrial fragmentation and improve skeletal muscle function, positioning Mfn2 as a potential therapeutic target for preventing skeletal muscle atrophy in ALS.
7. Summary and future perspectives
Mitochondrial dysfunction and impaired cellular bioenergetics underlie a diverse variety of cardiovascular and neurological disorders. Imbalances in mitochondrial dynamics, primarily excessive mitochondrial fission and deficient mitochondrial fusion, have been shown to contribute to the pathophysiology of these clinical conditions including AMI, various cardiomyopathies, and PAH in the cardiovascular system, and stroke, acute cerebral injury, AD, PD, HD, and ALS in the neurological system. As such, targeting the mitochondrial fission machinery using pharmacological inhibitors (such as peptides [P110], small molecules [mdivi-1, Drpitor1] and drug repurposing [cilnidipine]) have been shown to have beneficial effects in these diverse conditions. Furthermore, novel, more specific pharmacological inhibitors of the mitochondrial fission proteins are needed to reduce off-target effects and improve efficacy, so as to translate this therapeutic strategy into the clinic for patient benefit. Targeting the mitochondrial fusion proteins has been more challenging given their non-fusion pleiotropic roles. Interestingly, the data so far suggests that the mitochondrial fusion and fission proteins can be targeted using either peptides, small molecules or drug repurposing, and in this regard, the three different pharmacological approaches appear to be similalrly efficacious in the heart and the brain. In terms of translating the therapeutic approaches into the clinic, of course drug repurposing would be less challenging than testing novel molecules and peptides, the latter of which may also have challenges in terms of bioavailability and drug delivery to the diseased heart or brain.
Conflicts and disclosures
All authors have no conflicts or disclosures.
Acknowledgement
Sang-Ging Ong is supported by National Institutes of Health grants (R00 HL130416 and R01 HL148756). Chrishan Ramachandra is supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (NMRC/OFYIRG/0073/2018), the National Health Innovation Centre Singapore under its Innovation to Develop Grant (NHIC-I2S-1811007), and the SingHealth Duke-NUS Academic Medical Centre under its SingHealth Duke-NUS Academic Medicine Research Grant (AM/TP033/2020 [SRDUKAMR2033]). Sauri Hernandez-Resendiz is supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (OF-YIRG)–[NMRC/OFYIRG/0078/2018]. Derek Hausenloy is supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
References
Siavash Beikoghli Kalkhoran1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore.
Gustavo E. Crespo-Avilan1,2,3
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore. 3Department of Biochemistry, Medical Faculty, Justus Liebig-University, Giessen, Germany.
Sauri Hernandez-Resendiz1,2,4
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore. 4Tecnologico de Monterrey, Centro de Biotecnologia-FEMSA, Nuevo Leon, Mexico.
Chrishan J.A. Ramachandra1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore.
Sang-Ging Ong5,6
5Department of Pharmacology, University of Illinois College of Medicine, Chicago, Illinois, United States of America. 6Division of Cardiology, Department of Medicine, University of Illinois College of Medicine, Chicago, Illinois, United States of America.
Derek J Hausenloy1,2,7-9
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore. 7Yong Loo Lin Medical School, National University of Singapore, Singapore. 8The Hatter Cardiovascular Institute, University College London, London, UK. 9Cardiovascular Research Centre, College of Medical and Health Sciences, Asia University, Taiwan.
Corresponding author:
Professor Derek J. Hausenloy
Email: derek.hausenloy@duke-nus.edu.sg
In a new window | Download PPT
Figure 1: Pharmacological modulators of mitochondrial dynamics for cardioprotection and neuroprotection. This scheme depicts the main pharmacological agents for inhibiting mitochondrial fission or activating mitochondrial fusion to prevent mitochondrial dysfunction, and confer cardioprotection (in acute myocardial infarction, cardiomyopathy, pulmonary arterial hypertension), and neuroprotection (in stroke, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis).
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone, DDQ; Selective antagonist of mitofusin 1-β2PKC association, SAMßA; Epigallocatechin gallate, EGCG.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11729 | 65 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA