Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Classical chemokines, atypical chemokines, and MIF proteins in ischemic stroke: effects, mechanisms and roles in conditioning
Time:2021-04-05
Number:9305
Author Affiliations

Conditioning Medicine 2021. 4(1):39-57.
Abstract
Abstract: Cytokines and chemokines contribute to the inflammatory response in ischemic stroke both in the acute phase and in post-stroke inflammation. In addition to classical chemokines such as C-C motif ligand (CCL) 2, CCL5, or C-X-C motif chemokine ligand (CXCL) 12, and their receptors, more recent evidence suggests that atypical chemokines (ACKs) and alarmins/danger-associated molecular pattern (DAMP) may also play an important role in the pathogenesis of ischemic stroke. Atypical chemokines share functional similarities with classical chemokines and signal through classical chemokine receptors. The class of ACKs overlaps with the alarmin/DAMP class, with some members classified as both ACK and alarmin/DAMP. Here, we summarize the numerous effects of classical chemokines in ischemic stroke pathogenesis, including their therapeutic potential, and discuss the emerging role of ACKs and alarmins/DAMPs. Evidence from various experimental stroke models and in vitro studies is gathered and data from initial clinical studies weighed. Next to prominent alarmins/DAMPs such as peroxiredoxins and high-mobility group box protein 1, we focus on macrophage migration inhibitory factor (MIF) proteins as a prototypical example of ACKs engaging the CXC-motif chemokine receptors CXCR2, CXCR4, and CXCR7. A number of studies have explored MIF in ischemic stroke, but some findings have remained controversial, suggesting complex context-dependent mechanisms and a dichotomic role of MIF in ischemic stroke. Lastly, we summarize inter-relationships between the regulatory networks of these various protein classes in ischemic stroke and discuss promising examples suggesting that they may have important roles in remote ischemic conditioning (RIC) as a novel paradigm in protection from brain ischemia.
Keywords: Classical chemokines, atypical chemokines, macrophage migration inhibitory factor, DAMPs, chemokine receptors, inflammation, ischemic stroke, cardiovascular diseases, ischemic conditioning
Abstract
Abstract: Cytokines and chemokines contribute to the inflammatory response in ischemic stroke both in the acute phase and in post-stroke inflammation. In addition to classical chemokines such as C-C motif ligand (CCL) 2, CCL5, or C-X-C motif chemokine ligand (CXCL) 12, and their receptors, more recent evidence suggests that atypical chemokines (ACKs) and alarmins/danger-associated molecular pattern (DAMP) may also play an important role in the pathogenesis of ischemic stroke. Atypical chemokines share functional similarities with classical chemokines and signal through classical chemokine receptors. The class of ACKs overlaps with the alarmin/DAMP class, with some members classified as both ACK and alarmin/DAMP. Here, we summarize the numerous effects of classical chemokines in ischemic stroke pathogenesis, including their therapeutic potential, and discuss the emerging role of ACKs and alarmins/DAMPs. Evidence from various experimental stroke models and in vitro studies is gathered and data from initial clinical studies weighed. Next to prominent alarmins/DAMPs such as peroxiredoxins and high-mobility group box protein 1, we focus on macrophage migration inhibitory factor (MIF) proteins as a prototypical example of ACKs engaging the CXC-motif chemokine receptors CXCR2, CXCR4, and CXCR7. A number of studies have explored MIF in ischemic stroke, but some findings have remained controversial, suggesting complex context-dependent mechanisms and a dichotomic role of MIF in ischemic stroke. Lastly, we summarize inter-relationships between the regulatory networks of these various protein classes in ischemic stroke and discuss promising examples suggesting that they may have important roles in remote ischemic conditioning (RIC) as a novel paradigm in protection from brain ischemia.
Keywords: Classical chemokines, atypical chemokines, macrophage migration inhibitory factor, DAMPs, chemokine receptors, inflammation, ischemic stroke, cardiovascular diseases, ischemic conditioning
Introduction
Stroke is a major cause of death worldwide, accounting for 9% of the global death toll, and the leading cause of disability in adults. Most strokes result from cerebral ischemia and this accounts for about 85% of all cases; 15% of strokes are hemorrhagic (Hankey, 2017). Ischemic stroke is an acute vascular event caused by a disruption of blood flow supplied to a corresponding area of the brain that results in rapid oxygen and nutrient deficiency, and leads to the development of a focal neurologic deficit. According to the TOAST (Trial of Org 10172 in Acute Stroke Treatment) classification, ischemic strokes are caused by thrombosis or embolism due to atherosclerosis of a large artery (large artery or atherosclerotic strokes), a cardiac embolism, blockage of a small vessel (small vessel disease), or of undetermined cause (cryptogenic stroke) (Adams et al., 1993). Up to now therapeutic options are limited and thrombolysis by recombinant tissue plasminogen activator (rt-PA), often applied in conjunction with mechanical thrombectomy (Campbell et al., 2015), remains the only pharmacotherapy approved by the United States Food and Drug Administration (FDA) but with severe limitations such as a narrow therapeutic time window (Hacke et al., 2008; Hankey, 2017). While immediate thrombolysis or thrombus removal will remain the first line of treatment, there is a requirement to develop new therapeutic strategies for the post-acute phases of ischemic stroke (Marsh and Keyrouz, 2010; Moskowitz et al., 2010).
Inflammation contributes to the pathogenesis of ischemic stroke, but the mechanisms are incompletely understood and efficacious inflammation-targeting strategies have yet to be developed (Fu et al., 2015; Esenwa and Elkind, 2016; Catanese et al., 2017; Zhang et al., 2021). The depletion of brain oxygen and glucose levels following vascular occlusion during ischemic stroke leads to a drop in neuronal ATP levels and lactate acidosis, which in turn affect energy-dependent ion pumps and ion homeostasis in neurons and glial cells. The ensuing depolarization of the plasma membrane leads to a rapid release of excitatory amino acids such as glutamate into the extracellular matrix, manifesting a pathogenic state of excitotoxicity. Oxidative and nitrosative stress additionally contribute to brain injury after ischemia, particularly following the rapid increase in oxygen supply upon reperfusion. The inflammatory response is induced within a few hours after the onset of ischemic stroke and overlaps with the excitatory and oxidative stress response. It is involved in almost all processes of post-stroke inflammation. Initiated by the rapid activation of resident glial and endothelial cells and the infiltration of a wide range of peripheral immune cells, alarmins, cytokines, and chemokines (CKs) such as tumor necrosis factor-α (TNF-α), interleukin-1β, IL-6, and C-C motif ligand (CCL) 2 play pivotal roles in stroke-associated inflammation (Allan and Rothwell, 2001; Iadecola and Anrather, 2011; Walsh et al., 2014; Ransohoff and Trettel, 2015), but the specific inflammatory factors and their interaction networks mediating stroke risk have remained incompletely defined (Catanese et al., 2017).
Oxygen-related stress and inflammatory cytokines and CKs also are functionally connected through the phenomenon of ischemic conditioning, in particular remote ischemic conditioning (RIC), also often called remote ischemic preconditioning (RIPC), ischemic preconditioning (IPC), or hypoxic preconditioning (HPC). In this article, for reasons of simplicity, we refer to it as RIC. RIC is a non-invasive and practical method that involves several cycles of inflation and deflation of a blood pressure cuff on an arm or a single short hypoxic exposure prior to a myocardial infarction or to an ischemic stroke. Although large-scale multicenter clinical trials examining the efficacy of RIC in ischemic heart disease (IHD) have come out neutral (Hausenloy et al., 2015; Meybohm et al., 2015) and multicenter randomized RIC trials in patients with acute stroke are still ongoing (England et al., 2019; Blauenfeldt et al., 2020), RIC has been shown to stimulate endogenous protective pathways in various “remote” organs including heart and brain, and is considered a promising new paradigm in neuroprotection. The precise mechanism(s) by which the protective signal(s) is transmitted from the periphery to the brain is unclear, but next to nerve fibers and circulating immune cells, soluble inflammatory factors including oxidants, CKs, and the cytokine and growth factor erythropoietin have been suggested to play a role (Brines and Cerami, 2005; Weber, 2010; Stowe et al., 2012; Hess et al., 2015; Selvaraj et al., 2017). Similarly, RIC-initiated signal transmission to the heart in IHD has been suggested to involve classical cytokines and CKs, macrophage migration inhibitory factor (MIF), or erythropoietin (Brines and Cerami, 2005; Weber, 2010; Davidson et al., 2013; Bernhagen, 2019b; Bromage et al., 2019; Ruze et al., 2019; Stoppe et al., 2019), but their suitability as RIC cues with translational potential has yet to be established. In fact, despite its simplicity and feasibility, RIC protocols have been regarded as rather crude, leading to a broad activation of numerous factors and potential unfavorable side effects due to the ischemia per se. Thus, more specific molecular strategies, i.e. targeting cytokines or CKs, which mimic RIC protection, might bear a potential to “replace” RIC.
In addition to classical CKs (Bachelerie et al., 2014), atypical chemokines (ACKs) have more recently been shown to significantly expand the chemokine/chemokine receptor (CKR) network. ACKs do not contain a classifying N-terminal cysteine motif and a chemokine fold, but yet mimic certain structural CK elements, signal through CKRs, and exhibit CK-like activities (Oppenheim et al., 2005; Oppenheim and Yang, 2005; Schober et al., 2008; Chan et al., 2012; Pawig et al., 2015; Kapurniotu et al., 2019). Interestingly, several ACKs have been identified to exert critical effects on neuroinflammation and ischemic stroke. One prototypical ACK is the inflammatory cytokine MIF as well as its recently discovered homolog D-dopachrome tautomerase (D-DT)/MIF-2. MIF is a pivotal mediator of acute and chronic inflammatory diseases, autoimmunity, and cardiovascular pathogenesis including atherosclerosis and heart failure. It also has a dual – phase-dependent – role in cardiac ischemia and myocardial ischemia/reperfusion injury (Calandra and Roger, 2003; Tilstam et al., 2017; Kang and Bucala, 2019; Sinitski et al., 2019). MIF is up-regulated in atherosclerotic lesions and promotes atherogenesis by a variety of mechanisms, most importantly by orchestrating the atherogenic recruitment of leukocytes through non-cognate interaction with the chemokine receptors CXCR2 and CXCR4 (Burger-Kentischer et al., 2002; Bernhagen et al., 2007; Sinitski et al., 2019). Cardioprotective effects of MIF in cardiac ischemia and in the early phase of reperfusion are mediated by MIF signaling through its cognate receptor CD74/invariant chain and MIF-based redox effects (Miller et al., 2008; Luedike et al., 2012; Tilstam et al., 2017). An increasing number of studies also suggest that MIF may have a role in ischemic stroke, but whether this contributes to stroke exacerbation and the stroke-associated inflammatory response or may display protective activity has remained controversial.
Here, we discuss the controversial findings on the role of MIF proteins in ischemic stroke and more generally focus on classical CKs and the emerging role of ACKs in ischemic stroke. Moreover, the implicated mechanisms will also be discussed in the context of their potential role as (pre-)conditioning factors.
The MIF protein family
MIF and its receptors
MIF is an evolutionarily conserved 12.5 kDa protein containing 114 amino acids (David, 1966; Weiser et al., 1989; Bernhagen et al., 1993; Calandra and Roger, 2003; Michelet et al., 2019). As one of the first cytokines to be discovered over half a century ago, it was initially described as a lymphocyte-derived soluble immune factor that inhibited the random migration of macrophages out of capillary tubes (David, 1966). MIF has been shown to be broadly expressed not only in lymphocytes but fairly ubiquitously in a variety of other cell types (Asare et al., 2013b), including monocytes, macrophages, cardiomyocytes, fibroblasts, endothelial cells, neuroendocrine cells, neurons, glial cells, and tumor cells (Bernhagen et al., 1993; Mitchell and Bucala, 2000; Calandra and Roger, 2003; Schober et al., 2008; Zernecke et al., 2008; Asare et al., 2013a; Tillmann et al., 2013; Leyton-Jaimes et al., 2018; Bernhagen, 2019b; Kang and Bucala, 2019; Kapurniotu et al., 2019; Noe and Mitchell, 2020). Over the years, MIF has been redefined from a lymphokine and endocrine factor to a pleiotropic inflammatory cytokine and upstream regulator of innate immunity with unique structural properties, which functions as a prototypical ACK (Kang and Bucala, 2019; Kapurniotu et al., 2019; Sinitski et al., 2019). Some of these properties are shared by D-DT/MIF-2 (Merk et al., 2012). Moreover, MIF has emerged as a critical player in various cardiovascular diseases (CVDs), ranging from atherosclerosis and heart failure to acute vascular events such as acute myocardial infarction (AMI) and abdominal aortic aneurysm (Schober et al., 2008; Zernecke et al., 2008; Tilstam et al., 2017; Sinitski et al., 2019).
MIF effects are mediated by high-affinity binding to its receptors CD74 (also known as the invariant chain of major histocompatibility complex (MHC) class II) and the CXC CK receptors CXCR2, CXCR4, and CXCR7 (Leng et al., 2003; Bernhagen et al., 2007; Alampour-Rajabi et al., 2015). Dependent on the cell, microenvironment, tissue, or disease context, MIF engages one or more of these four receptors, which also have been suggested to form heteromeric receptor complexes (Leng et al., 2003; Shi et al., 2006; Bernhagen et al., 2007; Alampour-Rajabi et al., 2015). Moreover, CD74 recruits the accessory molecule CD44 for signal transduction (Shi et al., 2006). The biochemical and structural mechanisms underlying the interaction between MIF and its receptors have been discussed in a number of excellent recent review articles (Leng and Bucala, 2006; Tillmann et al., 2013; Pawig et al., 2015; Kapurniotu et al., 2019). MIF does not share classifying structural homology with CXC CKs or any other cytokine class, but, owing to its evolutionary conservation, shares remarkable architectural homology with bacterial tautomerases/isomerases and exhibits catalytic activity (Michelet et al., 2019). However, the relevance of this activity in mammalian systems has been questioned; instead, it appears that inhibition of the MIF tautomerase/isomerase catalytic site by small molecule inhibitors induces a conformational change that leads to an attenuation of MIF binding to CD74 and CXCR4 (Fingerle-Rowson et al., 2009; Rajasekaran et al., 2016; Pantouris et al., 2015).
The binding of MIF to its receptors induces several signaling pathways that regulate numerous physiological and pathophysiological processes. Briefly, MIF mediates the activation and migration of monocytes, neutrophils, and T cells by binding to CXCR2 and CXCR4 in inflammation, infection, cancer, and cardiovascular pathogenesis (Bernhagen et al., 2007; Cho et al., 2010; Dessein et al., 2010; Hsieh et al., 2014; Jager et al., 2020; Kontos et al., 2020; Perpina-Viciano et al., 2020; Rodrigues et al., 2020). The MIF/CD74 axis has been implicated in autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus (SLE) as well as in atherogenic B cell migration and survival (Morand et al., 2006; Zernecke et al., 2008; Lapter et al., 2011; Klasen et al., 2014; Schmitz et al., 2018; Kang and Bucala, 2019; Shin et al., 2019). In addition, MIF and its receptor CD74 are also involved in tumorigenesis inducing tumor cell proliferation, enhancing tumor angiogenesis and modulating tumor immunity (Chesney and Mitchell, 2015; O'Reilly et al., 2016). The MIF/CXCR7 axis mediates lymphocyte chemotaxis, platelet apoptosis, and tumor cell activation (Tarnowski et al., 2010; Chatterjee et al., 2014; Alampour-Rajabi et al., 2015). Intriguingly, by binding to CD74 and activation of the AMP kinase pathway, MIF conveys cardioprotection in the ischemic heart and protects the liver from steatohepatitis (Miller et al., 2008; Luedike et al., 2012; Heinrichs et al., 2014; Tilstam et al., 2017).
D-dopachrome tautomerase (D-DT)/MIF-2
D-DT, also known as macrophage migration inhibitory factor-2 (MIF-2), is a member of the MIF cytokine superfamily that shows a close structural and amino acid sequence similarity to MIF (Merk et al., 2011). Both, the Mif and D-dt/Mif-2 genes are located in close proximity on chromosome 22q11.23 and show a conserved intron-exon structure, and their coding regions are highly homologous (Merk et al., 2012). CD74 also serves as the cognate receptor for MIF-2 (Merk et al., 2011), but whether MIF-2 also binds to MIF chemokine receptors is unclear. MIF has a tenfold higher tautomerase activity than MIF-2 and also appears to be a more potent activator of CD74, consistent with the notion that its Koff is three times lower and its Kon 11 times lower than that of MIF-2 (Merk et al., 2011). The pathophysiological effects of MIF-2, as far as they have been addressed, partly overlap with those of MIF, e.g. in endotoxemia and sepsis (Merk et al., 2011), but clearly distinct activities have also been reported. For example, while MIF-2 is also involved in obesity, adipogenic inflammation, and wound repair, its effects appear to be opposite to those of MIF (Iwata et al., 2012; Kim et al., 2015; Kim et al., 2017; Kim et al., 2020). In myocardial ischemia/reperfusion injury and heart failure, similar or opposite effects for MIF and MIF-2 have been noted, dependent on the model or species studied (Qi et al., 2014; Stoppe et al., 2015; Ma et al., 2019; Voss et al., 2019). The discovery of 4-CPPC, a novel and reversible small molecule inhibitor of MIF-2 that shows 13-fold selectivity for MIF-2 over MIF (Pantouris et al., 2018; Tilstam et al., 2019), could help to better understand the distinct activities that MIF and MIF-2 have in various CVDs (Bernhagen, 2019a).
Intracellular activities of MIF
Another intriguing aspect of the ACK features of MIF is its emerging role as an intracellular regulator of immunity and cellular function. Intracellular cytokine activities are not a unique property of MIF proteins, but only very few other examples, including the interleukin (IL)-1 cytokines IL-33 and IL-37 are known (Martin and Martin, 2016). The Mif gene does not encode an N-terminal signal sequence (Calandra and Roger, 2003). Accordingly, MIF secretion does not follow the conventional endoplasmic-reticulum (ER)/Golgi pathway; instead translated MIF is deposited in intracellular - cytosolic - pools. It is increasingly being recognized that this pool does not only represent a storage compartment enabling rapid release of MIF through the unconventional protein secretion (UPS) pathway (Flieger et al., 2003; Hoffmann and Bernhagen, 2020), but represents “active” MIF protein that modulates cell homeostasis and cellular stress responses. For example, cytosolic MIF interacts with COP9 signalosome (CSN) subunit 5 (CSN5/JAB1) to modulate CSN signaling and apoptotic processes via p53 (Kleemann et al., 2000; Lue et al., 2007). Intracellular MIF also is involved in cellular redox homeostasis via interactions with the thiol-protein oxidoreductases (TPORs) thioredoxin and peroxiredoxin and mutant forms of superoxide dismutase (Thiele and Bernhagen, 2005; Koga et al., 2011; Israelson et al., 2015). Of note, these activities are suggested to be important in cardioprotection and neuroprotection mediated by MIF. The intracellular activities of MIF have been comprehensively discussed in a recent review article (Kapurniotu et al., 2019). The surprising discovery of nuclear MIF and its role in ischemic stroke will be discussed below.
Classical and atypical chemokines in ischemic stroke
Ischemic stroke and inflammation
The inflammatory response during ischemic stroke pathogenesis is induced within a few hours after the ischemic insult and is generally associated with all stages of the ischemic and post-ischemic cascade, including activation of a rapid microglia and astrocyte inflammatory response, endothelial cell inflammation, and the infiltration of peripheral immune cells including platelets and neutrophils into the ischemic lesion, in conjunction with an enhanced disruption of the blood brain barrier (BBB). All these brain cell types, infiltrating immune cells and damaged neuronal cells contribute to the production and release of inflammatory cytokines, CKs, and ACKs/alarmins. Neuroinflammation then exacerbates brain damage, which also encompasses reperfusion injury, at all ensuing stages over days and weeks, while reparative functions successively kick-in overlappingly at later phases.
Classical chemokines in ischemic stroke
Classical chemokines are small chemotactic cytokines with a classifying chemotactic activity for leukocytes, but also other cell types. They are well known drivers of inflammation in several diseases including ischemic stroke (Ransohoff and Trettel, 2015). There are almost 50 classical human CKs that interact with 20 classical CK receptors, as well as some ACK receptors. Based on the positioning of their N-terminal cysteine residues, CKs are classified into four subfamilies based on their structures: C-, CC-, CXC- and CX3C-motif CKs, which may have unique or overlapping receptor specificities, with many of them exhibiting promiscuous features (Bachelerie et al., 2014; Hughes and Nibbs, 2018). While, so far, no study has addressed the role of C-motif CKs in stroke, all other classical CK subclasses have been suggested to be involved in the pathogenesis of ischemic stroke.
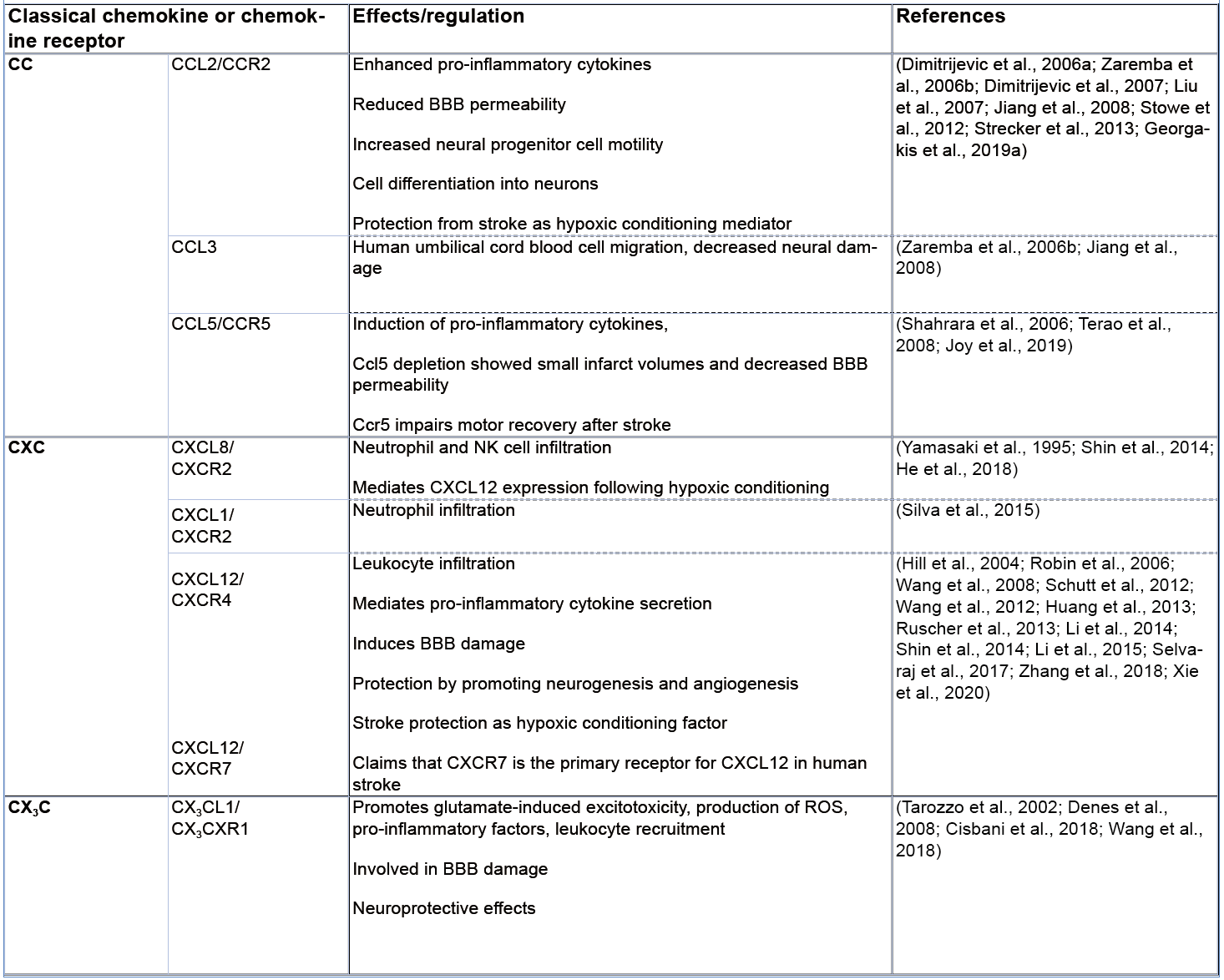
The relevance of CC-motif CKs in the pathogenesis of ischemic stroke is supported by numerous studies showing the involvement of several of its members including CCL2, CCL4, CCL5, CCL7, CCL20, and CCL21, and their corresponding receptors, in this disease (Tatara et al., 2009; Tokami et al., 2013; Joy et al., 2019). CCL2, also known as monocyte chemotactic protein-1 (MCP-1) and a cognate ligand of the CCR2 receptor, is considered one of the most prominent examples, highlighting a role of CC-family CKs in ischemic stroke. The expression of CCL2 is upregulated in neurons as well as in astrocytes after 12 hours and 2 days of ischemia, respectively, leading to the activation of inflammatory processes in the early stage of ischemic stroke (Che et al., 2001). Applying antisense oligonucleotides or neutralizing antibodies to block CCL2, Dimitrijevic and colleagues (2006a, 2007) showed that the CCL2/CCR2 axis has an important role in regulating BBB permeability in ischemic/reperfusion injury after stroke, while a causal role of the CCL2/CCR2 axis in this condition was further underscored by studies in Ccr2 gene knockout mice (Dimitrijevic et al., 2006a, 2007). This study also showed that Ccr2-deficiency attenuated damage by reducing brain edema, leukocyte infiltration, and inflammatory mediator expression (Dimitrijevic et al., 2007). Targeting CCL2 or its receptor CCR2 also resulted in a decrease in monocyte infiltration, decreased levels of pro-inflammatory cytokines, and reduced BBB permeability (Strecker et al., 2013). On the other hand, Liu and colleagues (2007) showed that CCL2 promotes neural progenitor cell motility and cell differentiation into neurons in mice after 7 days of middle cerebral artery occlusion (MCAO), implying a reparative role. Also, CCR2 has been suggested to prevent the transformation of an ischemic infarct into a hemorrhagic lesion by inducing bone marrow-derived monocyte/macrophage recruitment (Gliem et al., 2012). Intriguingly, Stowe et al. (2012) presented evidence for a role of endothelial CCL2 as an ischemia-induced protective preconditioning factor for stroke, which mediated a reduction in CCR2+ monocyte infiltration and infarct size. The emerging evidence from human genetic and clinical studies about a causal link between CCL2 and stroke (Georgakis et al., 2019a; Georgakis et al., 2019b; Georgakis et al., 2020) mainly refers to the role of circulating CCL2 and its more general role in increasing vascular risk and will not be discussed here in detail. Similarly, we will not further cover the important and well documented roles of several other circulating CKs and their receptors in driving atherosclerotic lesion formation in general, and instead refer to the excellent review articles that have comprehensively discussed this aspect (Charo and Ransohoff, 2006; Koenen and Weber, 2010; Weber and Noels, 2011).
CCL3, a common ligand of both CCR1 and CCR5, is elevated in the serum of ischemic stroke patients and was identified in rat brain tissue after cerebral ischemia (Kim et al., 1995; Zaremba et al., 2006b). CCL3 also promotes the recruitment of human umbilical cord blood cells to ischemic lesions resulting in a decrease in neural damage in rodents after MCAO (Jiang et al., 2008). In contrast, CCL5, which selectively binds three different CC-motif CK receptors, i.e. CCR1, CCR3 and CCR5, increases cerebral damage through induction of pro-inflammatory cytokines (Shahrara et al., 2006). In line, Ccl5-deficient mice were protected from ischemic stroke having smaller infarct volumes and decreased BBB permeability in the focal cerebral ischemia compared to control mice (Terao et al., 2008). Notably, a substantial upregulation of CCL5 is observed in the plasma of ischemic stroke patients as well as in murine neurons and plasma of mice after MCAO, but appears to have a neuroprotective role, resulting in an enhanced production of different neurotrophic factors in peri-infarct areas (Tokami et al., 2013). In contrast, Joy et al. (2019) identified the CCL5 receptor CCR5 as a potential therapeutic target for stroke recovery. They found that CCR5 was differentially upregulated in neurons after stroke, that deficiency of CCR5 induced motor neuron recovery and cognition after injury, and demonstrated that the FDA-approved drug maraviroc, a small molecule CCR5 inhibitor, induced recovery after stroke, demonstrating the principal utility of a therapeutic anti-CK axis strategy for stroke.
Several CXC-motif CK have also been studied in the context of ischemic stroke. CXCL8, a member of the glutamate-leucine-arginine (ELR)+ CXC-motif CK family, is known as a potent chemoattractant for the recruitment of neutrophils. In the context of ischemic brain injury, CXCL8 levels in both blood and brain strongly correlate with brain edema and cerebral neutrophil infiltration (Yamasaki et al., 1995). Blockade of the CXCL8 receptor CXCR2 with reparixin impairs neutrophil infiltration and leads to reduced infarct size in transient as well as permanent MCAO rat models (Villa et al., 2007). Similarly, CXCL1 (also called KC), an alternate CXCR2 ligand in humans and the only CXCR2 CK in mice, is mainly released during the acute stage of ischemia (Ormstad et al., 2011) and drives neutrophil infiltration into the ischemic lesions, leading to an excessive inflammatory reaction as well as neuronal cell death (Silva et al., 2015). The ELR- CXC-motif CK CXCL12 is produced by glial cells and neurons, is elevated in the serum of ischemic patients, and is associated with stroke severity. Huang et al. (2013) reported that inhibition of the CXCL12/CXCR4 axis attenuated leukocyte infiltration, secretion of pro-inflammatory cytokines, and BBB damage in the acute phase of stroke in MCAO models. In contrast, another study found that the CXCL12/CXCR4 axis promoted neurovascular recovery by strengthening neurogenesis and angiogenesis during the post-acute phase of ischemia (Li et al., 2014). Indeed, an increasing number of studies provide evidence that CXCL12 may exhibit both pro- and anti-inflammatory, i.e. exacerbating or regenerative, functions in ischemic stroke, dependent on the context, and may serve as a protective ischemic conditioning mediator (Robin et al., 2006; Wang et al., 2008; Wang et al., 2012; Li et al., 2015; Selvaraj et al., 2017; Xie et al., 2020). Additionally, other CXC-motif CKs such as CXCL5 and CXCL10, showed a correlation with poor clinical outcome in ischemic stroke patients (Zaremba et al., 2006a; Amin et al., 2017).
In the CX3C-motif sub-family, CX3C ligand1 (CX3CL1) is one of the widely studied CKs in ischemic stroke, partly because of the established role of its receptor CX3CR1 as a signature receptor for microglial cells and perivascular macrophages (Hanisch and Kettenmann, 2007). Both genetic depletion of Cx3cl1 or its receptor Cx3cr1 was associated with a reduction of harmful ischemic consequences, including excitotoxicity, production of ROS, inflammatory factor release, leukocyte recruitment and BBB damage after MCAO (Denes et al., 2008). Using a ferric chloride model of focal cerebral ischemia, Cisbani and colleagues showed that the genetic deletion of Cx3cr1 (as well as that of Ccr2) had a neuroprotective effect, while another study indicated that neuronal CX3CR1 mediates neuronal apoptotic cell death in ischemia, suggesting a role of the CX3CL1/CX3CR1 axis in both microglia and neurons (Cisbani et al., 2018; Wang et al., 2018). However, two studies also reported that the signaling mediated by the CX3CL1/CX3CR1 axis has neuroprotective effects in experimental ischemic stroke (Pimentel-Coelho et al., 2013) and correlates with an improvement of clinical outcome in ischemic patients (Donohue et al., 2012).
Taken together, classical CKs and their receptors exhibit both exacerbating and protective effects in stroke pathogenesis. Pathogenic activities are primarily linked to their ability to recruit immune cells and activate resident microglia (Vidale et al., 2017). Besides chemotaxis, CKs were demonstrated to be directly involved in BBB disruption (Dimitrijevic et al., 2006). Conversely, several CKs (CXCL12 maybe being the most prominent representative) also showed beneficial effects, e.g. related to the clearance of debris, progenitor cell activation, and post-stroke recovery (Le Thuc et al., 2015), but also in the context of RIC (Selvaraj et al., 2017).
Atypical chemokines in ischemic stroke
In addition to classical CKs, ACKs and alarmins have more recently been identified to exert an important impact on ischemic stroke as well. ACKs, sometimes also referred to as “innate CKs” or “CK-like function (CLF) CKs”, are a structurally diverse class of small 8-25 kDa multifunctional proteins that share a number of joint properties with danger-associated molecular patterns (DAMPs)/alarmins, as recently reviewed in detail by us (Kapurniotu et al., 2019). Compared to the four classical CK sub-categories, ACKs lack the typical CK-fold and the classifying N-terminal cysteine residues, but share bona fide chemotactic activity with classical CKs and bind to classical CK receptors. This activity is believed to be mediated by mimicry of certain conformational and surface charge motifs that ACKs share with classical CKs (Hoover et al., 2002; Weber et al., 2008; Kraemer et al., 2011; Kapurniotu et al., 2019). Typical ACKs are also often semi-constitutively expressed intracellular proteins that can be secreted to adopt additional extracellular functions as a cytokine/CK upon inflammatory or stress stimulation. Despite the absence of classifying structural homology with classical CKs, ACKs may engage in high-affinity interactions with classical CK receptors to drive inflammatory processes and cell migration.
High-mobility group box protein 1 (HMGB1) is a prototypical DAMP/alarmin due to its dual role as an abundant DNA-binding chromatin component and as an extracellular inflammatory mediator (Wang et al., 1999; Czura et al., 2001; Andersson et al., 2002; Yang et al., 2005; Bianchi, 2007; Yang et al., 2013; Venereau et al., 2015; Bianchi et al., 2017). HMGB1 also qualifies as an ACK due to its hetero-oligomerization with CXCL12 (Schiraldi et al., 2012; De Leo et al., 2019). Upon ischemic injury in stroke, neuronal HMGB1 is abundantly released into the extracellular space, both by active secretion and passive release, to accumulate in the ischemic core of the acute phase and to activate resident brain immune cells, leading to the recruitment of mononuclear cells that further amplify inflammation (Singh et al., 2016). The receptors of HMGB1 comprise the receptor for advanced glycation end (RAGE), Toll-like receptor 2 (TLR2) and 4 (TLR4), which are expressed in the brain (Carty and Bowie, 2011), and were independently found to mediate HMGB1-mediated neurite outgrowth in the rat embryonic nervous system (Hori et al., 1995), but also to contribute to brain damage and neurological deficits caused by stroke (Tang et al., 2007). In addition, the release of HMGB1 acts as a modulator that induces pro-inflammatory cytokine expression such as IL-1β and inducible nitric oxide synthase (iNOS) through toll-like receptor 4 (TLR4) (Yang et al., 2010). The HMGB1-induced recruitment of inflammatory cells to injured tissue has been shown to be driven by HMGB1/CXCL12 heterodimers via CXCR4 (Schiraldi et al., 2012). HMGB1-mediated signals have been suggested to be biphasic during stroke (Hayakawa et al., 2010). Besides the release of HMGB1 in the acute phase of stroke, secretion of HMGB1 is also observed in the delayed-recovery phase of stroke (Hayakawa et al., 2010) and plays a role in post-stroke inflammation and atherosclerosis (Liesz et al., 2015; Roth et al., 2018).
Human beta-defensin 2 (HBD2), a human antimicrobial peptide, is an ACK member with chemotactic activity through non-cognate binding to CC-motif CK receptor CCR6 and is highly elevated in the sera of stroke patients (García-Berrocoso et al., 2014). In the brain, HBD2 is mainly expressed in capillary endothelial cells and astrocytes. It is induced by infectious microorganisms including Pseudomonas aeruginosa as well as cytokines such as TNF-α, or hypoxic stimulation (Nickel et al., 2012). The copy number variant of the HBD2 gene variant DFEB4 and circulating protein concentrations have been investigated in stroke patients (Tiszlavicz et al., 2012). Although the number of studies on HBD2 in stroke is limited, they may suggest that it may be a potential biomarker in the acute phase of stroke for prediction of stroke outcome.
Similarly, thioredoxin (Trx), a small cellular protein (12 kDa) known to play a critical role in maintaining cellular redox homeostasis, that also has CK-like properties (Bertini et al., 1999; Tamaki et al., 2006; Nakamura, 2008; Nakamura et al., 2009; Koga et al., 2011), was recently designated as a new potential diagnostic and prognostic marker of ischemic stroke (Oraby and Rabie, 2020). The components of the Trx system comprise NADPH and thioredoxin reductase (Trx-R), and Trx is regenerated by Trx-R at the expense of reducing NADPH (Holmgren, 1985). In line with thioredoxin’s role as extracellular mediator and chemoattractant (and thus ACK-like protein), Hattori et al. (2004) demonstrated that the administration of recombinant Trx in mice showed protective effects on cerebral ischemia reperfusion. Peroxiredoxins (Prxs) are a large and highly conserved family of peroxidases reducing substrates with the help of glutathione and helping cells against oxidative stress (Rhee, 2016), and, like Trx, belong to the family of thiol-protein oxidoreductases (TPORs) with a signature CXXC redox-active motif (Ahsan et al., 2009). Peroxiredoxins were demonstrated to be released from ischemic neuronal cells into the extracellular space and to contribute to pathogenic cytokine responses (Garcia-Bonilla and Iadecola, 2012; Shichita et al., 2012). While intracellular Prxs have been suggested to exhibit protective effects in ischemically stressed neurons (Rashidian et al., 2009), extracellular released Prx family of proteins appears to primarily behave as DAMPs/ACKs and promote stroke pathogenesis. When released from necrotic brain cells in the ischemic core within 12 h after stroke onset, Prxs proteins trigger the expression of inflammatory cytokines such as IL-23 in macrophages through TLR2 and TLR4 that further promote neuronal cell death (Shichita et al., 2012; Shichita et al., 2017).
The involvement of both classical and ACKs in ischemic stroke as described in the chapter above is summarized in Figure 1, Table 1, and Figure 2.
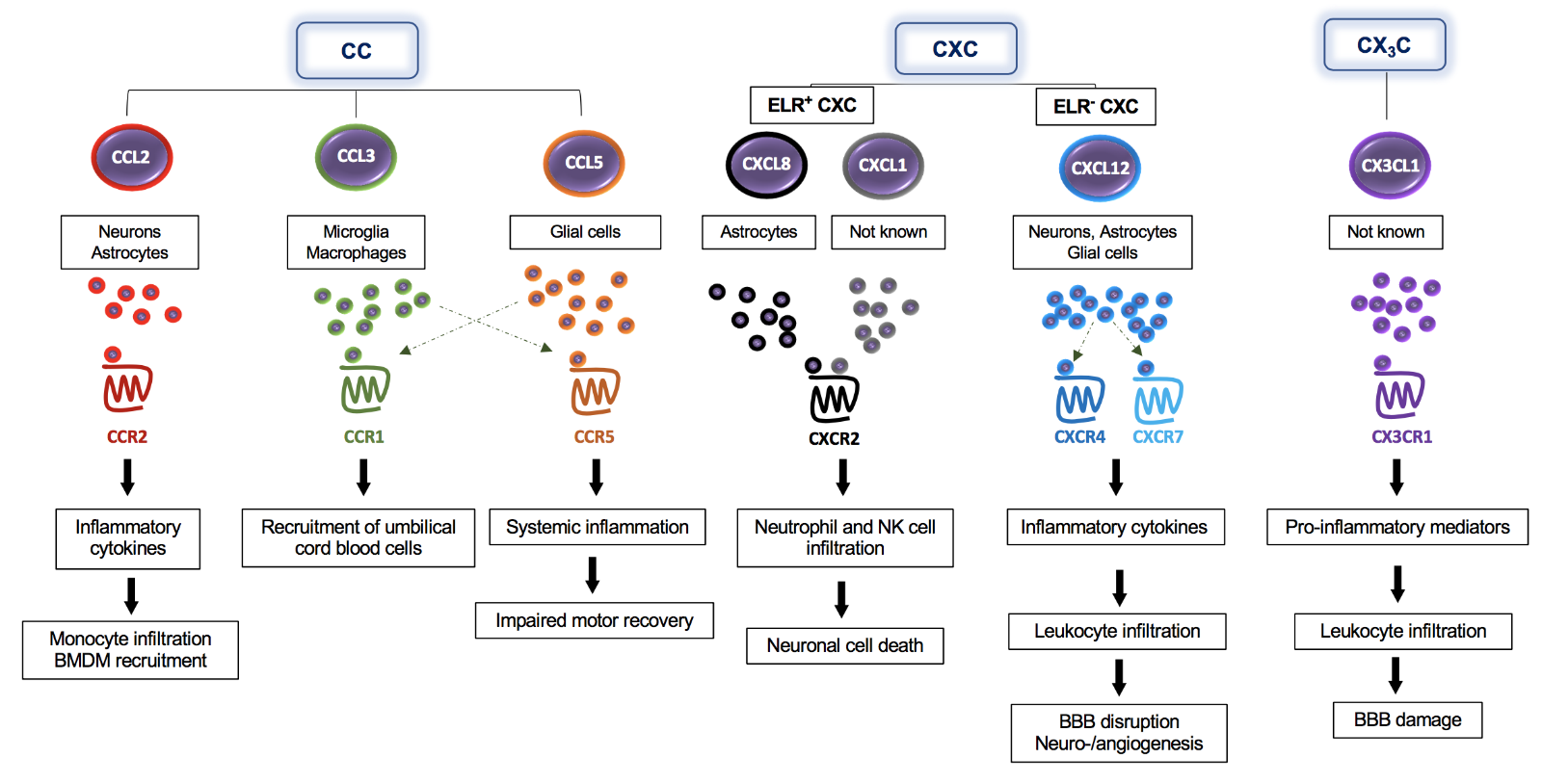
In a new window | Download PPT
Figure 1: The multifaceted impact of classical chemokines on the pathogenesis of ischemic stroke – classes, cells, receptors, and downstream effects. CC-, CXC-, and CX3C-chemokines are expressed by different brain cells including neurons, astrocytes, and microglia, as indicated in the figure. Following their release, these chemokines bind to their specific CC- (CCR2, CCR1, CCR5), CXC- (CXCR2, CXCR4), or CX3C-chemokine receptors (CX3CR1) to promote signaling responses, the secretion of additional inflammatory cytokines/chemokines, and direct recruitment effects including the infiltration of several immune cells into the brain. Chemokines also enhance BBB dysfunction and neuronal cell death.

In a new window | Download PPT
Figure 2: Expression and effects of atypical chemokines (ACKs) in ischemic stroke. The scheme illustrates differential expression and involvement of ACKs, i.e. HMGB1, β-defensin 2 (HBD2), thioredoxin, and peroxiredoxins in ischemic stroke. HMGB1, high-mo¬bi¬lity group box protein-1; IL-1β, interleukin-1β; CXCL12, C-X-C-motif chemokine ligand 12; iNOS, inducible nitric oxide synthase; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor-α.
Table 1: Summary of the involvement of classical chemokines in ischemic stroke.
MIF proteins in ischemic stroke
MIF proteins in brain
MIF is a multifunctional cytokine/CK. While MIF is generally regarded as a pro-inflammatory factor, it may, as discussed above, and depending on the disease context and engaged receptor pathway, exert pro- or anti-inflammatory properties. The role of MIF in ischemic brain diseases including ischemic stroke has only been studied fairly recently. The obtained findings turned out to be complex and in part controversial. In 1998, Bacher et al. (1998) first showed in rat brain that both MIF RNA and protein levels are abundant in different brain regions, namely in cortex, hippocampus, cerebellum, hypothalamus. At the cellular level, MIF was reported to be expressed in neurons, astrocytes, and microglial cells (Bacher et al., 1998; Ogata et al., 1998). Interestingly, MIF is upregulated and appears to be involved in the developing rat brain (Suzuki et al., 1999). Deletion of the Mif gene is associated with more anxiety and depression-like behaviors, as well as worse hippocampus-dependent memory (Conboy et al., 2011). Furthermore, in vitro studies suggested MIF may have a growth-promoting effect on certain neuronal cell populations such as neuronal stem/progenitor cells (Ohta et al., 2012). The physiological role of MIF in the brain could be related to the growth and maturation of brain cells and participation in the maintenance of normal brain functions.
MIF in preclinical ischemic stroke studies
Previous studies performed in rats demonstrated that both protein and mRNA levels of MIF were upregulated in the ischemic brain, particularly in neurons and astrocytes of the ischemic penumbra, as well as in microglia in the infarct core after stroke, correlating with stroke severity (Baugh et al., 2006; Welford et al., 2006; Wang et al., 2009; Li et al., 2016). Of note, the upregulation of MIF under hypoxic conditions is driven by the binding of hypoxia-inducible factor 1-alpha (HIF-1α) to the hypoxia responsive elements (HREs) present in the MIF promoter (Baugh et al., 2006; Welford et al., 2006; Wang et al., 2009). Indeed, MIF promoter activity was significantly upregulated under hypoxia having a protective effect on cortical neurons under oxygen and glucose deprivation (OGD) treatment (Zis et al., 2015). Additionally, microRNA-493 was demonstrated to regulate angiogenesis under ischemic conditions through its ability to directly modulate the expression of MIF, suggesting its involvement in ischemic stroke (Li et al., 2016). There are an increasing number of animal studies describing the in vitro and in vivo relevance of MIF in the pathogenesis of stroke, but the findings are divergent and controversial. In line with an assumed inflammatory capacity of MIF, Inácio et al. (2011c) found that Mif deficiency was neuroprotective in a transient MCAO model of ischemic stroke and was associated with higher galectin-3 immunoreactivity in the ischemic brain hemisphere and a better functional outcome after transient MCAO. MIF accumulated in neurons of the peri-infarct region and in cultured cortical neurons exposed to OGD, and inhibition of MIF by pharmacological blockade with the small molecule tautomerase inhibitor ISO-1 protected OGD-stressed neurons against cell death. Curiously, gene deletion of Mif did not affect brain or serum levels of the inflammatory cytokine interleukin (IL)-1β. This is surprising because both extra- and intracellular MIF activity was recently linked with NLRP3 inflammasome activation (Lang et al., 2018; Shin et al., 2019; Dankers et al., 2020; Hoffmann and Bernhagen, 2020). The disconnect between Mif deletion and the inflammatory cytokine status in brain after MCAO was confirmed in additional studies by the same group, showing that Mif deletion did not alter the levels of IL-2, interferon (IFN)-γ, IL-1β, and TNF-α, suggesting an alternative, perhaps intra- or inter-neuronal, mechanism for MIF in this model (Inacio et al., 2011b; Inacio et al., 2011c). To this end, experiments combining an “enriched environment approach” with an MCAO insult showed a downregulation of MIF protein levels in the penumbra, suggesting that MIF could be part of a signaling network involved in brain plasticity, wherein increased neuronal and/or astrocytic MIF levels after stroke could repress the recovery of sensory-motor function after stroke (Inacio et al., 2011a). In contrast, Zhang et al. (2014) noticed a significant MIF downregulation following activation of the NF-κB pathway under hypoxic conditions in neuronal cultures in vitro. Similarly, Mif gene disruption was associated with caspase-3 activation resulting in neuronal loss and increased infarct size after ischemic stroke, indicating a potential overall neuroprotective effect of MIF. Although most data in this study was performed in vitro, the author concluded that MIF could be important for preserving a longer time window for stroke treatment, and that strategies to maintain MIF expression at physiological levels could be beneficial for stroke patients. However, this hypothesis has not yet been followed up on. Interestingly, Turtzo et al. (2013) suggested that the impact of Mif deficiency on ischemic stroke is gender-dependent, and that genetic ablation of Mif resulted in microglial activation and worse stroke outcomes in females but not males. Remarkably, these effects were independent of estrogen levels and pro-inflammatory cytokines profiles in both genders. One other mechanism related to the involvement of MIF in ischemic stroke could be related to BBB integrity. Treatment of adult rat brain endothelial cells with recombinant MIF enhanced the disruption of tight junctions without affecting the viability of primary neuronal cells under OGD/reoxygenation stress (Liu et al., 2018). Consistently, administration of MIF to mice following ischemic stroke disrupted tight junctions of the BBB and increased infarct volumes in vivo, which could be reversed by a MIF antagonist (Liu et al., 2018). The involvement of intracellular neuronal MIF in ischemic stroke pathogenesis was studied by Wang et al. (2016). In a search for novel interactors of the protein apoptosis-inducing factor (AIF) in neuronal lysates, they identified MIF and found that following AIF-mediated nuclear import, it functions as a nuclease that enhances DNA cleavage and neuronal cell death. The nuclease activity of MIF was shown to be strictly dependent on the activation of poly-ADP-ribose-polymerase-1 (PARP-1), nuclear-derived PAR trees, the release of AIF from mitochondria, and its translocation together with MIF into the nucleus. The study links MIF to oxidative neuronal stress, glutamate excitotoxicity, and DNA damage. The mechanism has received wide attention in the field, but the structural nuclease motif of MIF has been questioned (bioRxiv preprint doi: https://doi.org/10.1101/085258) and the MIF nuclease and parthanatos activity has not yet been independently recapitulated by other studies.
MIF in clinical stroke studies
Besides the above discussed experimental studies, several clinical studies have addressed correlative relationships between MIF levels in peripheral blood and the pathological prognosis and severity in stroke patients. Wang et al. (2009) reported that both MIF protein plasma levels and its mRNA expression in peripheral blood mononuclear cells (PBMCs) were upregulated in ischemic stroke patients, correlating with the severity of ischemic damage based on the National Institute of Health Stroke Scale score. An increase in circulating MIF levels occurred in the acute phase (early stage, day 3) and decreased to normal levels towards a later stage (10 to 14 days after stroke) (Wang et al., 2009; Li et al., 2017; Yang et al., 2017; Wang et al., 2019). Li et al. (2017) observed a positive correlation between infarct size and MIF serum levels in ischemic stroke patients. The same authors also compared MIF levels in different stroke subtypes including large-artery atheroschlerosis and small artery occlusions and found an upregulation of MIF in plasma despite a diversity in etiology and pathophysiology of both subtypes (Li et al., 2017). Another study reported on an increase in MIF levels in plasma at admission, which were associated with higher risk of post-stroke depression during the following three months (Fang et al., 2018). Examining brain sections with an antibody against MIF, pronounced MIF immunoreactivity was detected in endothelial cells in the penumbra of 10 human ischemic stroke patients compared to control brain sections (Zis et al., 2015). Similar to MIF, the level of MIF receptor CD74 in PBMCs also increased and this peripheral upregulation is associated with a bigger infarct volume and worse neurological outcome of ischemic stroke (Yang et al., 2017).
Despite these various observations, the mechanisms underlying the upregulation of peripheral MIF levels in ischemic stroke patients remain unknown. Nevertheless, the aforementioned clinical studies insinuate that MIF may have utility as a potential biomarker and therapeutic candidate that may predict the severity and outcome of ischemic stroke. However, clinical studies with significantly larger cohorts will be needed to further substantiate this notion.
The MIF receptors in ischemic stroke
The recruitment and infiltration of circulating leukocytes into the brain following an ischemic stroke is orchestrated by the upregulation of various CKs and their receptors (Tarozzo et al., 2002; Gelderblom et al., 2009). MIF and its receptors CXCR2, CXCR4, and CXCR7, as well as its cognate receptor CD74 have been shown to promote the recruitment of inflammatory leukocytes in various settings of CVDs (Bernhagen et al., 2007; Sun et al., 2010; Gao et al., 2011; Klasen et al., 2014; Alampour-Rajabi et al., 2015; Schmitz et al., 2018). While the role of the MIF/receptor axes have not yet been specifically addressed in the context of leukocyte recruitment into the brain following an ischemic stroke, the same receptors have been studied in the context of their cognate CK ligands, i.e. CXCL1/8 and CXCL12, and have been noted to generally participate in cytokine- and CK-mediated inflammation in ischemic stroke (Schioppa et al., 2003; Schönemeier et al., 2008; Connell et al., 2015) as outlined in more detail below.
CXCR2 was found to be upregulated in brain (Brait et al., 2011) and in peripheral leukocytes (He et al., 2018) following ischemic stroke. Similarly, the concentrations of CXCR2 ligands CXCL1, CXCL2, and CXCL8 were found to be elevated in brain, peripheral blood, and cerebrospinal fluid during ischemic stroke (Kostulas et al., 1999; Chapman et al., 2009; Brait et al., 2011). However, the precise mechanism(s) and direct interplay between CXCR2 and its ligands and the relation to the severity of ischemic stroke remains poorly studied and partially controversial. Interestingly, Montaner et al. (2003) and Pedersen et al. (2004) noted no changes in circulating CXCL8 in the plasma of stroke patients. Yet, the dysregulation of CXCR2 and its ligands correlates with worse outcomes after ischemic stroke (Mirabelli-Badenier et al., 2011; He et al., 2018) and may play a crucial role in the infiltration of neutrophils during ischemic stroke (Villa et al., 2007; Herz et al., 2015). Besides neutrophils, CXCR2 is an important receptor for the recruitment of natural killer (NK) cells in ischemic stroke (He et al., 2018). Of note, targeting CXCR2 was associated with decreased neutrophil infiltration and reduced infarct size and thus protection from stroke (Villa et al., 2007; Connell et al., 2015). However, the CXCR2 antagonist SB225002 abrogated neutrophil infiltration but failed to reduce infarct volume or improve outcome after cerebral ischemia-reperfusion in mice (Brait et al., 2011; Copin et al., 2013). Similarly, Frieler et al. (2017) demonstrated that blockade of CXCR2 promotes a partial reduction of neutrophil recruitment to the brain, but this was not paralleled by a reduction in infarct size after MCAO.
In addition to CXCR2, CXCR4 and its cognate ligand CXCL12, also termed stromal cell derived factor-1α (SDF-1α), are widely expressed in the CNS and, among other effects, contribute to neuronal development (Tanabe et al., 1997; Pujol et al., 2005). Circulating CXCL12 levels in blood correlate with the severity and outcome of ischemic stroke, suggesting it may be a potential prognostic marker (Schutt et al., 2012; Huang et al., 2019). Following ischemic stroke, both CXCR4 and CXCL12 are upregulated in the penumbra and found to be associated with the migration and infiltration of numerous cells into the ischemic area including neural stem cells (Imitola et al., 2004), neural progenitor cells (Robin et al., 2006), microglial cells (Lipfert et al., 2013), endothelial progenitor cells (Fan et al., 2010; Bogoslovsky et al., 2011), and bone marrow-derived cells (Hill et al., 2004; Werner et al., 2020). Of note, using a Cre-ER mouse model and lineage tracing, Werner et al. (2020) also showed that CXCR4 distinguishes hematopoietic stem cells-derived monocytes from microglia and that the monocyte immune response is instrumental to experimental stroke. CXCR4 promoted initial monocyte infiltration but then mediated spatial restriction of monocyte-derived macrophages to infarct tissue. While Cxcr4 gene deficiency reduced monocyte infiltration and attenuated the expression of innate defense response genes in monocyte-derived macrophages, an altered microglial response and exacerbated stroke outcome was observed, implying an overall beneficial role of CXCR4 in cerebral ischemia (Werner et al., 2020). Similarly, the CXCL12/CXCR4 axis was also shown to have beneficial effects in stroke recovery by promoting neurogenesis and angiogenesis (Li et al., 2014; Le Thuc et al., 2015). Yet, other studies showed that blocking of CXCL12/CXCR4 signaling protected against BBB leakage, reduced leukocytes and M1 microglia infiltration, and infarct volume, overall improving the functional recovery in an MCAO model (Huang et al., 2013; Ruscher et al., 2013; Wu et al., 2017). Together, these various studies suggest a context- and disease model/stroke phenotype-dependent role of the CXCR4/CXCL12 axis that also is governed by temporal and spatial parameters in the ischemic brain tissue after stroke. It should be noted that most of the above-discussed studies did not take into account CXCR4-based signaling pathways through the non-cognate CXCR4 ligand MIF and have not accounted for scavenger or co-signaling effects of CXCR7.
Indeed CXCR7, also termed ACKR3, is a scavenger and signaling receptor for CXCL12, CXCL11, and MIF (Burns et al., 2006; Bachelerie et al., 2014; Alampour-Rajabi et al., 2015). It is expressed in the brain, specifically in neurons, microglia, and astrocytes (Thelen and Thelen, 2008). Similarly to CXCR4, CXCR7 was also found to be upregulated in the brain during ischemic stroke (Schönemeier et al., 2008). Inhibition of CXCR7 with a neutralizing antibody ameliorated neurogenesis in dentate gyrus and cognitive function post ischemic stroke, suggesting its involvement in disease pathogenesis in chronic ischemia (Dong et al., 2020). Conversely, Bakondi et al. (2011) demonstrated that the CXCL12/CXCR7 axis exhibits a protective effect by promoting neural progenitor cell survival during ischemic stroke. Interestingly, a receptor complex between CXCR7 and CXCR4 is expressed in microglia and was reported to be upregulated following activation enhancing microglia proliferation and migration (Lipfert et al., 2013). Additionally, recent studies demonstrated the importance of signaling pathways mediated by the CXCL12/CXCR4/CXCR7 axis in recruiting stem cells to ischemic lesion sites and in brain repair after ischemic stroke (Guyon, 2014; Cheng et al., 2017; Zhang et al., 2018).
The role of CD74, a common receptor for both MIF and MIF-2, in the pathology of ischemic stroke has been poorly investigated. CD74 expression was described to be increased in the hippocampal Cornu Ammonis 1 region and colocalized with the M1 microglia subtype after ischemic stroke (Hwang et al., 2017). In addition, CD74 was significantly upregulated after exposure to hyperbaric oxygen (HBO) prior to ischemia in a rat model that induced neuroprotection (Hirata et al., 2007). Interestingly, blocking CD74 signaling, most likely elicited by MIF, with the HLA-derived peptide DRα1-MOG-35-55 significantly abrogated T cell infiltration in the brain resulting in significant reduction of infarct size and improved functional outcome (Wang et al., 2017).
Figure 3 summarizes current evidence on MIF and its receptors in the inflammatory response and outcome of ischemic stroke. The receptors are also covered in Figure 1 and Table 1. While these data strongly point towards a role for the MIF/receptor axes in ischemic stroke pathogenesis, the precise mechanisms have remained unclear and most evidence on the MIF CK receptors leave open whether these act in concert with their cognate classical CK ligands, or MIF, or both.
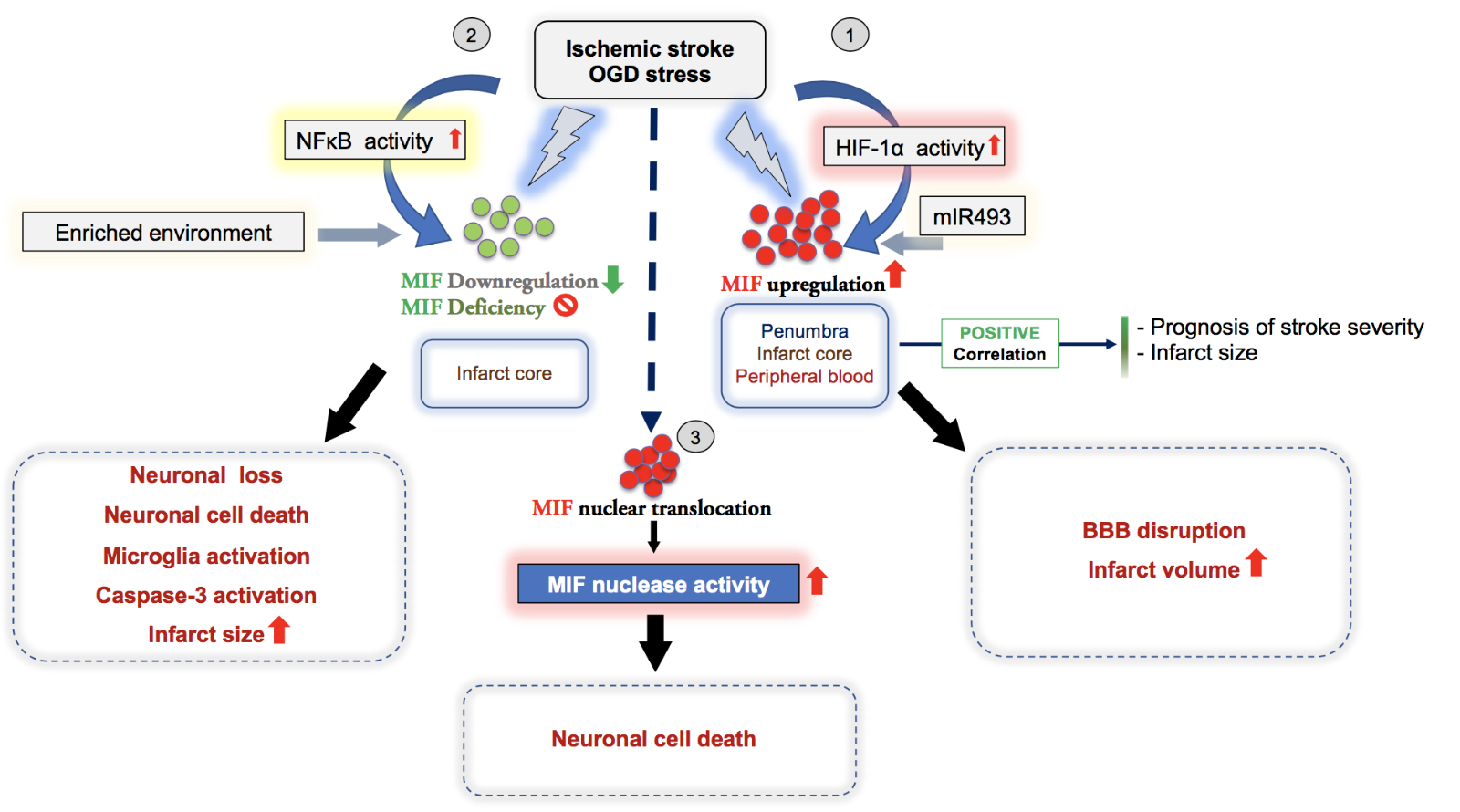
In a new window | Download PPT
Figure 3: Overview illustrating the dichotomic effects and suggested mechanisms of the atypical chemokine MIF in ischemic stroke as evidenced by experimental and correlative clinical studies. In most studies, the expression of MIF in stroke positively correlates with infarct size and the prognosis of stroke severity. (1-3) Depending on the context and stimulus, MIF has a detrimental or protective effect, overall suggesting a dichotomic role in ischemic stroke, although overarching systematic studies are elusive. (1) Following OGD stress or cerebral ischemia, HIF1-α activation drives the upregulation of MIF in brain (penumbra, infarct core), which promotes the disruption of the blood brain barrier (BBB) and an increase of the infarct volume, consistent with a detrimental effect of MIF in ischemic stroke. (2) In contrast, genetic deletion of Mif or MIF downregulation mediated by NFκB signaling activation under ischemic conditions enhances microglia activation, caspase-3 activation, neuronal cell death, and increases infarct size, suggesting a protective effect of MIF in stroke. (3) One study suggests that MIF has a novel intracellular function acting as nuclease promoting parthanatos and increasing ischemic stroke lesions. After its nuclear translocation, the MIF nuclease activity induces DNA damage to promote neuronal cell death.
Concluding remarks, opportunities for “conditioning”, and potential clinical implications
Based on data from a variety of experimental stroke models, gene knockout approaches and blocking strategies, as well as correlative clinical studies, several classical CKs and their receptors have been implicated in the pathogenesis of ischemic stroke. Out of an overall vastly complex network of 49 ligands and 23 receptors, 10-15 classical CKs or their receptors have been more or less extensively explored. Studies have either focused on the ligand or the receptor, but have seldom jointly addressed both the ligand and receptor, let alone the complexity associated with the promiscuity and multiplicity inherent to some ligand/receptor pairs. Studies on some of them, i.e. CCR5, CCL2, CXCR2, and CXCL12, have demonstrated a promising therapeutic potential, at least in preclinical models. For example, irbesartan, a CCR2 antagonist, abrogates signaling pathways mediated by the CCL2/CCR2 axis that promote ischemic brain damage (Tsukuda et al., 2011); the CCR5 inhibitor maraviroc, an FDA-approved drug, induces recovery after stroke and traumatic brain injury (Joy et al., 2019); the dual CXCR1/CXCR2 inhibitor reparixin was associated with protective effects after ischemic stroke in animal models (Garau et al., 2006; Sousa et al., 2013); and AMD3100, a clinically approved antagonist of CXCR4, was reported to exhibit protective effects after ischemic stroke (Huang et al., 2013). Of note, two of them, CXCL12 and CCL2, have so far been directly implicated in RIC (Stowe et al., 2012; Selvaraj et al., 2017).
At the same time, an increasing body of evidence became available on the emerging network of ACKs and DAMPs/alarmins and their potential role in neuroinflammation and ischemic stroke. The most compelling data are available for Prx proteins and HMGB1 (Garcia-Bonilla and Iadecola, 2012). MIF is also a prototypical ACK and exhibits a well-documented role in inflammation, neuroinflammation, atherosclerosis, and cardiac ischemia (Sinitski et al., 2019). However, although its role in ischemic stroke has been studied since 2011, the impact of MIF in the brain and particularly in ischemic stroke has remained unclear, with both exacerbating and beneficial effects observed that at least in part also appeared to be dependent on model, sex, and disease stage. Figure 3 summarizes this seemingly dichotomic involvement of MIF in ischemic stroke, which in part is reminiscent of its role in cardiac ischemia, outlines the currently suggested pathways and mechanisms, including proposed receptor-mediated and intracellular routes, and shows the correlations with infarct size and outcome. Notably, the participation of the MIF receptors CXCR2, CXCR4, CXCR7, and CD74 in the pathogenesis of ischemic stroke strengthens the potential role of MIF in these diseases. However, as the MIF CK receptors also engage in numerous interactions with their cognate classical CK ligands such as CXCL8 and CXCL12, future approaches will have to employ CK- or even preferably, pathway-specific blockade or interference approaches. A recent study performed in the context of atherosclerosis models suggests that it may be possible to selectively block the MIF/CXCR4 axis in stroke without interfering with the MIF/CD74 or CXCL12/CXCR4 pathways (Kontos et al., 2020). The MIF homolog MIF-2, which also engages CD74, has not yet been studied in ischemic stroke, but data from myocardial ischemia/reperfusion models suggest a similar dichotomic behavior.
Lastly, while our understanding of the role of classical CKs and ACKs including MIF proteins in ischemic stroke is still incomplete, it is clear that they could represent important players in RIC-based protection strategies in ischemic stroke. Toll-like receptors, DAMPs, and IFNs have been implicated in RIC in brain ischemia (Hess et al., 2015) and the accumulating evidence on the key roles of classical CKs, ACKs, and MIF proteins, as discussed in this review article, suggest that some of these mediators, while being interesting therapeutic targets by themselves, could also be interesting in the context of RIC (Hamner et al., 2015; McDonough and Weinstein, 2016, 2018, 2020). With emerging data for the classical CKs CXCL12 and CCL5 (and their receptors CXCR4 and CCR5, respectively), the anti-inflammatory cytokine IL-10, MIF, and erythropoietin in RIC in cardiac ischemia (Brines and Cerami, 2005; Weber, 2010; Davidson et al., 2013; Heusch et al., 2015; Bernhagen, 2019b; Bromage et al., 2019; Ruze et al., 2019; Stoppe et al., 2019), and initial evidence from focal ischemic stroke models suggesting that RIC-induced cerebral microvessel-triggered CXCL12 and CCL2 (or their receptors CXCR4 and CCR2) have protective effects on leukocyte infiltration and lesion size in a successive stroke insult, the cognate CXCR4 ligand CXCL12 (Hess et al., 2015; Selvaraj et al., 2017), the cognate CCR2 ligand CCL2 (Stowe et al., 2012), and potentially also the alternate CXCR4 ligand MIF, could be such candidates. In this regard, it is noteworthy to mention that in vitro studies showed that increases in astrocyte-derived CXCL12 levels in ischemic brain extracts were dependent on the CXCR2/CXCL1 or /CXCL5 axis (Shin et al., 2014). Although preliminary and not yet confirmed in vivo, this connects the CXCR2 and CXCR4 pathways with RIC and it may be hypothesized that the dual CXCR2/CXCR4 ligand MIF might be a connecting molecular cue. Interestingly, endothelial storage compartments such as Weibel-Palade bodies (WPBs) (Metcalf et al., 2008) as well as smaller secretagogue-sensitive vesicles have been shown to rapidly and massively release the CKs CXCL8, CCL26, CXCL1, and CCL2. Of these, CXCL8 and CCL26 are pre-stored in the classical WPB compartment and released upon inflammatory or thrombotic stimulation, while CXCL1 and CCL2 are stored in distinct, yet equally responsive, vesicular compartments (Utgaard et al., 1998; Wolff et al., 1998; Oynebraten et al., 2004; Oynebraten et al., 2005). Of note, hypoxia was shown to induce exocytosis of WPBs, a process involved in mobilizing stem cells and neutrophils and participating in post-ischemic repair (Pinsky et al., 1996; Kuo et al., 2008). This latter action has been linked to ischemic preconditioning in renal ischemia (Patschan et al., 2006), suggesting together that CK-storing endothelial compartments could play an important role in RIC.
In summary, inflammation exacerbates infarct development after ischemic stroke and post-stroke inflammation is associated with neuronal death and secondary tissue injury (Zhang et al., 2021). CKs and alarmins are key mediators driving inflammatory processes in this phase, thus contributing to an exacerbation of tissue injury and stroke-related morbidity and mortality. The rapidly increasing mechanistic knowledge on the roles of classical CKs, ACK, and alarmins, including MIF proteins in ischemic stroke as outlined in this review article, offers potential novel therapeutic targets for the phase of post-stroke inflammation. A variety of potential modalities such as antibodies, nanobodies, small molecule inhibitors, peptides, and nucleotide-based approaches are available in principle and some of them have been very successful in preclinical in vivo models, but the utility of such drug candidates will have to be carefully studied in future clinical trials. Future studies should also further test the CK/RIC hypothesis in vitro and in vivo, firmly establish whether classical or atypical ACK represent RIC cues in stroke, and explore whether other CKs, ACKs, or DAMPs are involved as well. While clinical studies are still lacking, this could eventually also lead to new molecule-specific targeting strategies in brain ischemia and ischemic stroke that may complement or substitute RIC as well as other anti-inflammatory approaches. In this respect, the outcome of the recently completed RICA trial and similar clinical studies pursuing this paradigm will be of high interest.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant SFB1123/A3 to J.B., by the DFG excellence initiative program LMUexc/strategic partnerships with Singapore to J.B., and by the Munich Cluster for Systems Neurology (EXC 2145 SyNergy-ID 390857198) to J.B. S.W., C.Z., Y.T., and Y.G. acknowledge support by the Chinese Scientific Council (CSC) fellowship program CSC/LMU.
References
Sijia Wang1*
1Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilian-University (LMU), 81377 Munich, Germany
Omar El Bounkari1*
1Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilian-University (LMU), 81377 Munich, Germany
Chunfang Zan1
1Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilian-University (LMU), 81377 Munich, Germany
Yuan Tian1
1Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilian-University (LMU), 81377 Munich, Germany
Ying Gao1
1Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilian-University (LMU), 81377 Munich, Germany
Jürgen Bernhagen1,2,3
1Chair of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilian-University (LMU), 81377 Munich, Germany; 2Munich Heart Alliance, 80802 Munich, Germany; 3Munich Cluster for Systems Neurology (SyNergy), 81377 Munich, Germany.
*These authors contributed equally to this work.
Corresponding author:
Jürgen Bernhagen
Email: Juergen.Bernhagen@med.uni-muenchen.de)
or
Omar El Bounkari
Email: omar.el_bounkari@med.uni-muenchen.de
In a new window | Download PPT
Figure 1: The multifaceted impact of classical chemokines on the pathogenesis of ischemic stroke – classes, cells, receptors, and downstream effects. CC-, CXC-, and CX3C-chemokines are expressed by different brain cells including neurons, astrocytes, and microglia, as indicated in the figure. Following their release, these chemokines bind to their specific CC- (CCR2, CCR1, CCR5), CXC- (CXCR2, CXCR4), or CX3C-chemokine receptors (CX3CR1) to promote signaling responses, the secretion of additional inflammatory cytokines/chemokines, and direct recruitment effects including the infiltration of several immune cells into the brain. Chemokines also enhance BBB dysfunction and neuronal cell death.
In a new window | Download PPT
Figure 2: Expression and effects of atypical chemokines (ACKs) in ischemic stroke. The scheme illustrates differential expression and involvement of ACKs, i.e. HMGB1, β-defensin 2 (HBD2), thioredoxin, and peroxiredoxins in ischemic stroke. HMGB1, high-mo¬bi¬lity group box protein-1; IL-1β, interleukin-1β; CXCL12, C-X-C-motif chemokine ligand 12; iNOS, inducible nitric oxide synthase; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor-α.
In a new window | Download PPT
Figure 3: Overview illustrating the dichotomic effects and suggested mechanisms of the atypical chemokine MIF in ischemic stroke as evidenced by experimental and correlative clinical studies. In most studies, the expression of MIF in stroke positively correlates with infarct size and the prognosis of stroke severity. (1-3) Depending on the context and stimulus, MIF has a detrimental or protective effect, overall suggesting a dichotomic role in ischemic stroke, although overarching systematic studies are elusive. (1) Following OGD stress or cerebral ischemia, HIF1-α activation drives the upregulation of MIF in brain (penumbra, infarct core), which promotes the disruption of the blood brain barrier (BBB) and an increase of the infarct volume, consistent with a detrimental effect of MIF in ischemic stroke. (2) In contrast, genetic deletion of Mif or MIF downregulation mediated by NFκB signaling activation under ischemic conditions enhances microglia activation, caspase-3 activation, neuronal cell death, and increases infarct size, suggesting a protective effect of MIF in stroke. (3) One study suggests that MIF has a novel intracellular function acting as nuclease promoting parthanatos and increasing ischemic stroke lesions. After its nuclear translocation, the MIF nuclease activity induces DNA damage to promote neuronal cell death.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 9305 | 34 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA