Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
MIF proteins in neurological disorders: dichotomic role and perspectives for conditioning medicine
Time:2021-12-28
Number:9890
Author Affiliations

Conditioning Medicine 2021. 4(5): 216-233.
Abstract
Many neurological diseases are characterized as chronic inflammatory and/or degenerative disorders involving brain, spinal cord, as well as peripheral nerves. Glial cells including microglia, astrocytes, and neurons, as well as infiltrating immune cells serve as key regulatory effectors during neuroinflammation. Cytokines, chemokines, and damage-associated molecular patterns (DAMPs) integrally contribute to neuroinflammation, orchestrating immune reactions and subsequent inflammatory responses within the central nervous system (CNS), and thus are pivotal participants in the pathogenesis of various neurological diseases. Macrophage migration-inhibitory factor (MIF) is a pleiotropic inflammatory cytokine and an atypical chemokine (ACK) that serves a critical driver of acute and chronic inflammatory processes. MIF has been implicated in the initiation and progression of several neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), multiple sclerosis (MS), and Alzheimer’s disease (AD), as well as acute CNS damage such as spinal cord injury (SCI), traumatic brain injury (TBI), and ischemic stroke (IS). Emerging experimental and clinical evidence suggests a dichotomic role of MIF in neuro¬inflammation and neurodegeneration, especially in different disease settings and stages, raising partially controversial and unexplored aspects of its role in neurodegenerative diseases that require further elucidation. To this end, MIF has been reported to exert mostly beneficial effects in PD and ALS, while it promotes disease progression in MS, SCI, and TBI. In AD and stroke, both beneficial and exacerbating effects have been observed. In this review, we provide a qualified update on the clinical and experimental evidence linking MIF and its receptors to various neurological disorders, focusing on the multifaceted roles of MIF, and discuss potential MIF-based therapeutic strategies, including perspectives of conditioning medicine stimulated by MIF’s recent implication as a remote ischemic conditioning (RIC) signaling cue.
Keywords: macrophage migration-inhibitory factor, cytokine, atypical chemokine/ACK, DAMP/alarmin, neuroinflammation, neurological disease/disorder, neurodegeneration, remote ischemic conditioning (RIC, RIPC)
Abstract
Many neurological diseases are characterized as chronic inflammatory and/or degenerative disorders involving brain, spinal cord, as well as peripheral nerves. Glial cells including microglia, astrocytes, and neurons, as well as infiltrating immune cells serve as key regulatory effectors during neuroinflammation. Cytokines, chemokines, and damage-associated molecular patterns (DAMPs) integrally contribute to neuroinflammation, orchestrating immune reactions and subsequent inflammatory responses within the central nervous system (CNS), and thus are pivotal participants in the pathogenesis of various neurological diseases. Macrophage migration-inhibitory factor (MIF) is a pleiotropic inflammatory cytokine and an atypical chemokine (ACK) that serves a critical driver of acute and chronic inflammatory processes. MIF has been implicated in the initiation and progression of several neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), multiple sclerosis (MS), and Alzheimer’s disease (AD), as well as acute CNS damage such as spinal cord injury (SCI), traumatic brain injury (TBI), and ischemic stroke (IS). Emerging experimental and clinical evidence suggests a dichotomic role of MIF in neuro¬inflammation and neurodegeneration, especially in different disease settings and stages, raising partially controversial and unexplored aspects of its role in neurodegenerative diseases that require further elucidation. To this end, MIF has been reported to exert mostly beneficial effects in PD and ALS, while it promotes disease progression in MS, SCI, and TBI. In AD and stroke, both beneficial and exacerbating effects have been observed. In this review, we provide a qualified update on the clinical and experimental evidence linking MIF and its receptors to various neurological disorders, focusing on the multifaceted roles of MIF, and discuss potential MIF-based therapeutic strategies, including perspectives of conditioning medicine stimulated by MIF’s recent implication as a remote ischemic conditioning (RIC) signaling cue.
Keywords: macrophage migration-inhibitory factor, cytokine, atypical chemokine/ACK, DAMP/alarmin, neuroinflammation, neurological disease/disorder, neurodegeneration, remote ischemic conditioning (RIC, RIPC)
Introduction
Neurological disorders remain among the greatest public health challenges, since most of them lack disease-modifying therapies (Birbeck et al., 2015; Nussbacher et al., 2019; Kampmann, 2020). Neuroinflammation is associated with many neurological disorders and is a recognized driver of disease onset and progression, but our knowledge of the actual causative molecular and cellular mechanisms remains limited (Glass et al., 2010; Ransohoff and Brown, 2012; Ransohoff et al., 2015; Ransohoff, 2016; Kneussel and Friese, 2021). The neuroinflammatory response is triggered and orchestrated by cytokines and chemokines, which are released from brain resident cells or infiltrated immunocytes. To decipher their integral contributions to the neuroinflammatory process is key to understanding the pathogenesis of many neurological diseases. Their roles and mechanisms have been summarized in a number of excellent comprehensive review articles (Charo and Ransohoff, 2006; Savarin-Vuaillat and Ransohoff, 2007; Glass et al., 2010; Ransohoff, 2016; Becher et al., 2017; Wang et al., 2021b).
More recently, damage-associated molecular patterns (DAMPs) and atypical chemokines (ACKs) have emerged as additional pivotal soluble mediators in the regulatory network of inflammation, including neuroinflammation (Bianchi, 2007; Venereau et al., 2015; Kapurniotu et al., 2019; Wang et al., 2021b). DAMPs, also termed alarmins, are abundant intracellular proteins, metabolites, or nucleic acids (including mtDNA) that additionally function as potent “sterile” ligands for innate immune or pattern-recognition receptors (PRRs), once released into the extracellular space (Venereau et al., 2015; Gong et al., 2020). In the brain, DAMPs can be released by neurons, glial cells, and infiltrating immune cells. DAMPs are typically released upon cell death, damage, and tissue injury, but regulated secretion of DAMPs has also been described (Venereau et al., 2015). Extracellular DAMP signaling in turn promotes the release of conventional proinflammatory cytokines and chemokines from resident cells or infiltrating immune cells. For example, high-mobility group box-1 (HMGB1), a chromatin constituent and prototypical proteinaceous DAMP that is released by necrotic brain tissue participates in the modulation of brain injury through signaling, i.e. via its receptors Toll-like receptor (TLR) 4 or receptor of advanced glycation endproducts (RAGE) (Liesz et al., 2015; Singh et al., 2016). Moreover, HMGB1 activated inflammatory cascades and aggravated neurological outcomes after experimental and clinical stroke (Singh et al., 2016). How DAMPs contribute to neurodegenerative disorders has been comprehensively reviewed by Wilkins and co-authors (2017) and Picca et al. (2020). Alzheimer's disease (AD), multiple sclerosis (MS), and Parkinson's disease (PD) provide good examples, in which various disease-exacerbating roles of DAMPs are operative (Varhaug et al., 2017; Earls et al., 2019; Venegas and Heneka, 2019; Ihnatovych et al., 2020; Paudel et al., 2020).
Atypical chemokines share multiple structural and functional features with proteinaceous DAMPs such as HMGB1 and secreted aminoacyl-tRNA synthetase (AaRS) fragments, e.g. bona fide intracellular functions, signaling through chemokine receptors, or release as leaderless secretory protein (LSP). Accordingly, some of them are designated as both DAMPs and ACKs (Kapurniotu et al., 2019). For example, released HMGB1 forms heterodimers with CXCL12 to signal via the CXC chemokine receptor CXCR4 (Schiraldi et al., 2012; De Leo et al., 2019), and secreted Tyr-RS signals via CXCR1 (Wakasugi and Schimmel, 1999).
Macrophage migration-inhibitory factor (MIF) is one of the first cytokines discovered 55 years ago (David, 1966). Contrary to its eponymous name, today MIF is known as a pleiotropic inflammatory cytokine and prototypical ACK (Calandra and Roger, 2003; Tillmann et al., 2013; Kapurniotu et al., 2019). In fact, MIF is an evolutionarily conserved multifunctional protein mediator and LSP with both intra- and extracellular activities, thus sharing key properties with proteinaceous DAMPs (Kapurniotu et al., 2019; Hoffmann and Bernhagen, 2020). Intracellular MIF has been associated with cellular redox homeostasis, chaperoning and nuclear activities, while released extracellular MIF exerts a multitude of inflammatory activities through interaction with its receptor cluster of differentiation 74 (CD74) and/or via interactions with the chemokine receptors CXCR2, CXCR4, and CXCR7 (Calandra and Roger, 2003; Leng et al., 2003; Bernhagen et al., 2007; Kapurniotu et al., 2019). Interestingly, MIF receptors can form complexes and induce signaling via AMP-activated protein kinase (AMPK), phosphoinositide 3-kinase/protein kinase B (PI3K/AKT), and nuclear factor-κB (NF-κB) pathways. Beyond their role in the regulation of the host immune response, these pathways have been identified as major pathogenic routes in chronic inflammatory and autoimmune diseases, cancer, as well as cardiovascular and neurological diseases; however, the MIF/CD74 axis also is involved in tissue protection in the ischemic heart and fatty liver (Calandra and Roger, 2003; Conroy et al., 2010; O'Reilly et al., 2016; Kang and Bucala, 2019; Sinitski et al., 2019; Noe and Mitchell, 2020). Of note, MIF harbors an evolutionarily conserved catalytic pocket with homology to bacterial isomerases/tautomerases. While the physiological substrate of the tautomerase site in humans has not been identified, it appears that several identified small molecule tautomerase inhibitors affect MIF’s receptor-mediated activities, at least indirectly via conformational changes (Kok et al., 2018; Sinitski et al., 2019). The aforementioned redox and nuclear activities of MIF have been suggested to be associated with an intrinsic Cys-Xaa-Xaa-Cys redox site and a nuclease-like activity (Kleemann et al., 1998; Wang et al., 2016).
In the CNS, MIF can be released by several cell types including neurons, microglia, and astrocytes and contributes to glial activation, neuroinflammation, and neurodegeneration at various levels (Lerch et al., 2014; Leyton-Jaimes et al., 2018; Vandenbark et al., 2019; Nasiri et al., 2020; Chen et al., 2021). Not surprisingly, MIF has been suggested to be involved in a number of neurological diseases including AD, ALS, MS, PD, ischemic stroke, SCI, autism-spectrum disorders, depression, and brain tumors. While this has raised an interest in MIF-based diagnostic and therapeutic strategies in neurological diseases, further studies are needed to clarify its exact mode of action in such conditions.
In this article, we provide an update on the role of MIF proteins in neurological diseases, discuss the current knowledge and open questions about the involved receptors and mechanisms, and comment on potential translational avenues, including a potential exploitation of MIF’s emerging role as a conditioning cue.
MIF and its receptors in inflammation
Identified in 1966 as a soluble immune mediator and migration-inhibitory factor secreted from T lymphocytes, MIF is among the first cytokines ever discovered along with type I interferon and interleukin-1, back then known as endogenous pyrogen (Isaacs and Lindenmann, 1957; David, 1966; Dinarello, 2010). In fact, first reports on a MIF-like activity even date back to 1932 (Rich and Lewis, 1932). Moreover, MIF is a highly conserved protein with its orthologs in other kingdoms and phylogenetic branches dating back over 800 million years (Michelet et al., 2019). Contrary to its eponymous name, today MIF is known as a pleiotropic inflammatory cytokine and prototypical ACK that signals through several receptors and has been suggested to have additional intracellular activities (Calandra and Roger, 2003; Tillmann et al., 2013; Kapurniotu et al., 2019). In line with its role as ACK and its wide cross-kingdom preservation, MIF is broadly detected in essentially all types of immune cells, as well as fibroblasts, endothelial cells, various parenchymal cells, and neurons. While MIF expression per se is quasi ubiquitous, its secretion from immune cells occurs in a regulated and inducible manner. Still baseline autocrine MIF release is observed for many cell types as well (Hoffmann and Bernhagen, 2020).
Many of the inflammatory activities of extracellular MIF, including those related to tumorigenesis, are mediated by MIF engagement of CD74 (Leng et al., 2003), a bifunctional single pass type II transmembrane protein that serves both as the invariant chain (Ii) of major histocompatibility class (MHC) II and as the cognate MIF receptor. The three-dimensional structure of MIF (Sun et al., 1996) exhibits similarities to that of the MHC protein (Kato et al., 1996; Meza-Romero et al., 2016) and several of MIF’s activities on monocytes/macrophages coincide with class II-related inflammatory profiles, but it is generally believed that the functions of CD74/Ii as a class II chaperone and MIF receptor are independent of each other (Borghese and Clanchy, 2011; Shachar and Haran, 2011). In hindsight, the historic migration-inhibitory activity of MIF has been suggested to reflect a desensitization and/or arrest effect on macrophages exposed to MIF-containing supernatants for an extended period of time (David, 1966; Kapurniotu et al., 2019). In fact, MIF was found to exhibit bona fide chemotactic and chemokine-like activities that are mediated by non-cognate interaction with the G protein-coupled receptor (GPCR)-based CXC motif chemokine receptors i) CXCR2, ii) CXCR4, and iii) CXCR7, orchestrating the recruitment and activation of i) monocytes/macrophages and neutrophils; ii) monocytes/macrophages, neutrophils, eosinophils, B lymphocytes, T lymphocytes, platelets, cancer cells, and progenitor and stem cells; and iii) B lymphocytes and cancer cells; respectively. MIF/chemokine receptor interactions have mostly been studied in the context of leukocyte recruitment processes in atherosclerotic pathologies, but have also been studied in cancer, thrombosis, acute infection, and skin inflammation (Bernhagen et al., 2007; Zernecke et al., 2008; Cho et al., 2010; Dessein et al., 2010; Tarnowski et al., 2010; Simons et al., 2011; Tillmann et al., 2013; Chatterjee et al., 2014; Hsieh et al., 2014; Klasen et al., 2014; Alampour-Rajabi et al., 2015; Brocks et al., 2017; Kapurniotu et al., 2019; Sinitski et al., 2019; Rodrigues et al., 2020; Schindler et al., 2021a; Schindler et al., 2021b). Many of MIF’s inflammatory and recruitment activities are considered to be upstream of other inflammatory cytokine pathways. For example, MIF triggers the production of interleukin (IL)-6, IL-8/CXCL8, and tumor necrosis factor (TNF)-α.
In addition to chronic inflammatory diseases, cancer, and atherosclerotic diseases, MIF is a mediator of autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus (SLE), as well as sepsis and acute infectious conditions (Calandra and Roger, 2003; Zernecke et al., 2008; Conroy et al., 2010; O'Reilly et al., 2016; Leyton-Jaimes et al., 2018; Kang and Bucala, 2019; Kapurniotu et al., 2019; Sinitski et al., 2019; Noe and Mitchell, 2020; Wang et al., 2021b).
Studies on MIF/receptor mechanisms in brain and neurological diseases have been relatively limited and have so far mostly focussed on the MIF/CD74 axis. In the following chapters, we will discuss effects and mechanisms of MIF and its receptors in neuroinflammation and neurodegenerative diseases and relate them to the various cell types in the brain and the CNS.
Functions of MIF on different brain cells in the context of neuroinflammation
The nervous system is mainly composed of two classes of cells, i.e. neurons and glial cells. Neurons are electrically excitable cells that communicate with other nerve cells as well as other cell types via synapses. They serve as essential information conduits and sensors for all organismal functions and also serve as communication cues in response to abnormal cerebral changes. Glial cells mainly exert supportive functions to maintain homeostasis, including holding, nourishing, insulating neurons, and cleaning up pathogens. The major types of glial cells in the CNS are astrocytes, microglia, oligodendrocytes, and ependymal cells, which exert numerous roles in brain homeostasis and can display a high degree of complexity and biological functionality in response to inflammation (Hickman et al., 2018; Bernaus et al., 2020; Linnerbauer et al., 2020).
Neuroinflammation is triggered by neuronal damage and driven by the induction of various immune and inflammatory processes involving microglial and astroglial activation as well as immune cell infiltration, contributing in aggregate to the initiation and progression of CNS disorders such as chronic neurodegenerative diseases, acute TBI, and SCI. Cytokines, chemokines, and ACKs play various pivotal roles in these pathologies as summarized in several excellent review articles (Bauer et al., 2007; Rostene et al., 2007; Savarin-Vuaillat and Ransohoff, 2007; Li et al., 2012; Ransohoff and Brown, 2012; Ransohoff et al., 2015; Ransohoff, 2016; Wang et al., 2021b).
MIF was first associated with brain disorders in 1993, when Bernhagen and colleagues surprisingly discovered MIF as a pituitary-derived cytokine that was located to corticotrophic and thyrotrophic cells of the pituitary gland and potentiated lethal endotoxemia (Bernhagen et al., 1993; Nishino et al., 1995). However, it was not until three years later that MIF expression was detected in ependyma, astrocytes, and neurons, i.e. in bona fide brain resident cells (Nishibori et al., 1996). Meanwhile, several studies have provided evidence that MIF is expressed in virtually all brain cell types, most abundantly in neurons (Leyton-Jaimes et al., 2018). In the following chapters, we will summarize the available data on the expression and activities of MIF and its receptors in different brain cell types. This is also illustrated in Figure 1.
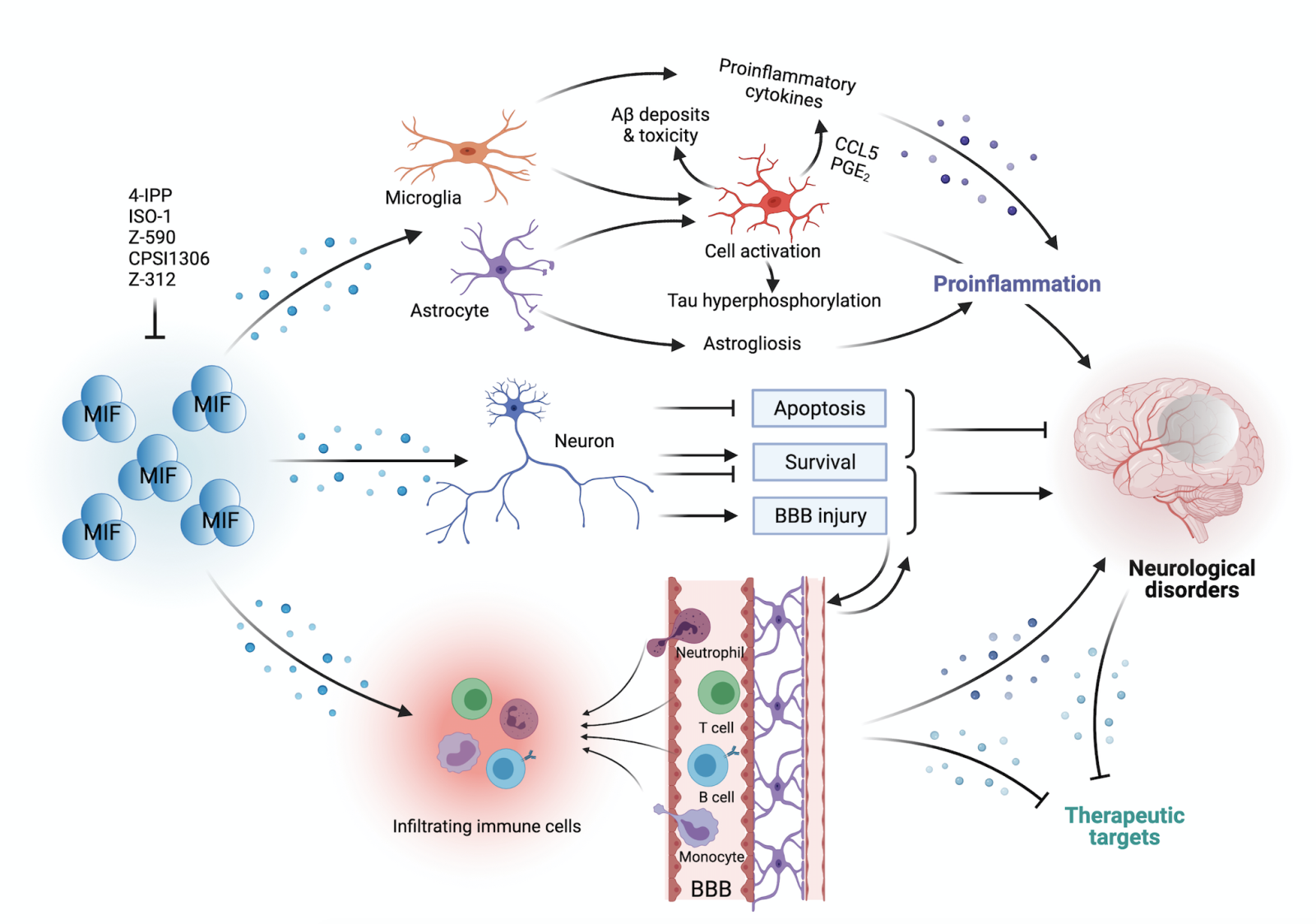
In a new window | Download PPT
Figure 1: MIF has been implicated in neuroinflammation through the regulation of microglia, astrocytes, neurons, and infiltrating immune cells. Glial cells including microglia and astrocytes dominate the inflammatory process, as indicated in the figure. MIF not only induces astrocyte and microglia activation, but also triggers an innate immune response by triggering the release of several potent proinflammatory cytokines such as IL-1β, IL-6 and TNF-α, at least partially dependent on glial activation, in the central nervous system. Besides inflammatory regulation in glial cells, astrocyte activation indirectly promotes tau hyperphosphorylation, and microglial activation enhances Aβ accumulation and toxicity. Additionally, MIF exerts neuroprotective effects on neurons via inhibition of apoptosis and improvement of neuronal survival, a notion that still is controversial and under active investigation. In addition to neuronal cells, infiltrating immune cells are also involved in neuroinflammation, and MIF behaves as a key cellular mediator promoting lymphocyte and monocyte/macrophage infiltration across the blood-brain barrier (BBB). Overall, MIF plays a dichotomic role in different cell types and disease entities and contributes to different neurological disorders through complex cellular mechanisms and signalling pathways.
MIF and glial cells
Among glial cells, microglia and astrocytes are major sources of MIF. Although developmentally derived from a yolk sac origin, microglial cells are brain-resident macrophage-like cells and have been widely implicated in the neuroinflammatory response both in the central and peripheral nervous system and are regarded as key modulators of neuroinflammation (Streit et al., 2004; Hickman et al., 2018), especially in neurodegenerative diseases. Microglia activation not only contributes to the initiation of proinflammatory cascades, but also furthers neuronal damage and loss, e.g. through release of pro-inflammatory cytokines and mediators such as complement proteins.
MIF was found to be involved in disease-associated microglia activation (Zhang et al., 2016). Applying a lipopolysaccharide (LPS)-activated BV-2-microglial cell system, Zhang et al. (2016) demonstrated that Z-590, a small molecule inhibitor of MIF tautomerase activity, reduced the secretion of TNF-α, reactive oxygen species (ROS), and nitric oxide (NO), and attenuated mRNA levels of inflammatory factors such as inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), TNF-α, IL-6, and IL-1β. Mechanistically, these effects were associated with the suppression of mitogen-activated protein kinase (MAPK) phosphorylation (i.e. extracellular signal-regulated kinase (ERK), p-38, and c-Jun N-terminal kinase (JNK)). In another study applying the tautomerase inhibitor ISO-1, microglia and astrocyte responses in rats after diffuse axonal injury were downregulated and associated with reductions in Toll-like receptor (TLR) 2/4 signalling, ERK, NF-κB, IL-1β, IL-6, and TNF-α, implying that MIF exacerbates neuronal and axonal damage at least partially through regulating glial inflammation (Zhao et al., 2020). Cox and colleagues (2013) demonstrated that Mif deficiency led to less inflammatory pathology in the CNS in a mouse model of MS, which was accompanied by more resting microglia and less activated microglia, suggesting a role of MIF in resident microglia activation. This activity was attributed to a mechanism mediated by CCAAT-enhancer-binding protein (C/EBP)-β. Moreover, microinjection of recombinant MIF into native spinal cord directly induced the release of microglia-derived proinflammatory cytokines, and restored inflammatory pathology in Mif-deficient mice (Cox et al., 2013). Similarly, Nasiri et al. (2020) showed that suppression of MIF resulted in a significant reduction in the production of cytokines in primary glial cells and a sporadic mouse model of AD, confirming a proinflammatory role of MIF in the brain both in vitro and in vivo.
Additionally, the impact of MIF on spinal microglia activation was revealed through in vitro experiments employing peripheral inflammatory stimulation (Wang et al., 2011). MIF upregulated COX-2 and prostaglandin E2 (PGE2) at both the mRNA and protein levels in primary rat microglia, which were strongly associated with neuroinflammatory severity. Mechanistically, the COX-2-PGE2 system was activated through the MIF-CD74 pathway, involving p38 and p44/p42 (Wang et al., 2011). Using a rat SCI model induced by compression, they also observed that spinal activated microglia near the lesioned site displayed an amoeboid morphology as well as a high expression of MIF three days after injury, as evidenced by in situ hybridization. Accounting for data of another study, the authors of this study speculated that MIF was primarily secreted by surrounding astrocytes to induce subsequent inflammation due to different peak points of MIF (Koda et al., 2004). In another inflammatory condition dominated by spinal MIF, spinal microglia were identified as the main source of MIF in the spinal cord dorsal horn, suggesting that microglia could be a key player in the hyperalgesia induced by formalin and that this could be associated with a MIF-activated MAPK pathway and an involvement of the N-methyl-D-aspartate receptor (NMDAR) (Wang et al., 2010). Collectively, these studies suggest that MIF exerts pro-inflammatory properties in the brain and spinal cord by either directly activating microglia or inducing proinflammatory cytokine secretion.
In addition to microglia, astrocytes act as an important CNS contributor to induce innate immune responses and further determine the pathological fates of neurological diseases. This involves multiple critical proinflammatory factors such as IL-1β, IL-6, IL-10, TNF-α, interferon (IFN)-γ, and classical chemokines such as C-C motif ligand (CCL)2, CCL5, CCL20, C-X3-C motif ligand (CX3CL)1, C-X-C motif ligand (CXCL)1, CXCL10, and CXCL12 (Ransohoff and Brown, 2012; Rossi, 2015). MIF was found to facilitate the production of CCL5 in astrocytes, and treatment with the small molecule MIF tautomerase inhibitor 4-IPP reversed this effect (Zhou et al., 2018). Furthermore, MIF was capable of triggering neuroinflammatory reactions in astrocytes after SCI and this effect was associated with phosphorylation of ERK1/2 via the interaction of MIF and CD74. Cholesterol-25-hydroxylase (CH25H) and phospholipase A2 (PLA2G2A) were identified as downstream targets of the MIF/CD74 axis in astrocytes based on a transcriptome analysis (Su et al., 2017). This association was further confirmed by Zhu and co-workers (2019). Moreover, MIF activated the COX-2/PGE2 signalling pathway in spinal astrocytes via interaction with CD74, fine-tuning the inflammatory microenvironment of spinal cord lesions (Wang et al., 2011). In line with these findings, 4-IPP treatment downregulated COX-2 and PGE2 in astrocytes (Zhang et al., 2019b). In addition, MIF dose-dependently facilitated the proliferation of primary cultured astrocytes in vitro (Su et al., 2017), which was consistent with in vivo data showing that MIF promoted proliferation and accumulation of astrocytes near the lesion after SCI (Koda et al., 2004). These two studies indicate that MIF has an astrogliosis-promoting property under inflammatory conditions. Subsequent reactive astrogliosis results in permanent glial scar formation and blocks neurological recovery in the context of acute injury and chronic neurodegenerative diseases. In conclusion, MIF-mediated astrocyte-specific inflammatory signalling is involved in several CNS pathologies.
MIF and NG2 glia
NG2 glia represent a special subtype of glial cells characterized by the expression of nerve/glial antigen 2 (NG2), an integral membrane proteoglycan found in diverse cell types, including oligodendrocyte progenitor cells (OPCs), chondroblasts, myoblasts, or pericytes, but most characteristically in NG2 glia. In fact, originally regarded as progenitors for oligodendrocytes and sometimes termed polydendrocytes, NG2 glia were newly classified as a fourth glia cell type in addition to astrocytes, microglia, and oligodendrocytes (Nishiyama et al., 2009). Special structural and functional features of these cells in healthy as well as diseased brain have been extensively reviewed by Dimou and Gallo, with a focus on the importance of NG2 glia in pathological conditions (Dimou and Gallo, 2015). Of interest, MIF was reported to promote the proliferation of NG2 glia in the context of traumatic grey matter (GM) injury, as revealed in a Mif deficient mouse model (Mattugini et al., 2018). This defined MIF as a contributor to NG2 glial proliferation.
MIF and neurons
Neurons themselves actively participate in neuroinflammatory regulation (Aktas et al., 2007; Kneussel and Friese, 2021). MIF has been reported to display neuroprotective properties through inhibition of apoptosis pathways, but can also be deleterious for neuronal survival through its proinflammatory activities and its role in parthanatos (Wang et al., 2016).
Bae et al. (2020) subjected a neuroblastoma cell line to oxygen-glucose deprivation (OGD) stress and found that MIF exhibited a neuroprotective role in such an ischemic environment, and promoted neuronal survival through an anti-apoptotic pathway (Bae et al., 2020). Mechanistically, MIF stimulation upregulated anti-apoptotic factors such as brain-derived neurotrophic factor (BDNF), B-cell lymphoma 2 (Bcl-2), and microtubule-associated protein 2 (MAP2), while it decreased the expression of pro-apoptotic markers such as caspase-3 and Bcl-2-associated X protein (Bax) in the OGD group, implying an anti-apoptotic effect induced by MIF as one mechanism of neuronal protection against hypoxic injury (Bae et al., 2020). Jung et al. (2021) further investigated the optimal conditions, including the concentration and timing of recombinant MIF (rMIF) application against neuronal apoptosis in an in vitro stroke model, and found 60 ng/mL of rMIF to be the most effective in activating BDNF expression, in line with previously reported inflammatory levels of MIF in circulation (Calandra and Roger, 2003; Jung et al., 2021). MIF additionally protects against neuronal apoptosis by regulating NF-κB signalling. This was confirmed in Mif-deficient mice, in which the knockout of Mif activated caspase-3 and accelerated neuronal loss as well as infarct progression during stroke (Zhang et al., 2014). Thus, in spite of the purported dual roles of NF-κB activation in neuronal survival, MIF-elicited NF-κB signaling appeared to play a neuroprotective role (Zhang et al., 2014). One study investigated the functional alterations of MIF on CA1 hippocampal neurons from murine brains. They showed that microinjection of MIF enhanced baseline Ca2+ activity and events, indicating a functional property of MIF in pyramidal neurons with regard to membrane excitability (Bancroft et al., 2020).
In contrast, some studies reported detrimental effects of MIF on neurons. MIF provokes inflammatory cascades through binding to its receptors on microglia, astrocytes, or infiltrating inflammatory cells, thereby indirectly affecting neuronal survival. In mouse models of transient middle cerebral artery occlusion (tMCAo), MIF was found to be highly expressed in neurons of the peri-infarct region, particularly in cortical parvalbumin-positive interneurons, and was associated with aggravated neuronal death and neurological deficits (Inácio et al., 2011c; Liu et al., 2018). Administration of ISO-1 not only reduced the infarct volume but also improved neuronal function, indirectly implying a detrimental role of MIF. Lastly, with respect to spinal neurons, MIF acted to compromise survival and viability. Stimulation with MIF resulted in increased cellular oxidative stress and intracellular calcium levels of spinal neurons, eventually leading to neuronal cell death (Chalimoniuk et al., 2006).
Interestingly, Wang and colleagues (2016) identified MIF as an unique apoptosis-inducing factor (AIF)-related nuclease that depends on poly(ADP-ribose) polymerase-1 (PARP-1) and is required for parthanatos, contributing to DNA damage and death of cortical neurons in vitro. Importantly, this phenotype was confirmed in vivo in a MCAo mouse model (Wang et al., 2016). The study by Wang et al. (2016) is discussed in more detail in the stroke chapter of this review. Additional effects of MIF on neuronal survival are discussed in the following disease-specific sub-chapters.
MIF and immune cell infiltration into the brain
When the blood-brain barrier (BBB) is compromised upon injury or inflammation, immune cells infiltrate into the parenchyma of the CNS to amplify neuroinflammation. Infiltrating cells are neutrophils, monocytes/macrophages, and lymphocytes, but also natural killer (NK) cells. In a mouse model of TBI induced by fluid percussion injury (FPI), the MIF antagonist ISO-1 suppressed TBI-induced upregulation of γδ T-cell and B-cell populations infiltrating into the brain, while increased B-cell numbers were observed in the spleen, as evidenced by flow cytometry using mouse brain tissue (Newell-Rogers et al., 2020). This indirectly reflects the property of MIF to contribute to the recruitment of peripheral lymphocytes into the brain. In addition, MIF also behaves as a potent driver in the activation and recruitment of inflammatory monocytes into the CNS, as demonstrated in an experimental autoimmune encephalomyelitis (EAE) mouse model. Less CD45+ cells, including lymphoid and myeloid cells, as well as activated microglia were found in the CNS of Mif –/– mice associated with decreased EAE severity. Mif deficiency also led to a significant reduction in CD45high/CD11b+ inflammatory macrophages/activated microglia in the brain (Cox et al., 2013). Furthermore, infiltrating monocytes/macrophages in the EAE model seemed to be deleterious and serve as a major source of MIF during neuroinflammation (Cox et al., 2013). These studies also suggested that MIF in the brain acts as a local alarmin or ACK released from injured neurons to drive the infiltration of circulating immune cells, which contributes to the orchestration of the neuroinflammatory response in the CNS. On one hand, infiltrating immune cells, especially monocytes and macrophages, can release MIF to promote the recruitment of additional inflammatory cells. On the other hand, injured neuronal cells may produce MIF to exacerbate inflammation. Moreover, plasma concentrations of MIF were significantly elevated in tMCAo mice and stroke patients compared with controls, indicating that CNS injury may also be accompanied with an upregulation of peripheral MIF levels (Liu et al., 2018). In line, Liu et al. (2018) injected rMIF into MCAo mice via the femoral vein and observed pronounced disruption of the BBB, providing evidence that peripheral MIF affects the BBB and thus brain injury (Liu et al., 2018). Moreover, since MIF is a small protein, it is reasonable to assume that peripheral MIF is able to cross the BBB to influence inflammation in the CNS, but further experimental evidence is needed to confirm this notion further.
Collectively, MIF is involved in neuroinflammation by affecting different cell types. As outlined in the following chapters, this is relevant for the diverse outcomes of MIF studies in various neurological disorders.
The dichotomic role of MIF in neurological diseases
A number of experimental studies and reports on clinical associations suggest an involvement of MIF in neurological disorders such as PD, ALS, SCI, AD, TBI, and stroke. However, the precise mechanisms mediated by MIF remain unsettled, and the effects on overall outcome are suggestive of a complex and context-dependent role, with both protective and exacerbating effects observed. The multifaceted roles of MIF in neurological disorders are described and summarized in Table 1 and Figure 2.
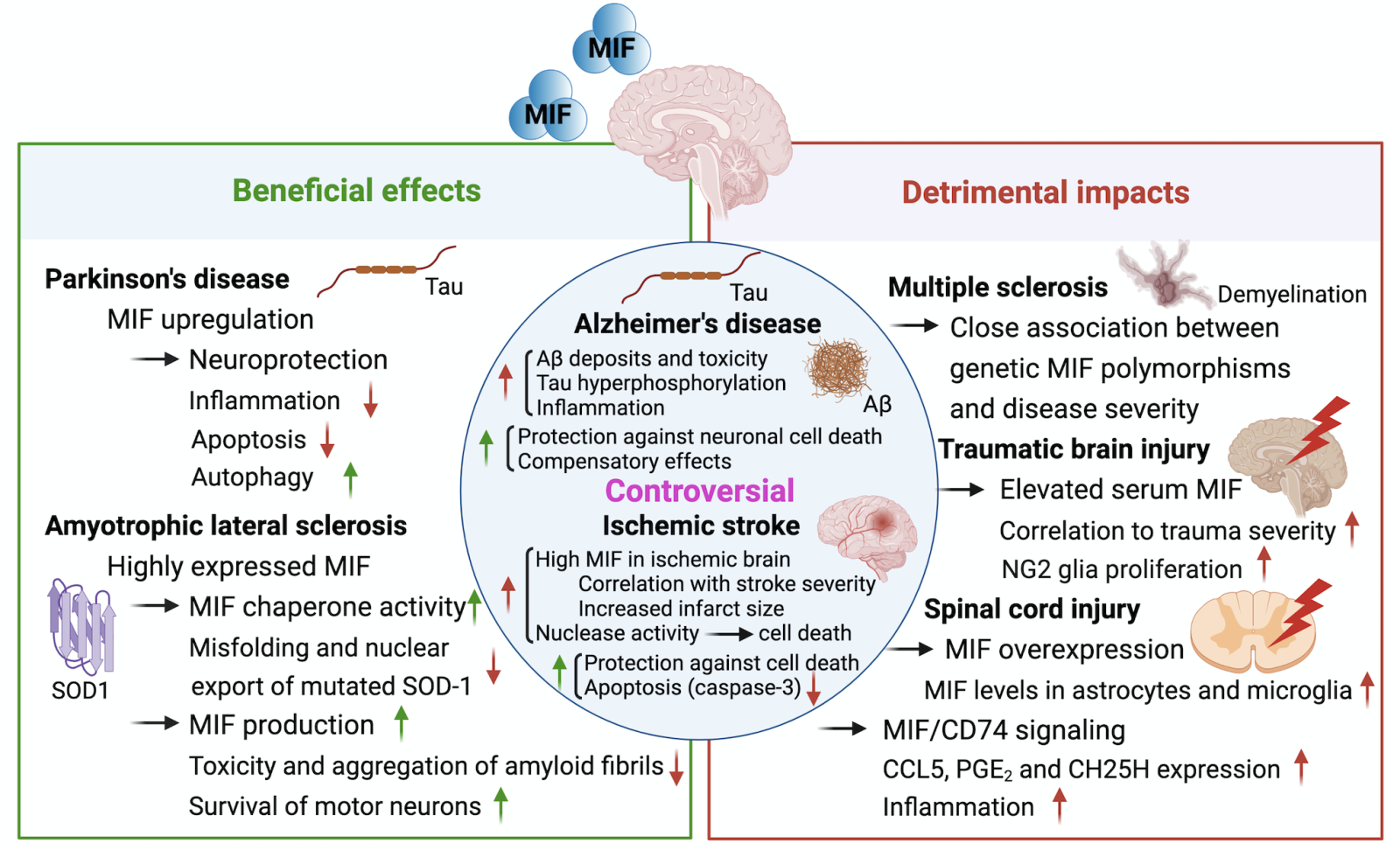
In a new window | Download PPT
Figure 2: The multifaceted impact of MIF on several neurological diseases. Based on current preclinical and clinical studies, MIF plays a mostly beneficial role in Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS)/familial ALS (FALS), whereas a deleterious role for MIF has been suggested in multiple sclerosis (MS), spinal cord injury (SCI), and traumatic brain injury (TBI). The functions of MIF in Alzheimer’s disease (AD) and ischemic stroke (IS) are controversial with both detrimental and protective effects observed. Major mechanisms of MIF regulating pathological characteristics of different neurological diseases are indicated in the figure. SOD1, superoxide dismutase type 1; CD74, cluster of Differentiation 74; CCL5, chemokine (C-C motif) ligand 5; PGE2, prostaglandin E2; Aβ, amyloid-beta; CSF, cerebrospinal fluid; tMCAo, transient middle cerebral artery occlusion.
Predominant protective role of MIF in Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS)
MIF in Parkinson’s disease (PD)
Among the common neurodegenerative disorders, PD is the second most frequent neuroinflammatory disease with an estimated prevalence of 9 million people worldwide by 2030 (Dorsey et al., 2007). Around 1% of aged adults (≥ 65 years old) are suffering from this disease (Kalia and Lang, 2015). The underlying causes behind the disease are still unclear. Possible mechanisms have been systematically reviewed by Abeliovich and Gitler (2016) and others.
MIF serum levels in PD patients were found to be upregulated compared with healthy controls, but no significant changes in mRNA levels in peripheral blood cells were detected (Nicoletti et al., 2011). This study also provided data suggesting that elevated MIF levels do not correlate with PD severity, suggesting that the main source of elevated MIF in PD may be neuronal cells, not peripheral immune cells. With respect to its causal role, the authors speculated that MIF may play a protective role in PD due to its involvement in catecholamine catabolism, even though MIF would be predicted to amplify immune reactions by inducing the secretion of various proinflammatory factors (Nicoletti et al., 2011). In fact, the results of a previously conducted transplantation study using a 6-hydroxydopamine rat model of PD showed that administration of rMIF inhibited microglia/macrophage activation in grafts (Schwarz et al., 1998), while no effects on the function and survival of grafts were observed. Consistently, Li and co-workers (2019) provided experimental evidence for a neuroprotective effect of MIF in PD. They observed that MIF was not only highly expressed in acute and chronic PD mouse models, but was also upregulated in SH-SY5Y PD-like neuronal cells, with a positive correlation with cell viability. Mechanistically, the protective effect of MIF was associated with a suppressed inflammatory profile, inhibition of apoptosis, and an increase in autophagy (Li et al., 2019). Additionally, two studies indirectly showed that MIF might exert a protective role in PD (Matsunaga et al., 1999; Nakahara et al., 2019). However, both studies were performed in the setting of neuroinflammation, and not in a genuine PD model. Based on its enzyme activity, Matsunaga et al. (1999) similarly found that MIF promotes the production of oxidized catecholamines, and then speculated MIF might have beneficial functions in PD. Nakahara et al. (2019) showed that MIF-mediated upregulation of BDNF contributed to neuronal survival through S-nitrosylation-related signaling, proposing a new mechanism for neurodegenerative diseases such as PD.
However, a couple of recent studies debated the role of MIF in PD, as recently reviewed by Basile and co-authors (2020). 20Zheng et al. (2021) utilised a novel inhibitor of MIF, Z-312, in a PD mouse model, and showed that inhibition of MIF attenuated microglial activation and neuronal loss, suggesting a rather detrimental role for MIF in PD. Cheng et al. (2020) emphasized the importance of autophagy in PD and observed that an autophagy defect in microglia led to PD-like symptoms, accompanied by elevated MIF levels and abnormal NLRP3 inflammasome activation, indirectly implicating MIF as a contributor to PD disease progression.
Taken together, while most studies indicate that MIF plays a beneficial role in PD, a couple of recent studies also indicated that it may have a more complex role in this disease, together calling for more experimental and clinical evidence to clarify its role, as well as that of MIF-2 and the MIF receptors.
MIF in amyotrophic lateral sclerosis (ALS)
In contrast, the evidence suggesting a neuroprotective role for MIF in ALS, also known as Lou Gehrig’s disease, has been unanimous. ALS is a progressive and devastating neurodegenerative disorder with unclear pathogenesis (Mitchell and Borasio, 2007). Updates on the epidemiology, pathology, clinical features, diagnosis, and therapeutics of ALS have been continuously summarized in mini-reviews in The Lancet (Mitchell and Borasio, 2007; Kiernan et al., 2011; van Es et al., 2017). At the cellular level, the central characteristic of this disease is neuronal cell death, especially the functional loss of motor neurons, leading to different clinical manifestations and difficult diagnosis (van Es et al., 2017). At the molecular level, the leading pathological hallmarks of ALS patients are hereditary mutations in the enzyme superoxide dismutase type 1 (SOD-1), resulting in its misfolding and aberrant accumulation in affected motor neurons (Kiernan et al., 2011).
In 2015, Israelson and colleagues (2015) found that purified MIF directly suppressed the misfolding of mutant SOD1 and identified a previously unrecognized intracellular chaperone activity of MIF in neuronal cells, suggesting that MIF is neuroprotective in motor neurons. This inhibitory effect of MIF on mutant SOD1 misfolding was associated with functional alterations in endoplasmic reticulum and mitochondria in motor neuron-like (NSC-34) cells. Moreover, elevated MIF levels in motor neurons abrogated the aggregation of misfolded mutant SOD-1 in those cells, thereby enhancing survival, and pointing towards an essential regulatory role of MIF in ALS (Abu-Hamad and Israelson, 2015; Israelson et al., 2015). Next, Leyton-Jaimes and co-workers systemically evaluated the role of MIF in ALS progression in vivo using an SOD1G85R Mif–/– mouse model (Leyton-Jaimes et al., 2016). They found a pronounced aggregation of SOD1 in the spinal cord of SOD1G85R-Mif–/– mice as compared to control mice. Consequently, MIF ablation resulted in the acceleration of ALS severity and shortening of lifespan in these mice. The same research group also demonstrated an impact of MIF on misfolded SOD1 amyloid aggregates in vitro (Shvil et al., 2018). On the cellular level, overexpressed MIF abolished the clearance and nuclear export of mutant SOD1 in NSC-34 cells and promoted the accumulation of disordered amyloid fibrils, while decreasing misfolded SOD1 toxicity through direct binding. Using a locked-trimeric mutant of MIF (MIFN110C), they also showed that blockade of MIF trimerization led to a decrease in MIF’s CD74-mediated cytokine activity, while the chaperone activity was increased, leading to a suppression of mutant SOD1 amyloid fibril formation. Moreover, in a mouse model of adeno-associated virus (AAV)-induced overexpression of MIF, a substantial delay of disease outbreak in loxSOD1G37R mice injected with AAV2/9-MIF compared with noninjected mice was observed. High MIF expression was associated with reduced accumulation of misfolded SOD1 and a higher survival rate of motor neurons in the spinal cord (Leyton-Jaimes et al., 2019).
Inflammatory role of MIF in spinal cord injury (SCI), multiple sclerosis (MS), and traumatic brain injury (TBI)
As discussed above, MIF appears to possess a neuroprotective effect on the initiation and progression of ALS, and likely also PD. Identified mechanisms encompass the regulation of inflammatory cascades, apoptosis, autophagy and mutated SOD-1 formation. The following paragraphs summarize studies on MIF’s role in SCI, MS, TBI, and neurological diseases for which essentially all available preclinical and clinical studies have described MIF as a detrimental mediator.
MIF in spinal cord injury (SCI)
Koda et al. (2004) observed a significant upregulation of MIF in injured rat spinal cords induced by compression. MIF mRNA levels peaked at day 3 after injury. The authors speculated that elevated MIF levels might stem from microglial cells that accumulate at injury sites, and that in turn, overexpressed MIF would exert proliferation-promoting properties, leading to the proliferation of astrocytes and an aggravation of inflammation. In line with this notion, an in vivo study in Mif–/– mice displayed a dramatic decrease in apoptotic neurons and a better functional recovery at 24 and 72 h after SCI compared to wild type (WT) mice, confirming a pro-inflammatory role of MIF that would affect neuronal survival in SCI models (Nishio et al., 2009). MIF deletion, at least partially, decreased deleterious secondary responses from primary SCI. In addition to this evidence from experimental models, initial correlative clinical studies indicated that circulating MIF levels were not only elevated in acute SCI, but also in chronic SCI patients, when compared to uninjured subjects (Stein et al., 2013; Bank et al., 2015). However, while MIF expression correlated with hepatocyte growth factor (HGF) and IL-16 in acute SCI patients (Bank et al., 2015), high MIF levels did not correlate with clinical or demographic characteristics in chronic SCI patients (Stein et al., 2013). Two studies suggested that MIF mediates inflammatory processes after SCI mainly through the release of other inflammatory cytokines or factors such as CCL5 and PGE2 released from astrocytes in the injured spinal cord (Zhou et al., 2018; Zhang et al., 2019b). In the contusion rat SCI model, MIF was able to expedite the production of CCL5 from astrocytes via its interaction with CD74 and activation of JNK signaling, and CCL5 further induced monocyte recruitment and amplified the initiated inflammatory response (Zhou et al., 2018). The same team also demonstrated that MIF, COX-2, and PGE2 were significantly upregulated after SCI through a pathway involving MAPK/COX-2/PGE2 signaling. PGE2 induced by MIF stimulation was capable of eliciting more IL-1β and IL-6, whereas less TNF-α was detected in LPS-activated macrophages (Zhang et al., 2019b). Moreover, MIF enhanced the expression and secretion of CH25H, a marker of CNS injury, in astrocytes via CD74 to promote cell migration, which might be an important mechanism through which MIF exacerbates progressive neuropathology after SCI injury (Zhu et al., 2019). Through bioinformatics analysis, they further identified cholesterol metabolism-related pathways involved in MIF/CD74 regulation, including inflammatory responses and leukocyte chemotaxis. Additionally, some researchers investigated the role of MIF in parthanatos, a pattern of cell death induced by oxidative stress after SCI as well as stroke, and showed that Mif deficiency led to protection of neurons from parthanatos in the injured brain and spinal cord (Wang et al., 2016; Yang et al., 2020). Interestingly, Saxena and colleagues (2015) applied a nanocarrier-mediated drug delivery approach to inhibit MIF in a rat SCI model. The administered small molecule inhibitor, a compound called Chicago sky blue (CSB) or p425, which blocks MIF oligomerization, resulted in improved vascular and white matter integrity after SCI. At the same time, mice treated with CSB showed improved wound healing, elevated arginase levels, and other pro-healing transcripts predictive of wound healing resolution. One of the main complications of SCI is deep vein thrombosis (DVT) (Chung et al., 2014). In this context, Wu and colleagues (2019) revealed a strong association between elevated MIF expression and the risks of developing DVT in SCI patients (Wu et al., 2019). This suggests that MIF may serve as a potential biomarker for early intervention of complications after SCI. Taken together, these studies demonstrate the relevance of MIF in the pathogenesis of SCI, providing evidence for MIF as a novel potential therapeutic target in spinal cord inflammation.
MIF in multiple sclerosis (MS)
MIF has been characterized as a key mediator that aggravates the progression of MS (Nishihira and Ogata, 2001; Benedek et al., 2017; Castañeda-Moreno et al., 2018; Fagone et al., 2018; Vandenbark et al., 2019). Benedek and coworkers demonstrated that not only MIF but also D-dopachrome tautomerase (D-DT)/MIF-2, a close structural homolog of MIF, was significantly upregulated in the serum of male MS patients compared to female MS subjects. Of note, this was accompanied by elevated CD74 levels and high MS severity (Benedek et al., 2017). This gender-specific phenotype was associated with two genetic MIF polymorphisms, a -794 CATT5-8 (rs5844572) and a -173 G/C SNP (rs755622) (Benedek et al., 2017; Castañeda-Moreno et al., 2018), which contribute to the modulation of MIF promoter activity and regulation of MIF expression to drive high- versus low-MIF expresser phenotypes (Baugh et al., 2002). Moreover, they revealed a strong correlation between MIF and D-DT in both plasma and CNS in MS subjects, as evidenced by human genetic data (Benedek et al., 2017). An association among MIF polymorphisms, MIF expression, and disease severity was also verified in a western Mexican population recruiting 230 MS patients and 248 healthy controls, indicating that MIF polymorphisms could represent gender-specific disease modifiers that promote MS progression (Castañeda-Moreno et al., 2018). Interestingly, the high-MIF expression profile in MS patients is similar to the regulatory patterns of other well-known inflammatory biomarkers of MS, for example TNF-related apoptosis inducing ligand (TRAIL) and Fas ligand (FasL) that are elevated in the sera of MS subjects (Rinta et al., 2008; Hagman et al., 2011). MIF has therefore been classified as a valuable marker to predict clinical worsening of MS.
In addition to MIF and D-DT/MIF-2, Fangone et al. (2018) also studied the MIF receptors CD74 (in conjunction with its co-receptor CD44), CXCR2, and CXCR4, and found them to be increased in encephalitogenic T cells from MS mice. Moreover, they observed that inhibition of MIF by ISO-1 attenuated clinical features of MS both in vivo and ex vivo (Fagone et al., 2018), suggesting a causal involvement of the MIF/D-DT pathway in MS pathogenesis. In an extension of these findings, elevated MIF derived from CD4-positive T cells was suggested to accelerate the conversion of clinically isolated syndrome (CIS) to clinically defined MS (Cavalli et al., 2019). Collectively, with currently only a few initial mechanistic studies available, most studies about MIF and MS are based on clinical data, suggesting MIF could be a detrimental mediator as well as a potential biomarker for predicting clinical outcome and prognosis of MS.
MIF in traumatic brain injury (TBI)
Lastly, MIF has unanimously been suggested to be a disease-driving factor in TBI. Clinical data showed that serum MIF was highly upregulated and closely associated with trauma severity and clinical outcomes of TBI patients (Yang et al., 2017a). Moreover, MIF levels correlated with serum IL-6, TNF-α, and C-reactive protein (CRP) concentrations in TBI patients, underscoring a proinflammatory role of MIF in TBI and suggesting a potential role for MIF as a prognostic biomarker for TBI. This notion was further confirmed by an experimental TBI study performed in C3–/– and C5–/– mice, which showed that complement components C3 and C5 contribute to the production of brain inflammatory mediators including CCL2/MCP-1, MIF and CCL5/RANTES, whereby inhibition by a C5a receptor antagonist improved the prognosis of TBI in a mouse model (Sewell et al., 2004). This indirectly confirmed a proinflammatory role of MIF in TBI. Additionally, Mif-deficient mice displayed less NG2 glial proliferation after traumatic GM injury, whereas no changes on astrocytes and microglia proliferation were observed, implicating MIF as a regulator of NG2 glial functionality (Mattugini et al., 2018), as discussed above.
Dichotomic role of MIF in Alzheimer’s disease (AD) and ischemic stroke
MIF in Alzheimer’s disease (AD)
Alzheimer’s disease is the most common neurodegenerative disease and causes heavy social and economic burden. Pathologically, it is defined as a major cause of cognitive impairment induced or favored by the formation and deposition of tau-containing neurofibrillary tangles (NFTs) and β-amyloid plaques in the brain (Busche and Hyman, 2020; Lauwers et al., 2020; Scheltens et al., 2021).
Oyama et al. (2000) first provided experimental evidence for the involvement of MIF in the pathogenesis of AD, when they, in addition to other interactors, identified an interaction between MIF and amyloid-β (Aβ) protein in the soluble fraction of the cerebral cortex of AD, as shown by immunoprecipitation (Oyama et al., 2000). They speculated that these binders, i.e. MIF, the α-chain of hemoglobin, and glutamine synthetase, are involved in the clearance or accumulation of Aβ. In line with the latter notion, Lashuel and coworkers (2005) observed amyloidogenic properties of the MIF protein. First clinical data were provided by Popp and colleagues (2009) suggesting a significant upregulation of MIF protein in CSF in both the mild cognitive impairment (MCI) group and in the AD group, with a moderate correlation between MIF and TNF-α in AD patients, indicating a proinflammatory role of MIF in AD pathogenesis. However, these data were based on relatively small patient numbers (31 patients with AD, 28 patients with amnestic MCI, and 19 subjects without cognitive deficits), and are thus of limited clinical significance.
Subsequently, MIF positivity was detected in microglia of amyloid precursor protein (APP) 23 transgenic mouse brain through immunostaining, and was associated with Aβ plaques. Functional in vitro experiments in neuronal cell lines in which MIF was targeted by ISO-1, showed an attenuation of Aβ toxicity (Bacher et al., 2010). Similarly, they observed an upregulation of MIF in the CSF of AD patients in comparison with the age-matched controls, consistent with previous findings from Popp and co-authors (2009). With regard to receptors, Yoshiyama et al. (2000) obtained immunohistochemical data on the MIF receptor CD74 and found that it was elevated in the microglia of AD patients. CD74 was also found to be increased in neurons of AD patients, an indication that MIF/CD74 signaling could contribute to neuronal damage in AD. This notion is further confirmed by an observed increase in CD74 levels in NFTs and Aβ aggregates (Bryan et al., 2008).
Moreover, MIF was found to affect neuronal cell death and tau hyperphosphorylation, another major pathological characteristic of AD (Goedert, 2015), and in another experimental study, an association among Mif deficiency, decreased astrocyte activation, and tau hyperphosphorylation was determined, employing two different AD mouse models on a Mif–/– background, indicating that Mif gene deletion signi¬ficantly ameliorated tau hyperphosphorylation by deactivating astrocytes and that the MIF/astrocyte axis could be a target in AD (Li et al., 2015). Mechanistically, the study suggested that MIF did not directly regulate tauopathy, but enhanced tau hyperphosphorylation in neurons by activating astrocytes to release intermediate stimulatory mediators (Li et al., 2015). Applying a human neuroblastoma SH-SY5Y cell model, Liang and co-authors (2018) noted that MIF promoted neuronal apoptosis through induction of pro-apoptotic Bcl-2-antagonist of cell death, BAD, similar to Aβ1-42. Of note, Fufang Danshen, a traditional Chinese medicine (TCM) drug, consisting of at least three different herbal effectors, displayed neuroprotective properties by activating MIF and downregulating BAD (Liang et al., 2018). Clinical data from Oikonomidi and colleagues (2017) point towards a relation between MIF, amyloid pathology, tauopathy, and neuronal injury, indicating that elevated MIF levels in the CSF may correlate with exacerbated cognitive decline in MCI and dementia. The study confirms the above conclusions for a role of MIF in the early clinical stages of AD, but employing 97 subjects with MCI or mild dementia and 52 healthy volunteers with normal cognition is still relatively weakly powered. Another mechanism of MIF contributing to tauopathy might stem from a role of MIF in chronic cytokine release upon neuroinflammation and neurodegeneration (Nasiri et al., 2020), along with an increase in biomarkers associated with tauopathy and neuronal injury. Moreover, two recent in vitro studies that have addressed aspects of glucose oxidation and modification indicate that MIF could be a link between metabolic dysfunctions and inflammation in AD (Kassaar et al., 2017; Yu et al., 2019). Collectively, the above summarized studies are suggestive of a detrimental role of MIF in AD progression that is associated with the most common pathological hallmarks of AD, such as promoting Aβ plaques and NFT accumulation, and aggravating inflammatory injury.
In contrast, other studies propose a protective role for MIF. Using a heterozygous Mif+/– APP23 AD mouse model and the SH-SY5Y neuronal cell line, Zhang et al. (2019a) found that MIF is secreted from Aβ-stimulated neurons, that it binds to Aβ plaques and protects against neuronal cell death from Aβ-mediated cytotoxicity. Moreover, high MIF levels were detected in the CSF of AD patients but not those patients with MCI and vascular dementia (Zhang et al., 2019a). In vivo, they demonstrated that Mif deficiency aggravated cognitive functions in APP23 mice, as evidenced by Morris water maze test displaying impaired spatial learning ability. They overall concluded that neuronal secretion of MIF might serve as a defense mechanism to compensate for cognitive decline in AD. In another experimental study, MIF was observed to decrease the activation of caspase-3 and hydrogen peroxide (H2O2)-induced cell death, and to thus be protective against neuronal loss under hypoxia (Zhang et al., 2014).
In aggregate, there still is a limited number of experimental and clinical data available that have addressed the role of MIF in AD, strongly arguing for the necessity of additional mechanistic studies as well as large-scale clinical trials to clarify the role of MIF and its receptor pathways in AD.
MIF in ischemic stroke
We recently comprehensively reviewed the available data on MIF in ischemic stroke (Wang et al., 2021b). However, as stroke is a second major neurological condition, for which both protective and exacerbating effects have been reported for MIF, we will briefly discuss the evidence in the context of the current review and as compared with MIF’s role in other neurological diseases.
MIF was found to be upregulated in peripheral blood mononuclear cells (PBMCs) or serum from patients with acute stroke, and upregulation of MIF had a positive correlation with infarct volume and long-term clinical outcome of patients, together stressing the clinical significance of MIF in stroke (Wang et al., 2009; Li et al., 2017b; Wang et al., 2019b; Wang et al., 2019c). Similarly, MIF’s cognate receptor CD74 was significantly elevated in PBMCs and also positively correlated with infarct volume and neurological outcome of stroke (Yang et al., 2017b). Notwithstanding the clear overall clinical association between MIF expression and stroke pathogenesis, the precise cellular and molecular role of MIF in stroke turns out to be more complex and in part controversial, calling for systematic experimental studies in the future.
Three studies performed in mice and rats demonstrated a detrimental role of MIF in experimental stroke (Inácio et al., 2011b; Inácio et al., 2011c; Inácio et al., 2011a). Mechanistically, MIF worsened neurologic deficits by aggravating neuronal death, as displayed by in vivo evidence from a tMCAo mouse model and in vitro data from cultured cortical neurons under OGD treatment. Additionally, MIF deficiency was strongly linked to higher immunoreactivity of galectin-3, indicating possible effects of MIF on molecular components of the microglia response after stroke (Inácio et al., 2011c). Furthermore, the same team showed that lack of MIF did not change the expression of inflammatory cytokines in the following 7 days after stroke, arguing for a neuronal rather than pure inflammatory mechanism of MIF (Inácio et al., 2011b; Inácio et al., 2011c). Another BBB-related mechanism supporting the deleterious impact of MIF in stroke was identified by Liu et al (2018), who showed that MIF administration worsened BBB integrity, while MIF inhibitor ISO-1 reversed this effect. Interestingly, a previously unrecognized intracellular function of MIF acting as a nuclear-targeted nuclease related to neuronal death was revealed (Wang et al., 2016). The Vandenbark group has developed a partial MHC class II/peptide construct termed DRα1-MOG-35-55 that showed a therapeutic effect on infarct volume in a MCAo model, presumably by blocking MIF/CD74 binding (Benedek et al., 2014; Wang et al., 2017).
By contrast, other studies suggested that a neuroprotective impact of MIF may predominate after experimental stroke, some of which may be sex-specific. To this end, while no effect of Mif deficiency was seen in male mice, a worse infarct outcome was observed in Mif knockout mice in females, an effect that was independent of estrogen levels and proinflammatory cytokines (Turtzo et al., 2013). Zhang and co-workers reported that male Mif–/– mice developed larger infarct sizes compared to control mice after experimental stroke (Zhang et al., 2014). These findings thus indicate a protective role of MIF against neuronal cell death following stroke. In aggregate, current studies on the role of MIF in stroke call for additional in-depth studies to clarify the partially contradictory outcomes and to better understand the underlying mechanisms. Such studies should also include MIF homolog MIF-2 and MIF’s receptors, but also address potential intracellular mechanisms.
Conditioning considerations and relevance for MIF’s role in neurological diseases
The concept of ‘‘ischemic preconditioning’’ was first coined by Murry et al. (1986) in 1986 for the ischemic myocardium. In the context of neurological conditions, remote ischemic conditioning (RIC) or preconditioning (RIPC) refers to an ischemic stimulus distant from the brain (for example transient limb ischemia) that can produce a neuroprotective effect after a neurological insult, e.g. after a stroke. The large-scale phase II RIC After Stroke Trial (RECAST) clinical trial recently demonstrated that RIC after acute stroke could improve neurological outcome and reduce vascular event rates, with possible molecular mechanisms involving released heat shock protein 27 (HSP27) (England et al., 2017). In addition to acute vascular ischemia (AVI) such as acute myocardial infarction (AMI) and stroke, RIC may also work in chronic neurodegenerative conditions, as comprehensively reviewed by Hess and coauthors (2015a) and You et al. (2019). One of the reasons is that neurovascular dysfunction significantly contributes to cognitive impairment and dementia (CID) in neurodegenerative diseases, especially in AD, as discussed in two comprehensive reviews (Bell and Zlokovic, 2009; Iadecola, 2010).
An increasing body of evidence has emerged that clarifies the mechanisms of RIC on stroke outcomes, as nicely summarized in recent reviews (Pan et al., 2016; Landman et al., 2019). Classical immunological, neuronal, and humoral pathways are believed to orchestrate this regulation (Weber, 2010; Pan et al., 2016). Wang et al. (2021a) showed that RIC upregulated 2,3-biphosphoglycerate (2,3-BPG) expression in red blood cells, which could explain how RIC affected an improved oxygen supply under ischemia. Additionally, RIC displayed a protective effect on blood flow by inhibiting collateral collapse as shown in a rat MCAo model (Ma et al., 2017). Another study confirmed the protection by RIC against stroke in aged rats by suppressing signaling through the TLR4/MyD88 pathway (Lv et al., 2021). With regard to chronic conditions, Khan et al. (2018) demonstrated that RIC promoted neurovascular remodeling in a vascular CID mouse model, whereby one month of RIC achieved the best outcome. Ren et al. (2018) found that RIC enhanced angiogenesis by regulating endothelial NOS/NO in a model of chronic cerebral hypoperfusion (CCH) in rats. Li et al. (2017a) applied the same model and showed that RIC accelerated myelination by activating the phosphatase and tensin homolog (PTEN)/Akt/mammalian target of rapamycin (mTOR) pathway. Collectively, these studies suggest that RIC favors a protective role in acute and chronic neurological disorders through different mechanisms.
In addition to RIC, there are other conditioning paradigms that have been applied to neurological disorders, expanding the scope of neuroprotective stress factors to hypoxia, temperature, certain chemicals, or neuroinflammatory mediators (Stetler et al., 2014). To this end, Dirnagl and colleagues suggested the so-called ‘‘sensor-transducer-effector’’ principle (Dirnagl et al., 2003). For example, moderate ethanol preconditioning (MEP) and oleanolic acid preconditioning (OAP) protect against Aβ toxicity, which could be linked to the treatment of AD (Mitchell et al., 2009; Zhang et al., 2018). Mechanistically, MEP suppressed Aβ-associated neurotoxicity in mixed cerebellar cultures and this neuroprotection was dependent on NMDAR activation, as shown by dual fluorescence staining (Mitchell et al., 2009). Another paradigm of conditioning applied in AD was intermittent hypoxic conditioning (IHC), similarly showing remarkable improvement in a transgenic AD mouse model (Correia et al., 2021).
Of note and unlike in preconditioning inducing neuroprotection, ‘‘negative conditioning’’ has also been reported and is defined as a situation when detrimental effects caused by exposure to a factor are greater than those induced by deprivation of this factor (Sun et al., 1996). Loss of gonadal steroids and targeting mitochondrial dysfunction have been discussed as negative conditioners in neurological diseases, especially for neurodegenerative conditions, as reviewed in the field of conditioning medicine (Selvaraji et al., 2019; Sun et al., 2019).
The role of MIF as a conditioning cue has recently been reviewed in an article published in a previous volume of Cond Med (Wang et al., 2021b). As such, the potential role of MIF in RIC has so far mostly been studied in the context of hepato- and cardioprotection (Goetzenich et al., 2014; Emontzpohl et al., 2019; Ruze et al., 2019; Wang et al., 2019a), as summarized in Table 2 and commented on by Bernhagen in 2019 (Bernhagen, 2019). Emontzpohl and colleagues (2019) applied RIC in rats after orthotopic liver transplantation (OLT) and demonstrated that RIC including remote ischemic preconditioning (RIPC/IPC) and post-conditioning (RIPOST/RIPostC) ameliorated hepatocellular injury, with a significant downregulation in MIF and transaminases in serum and liver, suggesting the involvement of MIF in hepatoprotective conditioning. MIF was even more prominently implicated in cardioprotection. Ruze and colleagues (2019) showed that MIF participated in RIPC-induced cardioprotection after myocardial ischemia-reperfusion (I/R) injury. RIPC (5 min ischemia plus 5 min reperfusion for three cycles) was performed in WT mice before an ischemic insult of 40 min and improved cardiac dysfunction, as qualified by echocardiography, as well as other assessment indices such as infarct size and enzymatic markers. By contrast, Mif deficiency abolished the cardioprotective effect induced by RIPC. This was associated with activation of the reperfusion injury salvage kinase (RISK) and AMPK signaling pathways, pointing towards an underlying mechanism of MIF in RIPC-mediated cardioprotection (Ruze et al., 2019). In addition to RIPC, MIF was implicated in cardioprotection conferred by RIPostC (5 min ischemia plus 5 min reperfusion for four cycles, after an ischemic insult of 20 min) in a HIF-1α-dependent manner (Wang et al., 2019a). Moreover, this effect elicited by MIF was confirmed in an inhibition approach using ISO-1 and mechanistically associated with the activation of the cardiac AMPK pathway (Wang et al., 2019a). Clinical data reanalyzed from the RIPHeart study showed elevated serum and tissue MIF levels in cardiac surgery patients after RIPC, confirming MIF could be a possible mediator of RIPC (Ney et al., 2018). Yet another form of conditioning is anesthetic–induced preconditioning (AIP or AIPC). One MIF/AIP study validated the protective property of MIF in the injured heart in the context of AIP (Goetzenich et al., 2014). They utilized isoflurane to induce preconditioning in primary ventricular myocytes isolated from rats in vitro, and showed that MIF was elevated and that this was associated with the activation of AMPK and PKCε. Taken together, these studies indicate that MIF could be a protective conditioning target.
Despite the disappointment that the entire RIC/RIPC field has experienced due to the neutral outcome of two independent large-scale multicenter clinical trials on cardiac protection in 2015 (Hausenloy et al., 2015; Meybohm et al., 2015), conditioning, and especially RIC/RIPC, has continued to be an appealing paradigm in neuroprotection. This concept is substantiated by more and more preclinical and clinical evidence (Li et al., 2017a; Ma et al., 2017; Khan et al., 2018; Ren et al., 2018; Lv et al., 2021; Wang et al., 2021a), and was recently reviewed in several excellent articles (Hess et al., 2015b; Hess et al., 2015a; Pan et al., 2016; Landman et al., 2019). Although direct experimental evidence is still lacking, MIF, as a prototypical inflammatory cytokine and ACK (Kang and Bucala, 2019; Kapurniotu et al., 2019; Sinitski et al., 2019), may qualify as a potential RIC signaling cue according to criteria suggested by Stoppe and coauthors (2009). A potential role in neuroprotective conditioning is further supported by evidence for MIF in protective cardiac and hepatic conditioning.
Conflict of Interest
J.B. is an inventor on patent applications related to anti-MIF strategies in inflammatory and cardiovascular diseases, but authors declare no competing interests regarding the specific topics in this article.NOTE FROM EDITORIAL OFFICE: Please add conflict of interest statement.
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant excellence initiative program LMUexc/strategic partnerships with Singapore to J.B., by DFG grant BE 1977/14-1 to J.B., and by the Munich Cluster for Systems Neurology (EXC 2145 SyNergy-ID 390857198) to J.B. and O.G.; C.Z., S.W., and L.L. acknowledge support by the China Scholarship Council (CSC) fellowship program CSC/LMU. Figures were created with BioRender.com.
References
Chunfang Zan1
1Division of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilians-University (LMU), 81377 Munich, Germany.
Sijia Wang1
1Division of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilians-University (LMU), 81377 Munich, Germany.
Lu Liu2
2Systems Neuroscience Group, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilians-University (LMU), 81377 Munich, Germany.
Ozgun Gokce2
2Systems Neuroscience Group, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilians-University (LMU), 81377 Munich, Germany.
Omar El Bounkari1
1Division of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilians-University (LMU), 81377 Munich, Germany.
Jürgen Bernhagen1,3,4#
1Division of Vascular Biology, Institute for Stroke and Dementia Research (ISD), LMU Klinikum, Ludwig-Maximilians-University (LMU), 81377 Munich, Germany. 3Munich Heart Alliance, 80802 Munich, Germany. 4Munich Cluster for Systems Neurology (SyNergy), 81377 Munich, Germany.
Corresponding author:
Dr. Jürgen Bernhagen
Email: Juergen.Bernhagen@med.uni-muenchen.de
In a new window | Download PPT
Figure 1: MIF has been implicated in neuroinflammation through the regulation of microglia, astrocytes, neurons, and infiltrating immune cells. Glial cells including microglia and astrocytes dominate the inflammatory process, as indicated in the figure. MIF not only induces astrocyte and microglia activation, but also triggers an innate immune response by triggering the release of several potent proinflammatory cytokines such as IL-1β, IL-6 and TNF-α, at least partially dependent on glial activation, in the central nervous system. Besides inflammatory regulation in glial cells, astrocyte activation indirectly promotes tau hyperphosphorylation, and microglial activation enhances Aβ accumulation and toxicity. Additionally, MIF exerts neuroprotective effects on neurons via inhibition of apoptosis and improvement of neuronal survival, a notion that still is controversial and under active investigation. In addition to neuronal cells, infiltrating immune cells are also involved in neuroinflammation, and MIF behaves as a key cellular mediator promoting lymphocyte and monocyte/macrophage infiltration across the blood-brain barrier (BBB). Overall, MIF plays a dichotomic role in different cell types and disease entities and contributes to different neurological disorders through complex cellular mechanisms and signalling pathways.
In a new window | Download PPT
Figure 2: The multifaceted impact of MIF on several neurological diseases. Based on current preclinical and clinical studies, MIF plays a mostly beneficial role in Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS)/familial ALS (FALS), whereas a deleterious role for MIF has been suggested in multiple sclerosis (MS), spinal cord injury (SCI), and traumatic brain injury (TBI). The functions of MIF in Alzheimer’s disease (AD) and ischemic stroke (IS) are controversial with both detrimental and protective effects observed. Major mechanisms of MIF regulating pathological characteristics of different neurological diseases are indicated in the figure. SOD1, superoxide dismutase type 1; CD74, cluster of Differentiation 74; CCL5, chemokine (C-C motif) ligand 5; PGE2, prostaglandin E2; Aβ, amyloid-beta; CSF, cerebrospinal fluid; tMCAo, transient middle cerebral artery occlusion.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 9890 | 35 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA