Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Protecting coronary microvascular integrity in reperfused acute myocardial infarction to improve clinical outcome
Time:2022-12-04
Number:6487
Author Affiliations

Conditioning Medicine 2022. 5(4): 144-154.
Abstract
Acute myocardial infarction is a life-threatening condition caused by an abrupt obstruction of a coronary artery, which warrants timely reperfusion by primary percutaneous coronary intervention to salvage viable myocardium. Paradoxically, reperfusion itself often causes additional damage to cardiac cells, distinct from the ischemic damage. This form of reperfusion injury causes additional damage to cardiomyocytes, but importantly, also impairs the coronary microvascular endothelial cells. Although injury to the microvasculature contributes to poor long-term patient prognosis, relatively little work focuses on protecting the microvascular endothelial cells and endothelial barrier integrity. This review discusses how endothelial barrier function is affected in reperfused acute myocardial infarction, discusses recent studies that explored therapeutic options to preserve microvascular integrity, and suggests options for future research in this field.
Keywords: Acute myocardial infarction, Reperfusion injury, Microvascular injury, Endothelial barrier function, No-reflow, Cardioprotection
Abstract
Acute myocardial infarction is a life-threatening condition caused by an abrupt obstruction of a coronary artery, which warrants timely reperfusion by primary percutaneous coronary intervention to salvage viable myocardium. Paradoxically, reperfusion itself often causes additional damage to cardiac cells, distinct from the ischemic damage. This form of reperfusion injury causes additional damage to cardiomyocytes, but importantly, also impairs the coronary microvascular endothelial cells. Although injury to the microvasculature contributes to poor long-term patient prognosis, relatively little work focuses on protecting the microvascular endothelial cells and endothelial barrier integrity. This review discusses how endothelial barrier function is affected in reperfused acute myocardial infarction, discusses recent studies that explored therapeutic options to preserve microvascular integrity, and suggests options for future research in this field.
Keywords: Acute myocardial infarction, Reperfusion injury, Microvascular injury, Endothelial barrier function, No-reflow, Cardioprotection
Introduction
Acute myocardial infarction (AMI) arises from an immediate epicardial coronary artery occlusion, which causes irreversible injury starting in the subendocardium and expanding toward the epicardium with ongoing occlusion (Reimer et al., 1977). Therefore, AMI requires timely restoration of blood flow by percutaneous coronary intervention (PCI) to salvage the myocardium (Ibanez et al., 2018). Paradoxically, in approximately half of PCI-treated AMI patients (van Kranenburg et al., 2014; de Waha et al., 2017), reperfusion of previously ischemic tissue inflicts incremental cardiac damage distinct from ischemic damage, hampering restoration of myocardial perfusion. This form of ischemia-reperfusion (IR) injury affects the coronary microvasculature and is known as microvascular obstruction or microvascular injury (MVI) (Robbers et al., 2013; Hollander et al., 2016; Konijnenberg et al., 2020). Independent of infarct size, MVI contributes to worse patient prognosis (van Kranenburg et al., 2014; de Waha et al., 2017). MVI is characterized by a cascade of pathophysiological changes, including loss of microvascular integrity, endothelial cell dysfunction, endothelial cell damage, and the development of intramyocardial hemorrhage (IMH) (Robbers et al., 2013; Hollander et al., 2016; Nair et al., 2020; Liu et al., 2022). This highlights that targeting MVI, in addition to current care, may represent a clinically relevant area of study. Although currently no widely approved therapeutic options for MVI exists in AMI patients, some preclinical studies have reported promising results. Targeting coronary endothelial barrier function, with the purpose of limiting the extent of MVI, could be a promising adjuvant strategy in addition to current care to improve patient outcome. This review discusses how microvascular endothelial cells are affected in reperfused AMI, discusses recent (pre)clinical studies on therapeutic options on top of current care to attenuate MVI, and highlights future directions.
The coronary microvasculature
The coronary microvasculature is a heterogenous network of vessels with a diameter smaller than 100 µm, including arterioles, capillaries, and venules. The microvasculature adapts to physiological and pathophysiological stimuli and has a key role in regulating vascular tone, blood flow, and oxygen transport. Blood that enters the coronary microcirculation starts in arterioles, which consist of endothelial cells and basement membrane, and are surrounded by vascular smooth muscle cells. Arterioles are the main regulators of myocardial blood flow. Subsequently, blood flows into the capillaries, which consist of a single layer of endothelial cells surrounded by basement membrane and are closest to the cardiomyocytes. Capillaries are crucial in the exchange of nutrients, oxygen, and waste products to the cardiomyocytes. Finally, capillaries converge in postcapillary venules that allow deoxygenated blood return to the venous system (Fonseca et al., 2016).
In the context of reperfusion-induced microvascular injury, several components of the coronary capillaries are important to highlight: the glycocalyx, the endothelial cell and its cell junctions, and the basement membrane. The glycocalyx covers the apical surface of the endothelial cell and consists of a network of mainly glycoproteins, proteoglycans, and plasma and endothelium-derived soluble components. The glycocalyx acts as the first line of defense for the cell against MVI (Reitsma et al., 2007). Endothelial cells line the interior surface of the vessel and are connected by cell-cell junctions, including adherens junctions and tight junctions, which permit passage of water and other molecules across the endothelium (Komarova et al., 2017). These cell-cell junctions form a crucial part in maintaining and preserving the integrity of the microvasculature. The basement membrane is attached to the basolateral side of the endothelium and consists of networks of mainly laminin and type IV collagen (Jayadev and Sherwood, 2017).
Determinants of microvascular injury during reperfusion
During myocardial ischemia, oxygen transport to the myocardium and endothelial cells is significantly restricted. Prolonged ischemia results in reduced cytosolic adenosine triphosphate production, affects ion-exchange channels, and induces anaerobic metabolism and metabolic acidosis (Pike et al., 1993). In experimental models, a longer duration of ischemia seemed to be an important determinant of the severity of MVI to a large extent (Kloner et al., 1974; Reffelmann et al., 2002). Clinical trials, however, show conflicting results regarding the association between time to reperfusion and MVI severity (Husser et al., 2013; Amier et al., 2017; Ferré-Vallverdú et al., 2021). One potential reason is that endothelial cells are relatively resistant to hypoxia (Quintero et al., 2006; Baldea et al., 2018). Anaerobic glycolysis can provide sufficient energy for endothelial cells (Culic et al., 1997), making them less prone to hypoxic injury compared to cardiomyocytes (Mertens et al., 1990). Although endothelial cells tolerate hypoxic conditions relatively well, endothelial cells are particularly vulnerable to reperfusion injury (Maxwell and Gavin, 1991; Hollander et al., 2016). Indeed, reperfusion results in an immediate, progressive deterioration of microvascular integrity (Hollander et al., 2016; Sezer et al., 2022), underlining the importance of early intervention. Upon presence of IR injury, key findings at the ultrastructural level include occurrence of swelling and blebbing of endothelial cells, presence of membrane-bound vesicles and cellular debris in the perivascular space (Maxwell and Gavin, 1991). In more severe cases, reperfusion injury causes thinning and rupture of endothelial cells, reduction in endothelial cell-junctions, and extravasation of erythrocytes (Hollander et al., 2016), whereas ischemia alone induces only mild morphological changes to the coronary microcirculation (Maxwell and Gavin, 1991; Hollander et al., 2016). These changes in endothelial cell function and structure upon reperfusion lead to increased vascular permeability and additional cellular damage. Mechanistically, various components modulate the extent of vascular permeability (Figure 1). Identifying the most important factors within this process could result in the development of novel therapeutic approaches, specifically targeted at these factors, in an attempt to further limit MVI (Table 1).
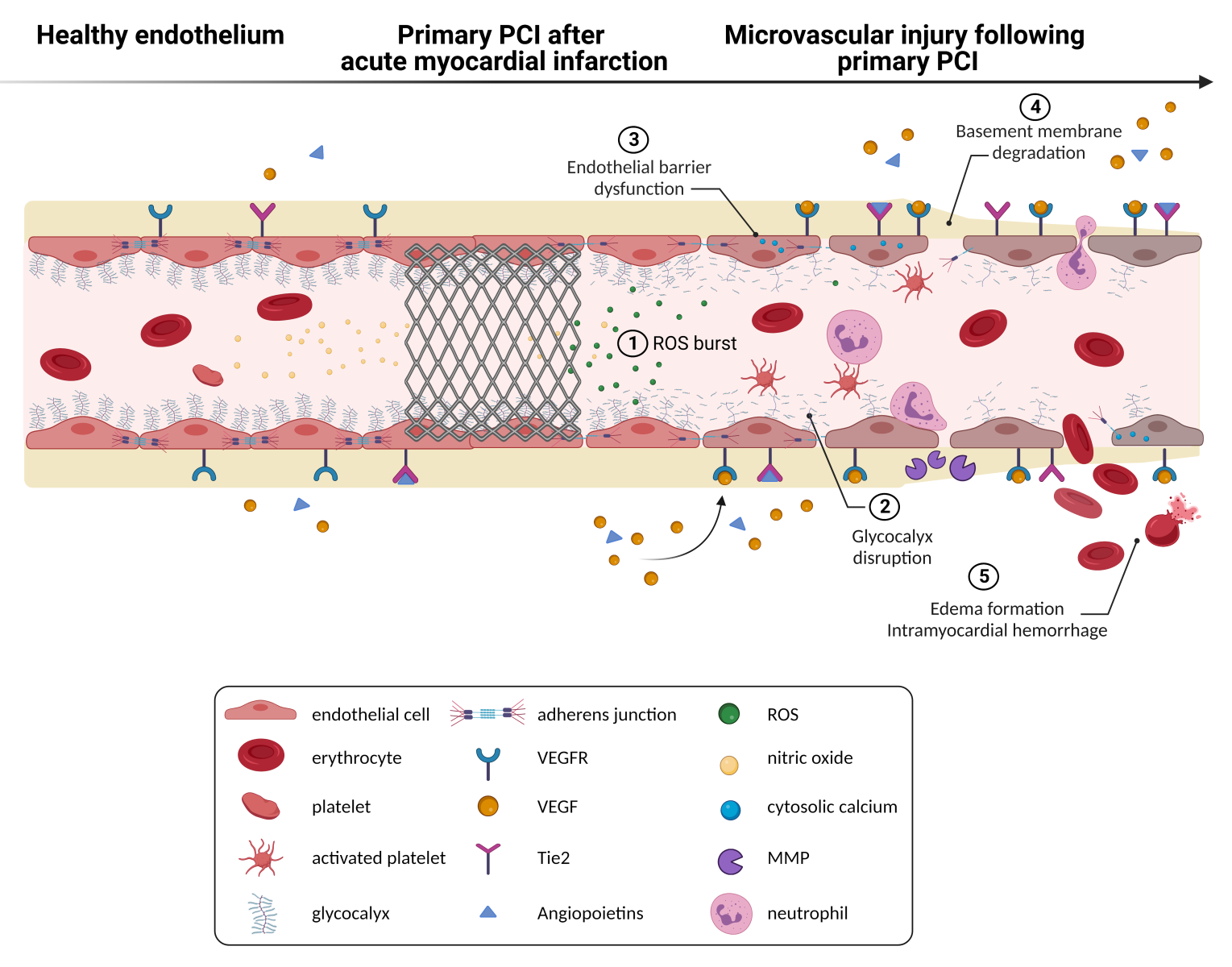
In a new window | Download PPT
Figure 1: Reperfusion of an occluded coronary artery is often accompanied by microvascular injury. Microvascular injury is reflected by a sequeala of pathophysiological changes, including increased production of reactive oxygen species, disruption of the glycocalyx, endothelial barrier dysfunction with increased cytosolic calcium levels, loss of endothelial cell-cell junctions, degradation of the basement membrane, and in more severe cases formation of edema and intramyocardial hemorrhage. In turn, hemolysis of erythrocytes can expose cardiomyocytes to cytotoxic heme. Preclinical studies suggest that pharmacologically targetting components of the coronary microvasculature may have potential as an adjuvant therapy in acute myocardial infarction patients. MMP, matrix metalloproteinases; PCI, percutaneous coronary intervention; ROS, reactive oxygen species; Tie2, receptor tyrosine kinase; VEGF, vascular endothelial growth factor; VEGFR, VEGF-receptor.
Strategies to minimize coronary microvascular injury
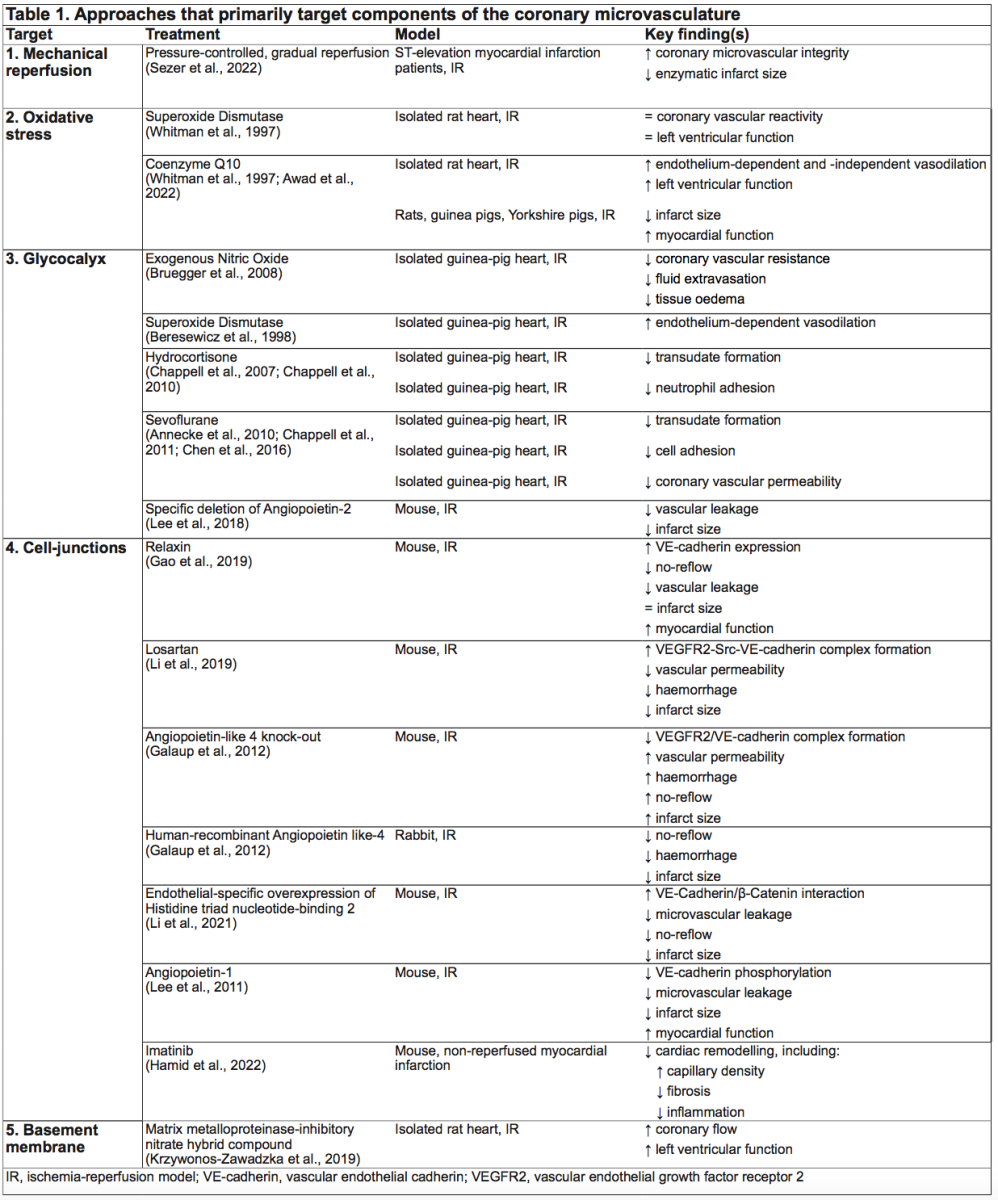
Prevention of MVI may have strong clinical potential, especially since MVI is related to clinical outcomes and MVI is currently not directly targeted. For this purpose, we have summarized studies that focused on potential targets of MVI; i.e. gradual reperfusion, oxidative stress, glycocalyx, endothelial barrier, and basement membrane.
Mechanical reperfusion by primary percutaneous coronary intervention
Primary PCI causes an abrupt restoration of intracoronary blood flow and pressure, leading to an imbalance between hydrostatic and colloid osmotic forces, and consequently increased vascular permeability and intramyocardial edema. Gradual or staged reperfusion could be an alternative approach to preserve coronary microvascular integrity. Gradual reperfusion has shown favorable effects in various experimental models (Okamoto et al., 1986; Takeo et al., 1995; Sato et al., 1997; Bopassa et al., 2005; Musiolik et al., 2010; Ferrera et al., 2015), including reduction in final myocardial infarct size (Okamoto et al., 1986; Sato et al., 1997; Bopassa et al., 2005; Musiolik et al., 2010; Ferrera et al., 2015) and better-preserved microvascular integrity (Okamoto et al., 1986). Several cardioprotective mechanisms of gradual reperfusion have been proposed; reduction of reactive oxygen species (ROS) production (Bopassa et al., 2005), preservation of endothelial function (Sato et al., 1997), and lowering reperfusion-induced sodium and calcium overload (Takeo et al., 1995). In contrast to the promising effects in preclinical work, gradual reperfusion in clinical trials has shown mixed results (Carrick et al., 2014; Kelbæk et al., 2016; Sezer et al., 2022). In patients with ST-elevation myocardial infarction (STEMI), gradual reperfusion by deferred stent implantation approximately nine hours after the index procedure reduced the extent of no-reflow and increased myocardial salvage at six month follow-up (Carrick et al., 2014). In contrast, deferred stent implantation approximately three days after the index procedure did not show any improvement in patient prognosis at the approximately 3.5 year follow-up (Kelbæk et al., 2016). Interestingly, a recent pilot study showed that pressure-controlled, gradual reopening of the culprit artery with deferred stent implantation 30 minutes after initial reperfusion resulted in improved microcirculatory response to reperfusion and enzymatic smaller myocardial infarct size compared to PCI with intermediate stenting (Sezer et al., 2022). Being in its infancy, clinical studies that aim to reduce MVI through (pressure-controlled) gradual reopening of the infarct-related artery could have benefit over immediate stenting. We suggest future work is required, including larger sized studies with longer follow-up, but also work around improving strategies around reopening the infarct-related artery.
Oxidative stress
ROS are generated at high levels by the re-introduction of oxygen. The role of ROS in reperfusion-induced microvascular dysfunction has been extensively reviewed by Yu et al (2019). Sources of ROS production include uncoupled endothelial nitric oxide synthase (eNOS), mitochondria, infiltrating leukocytes, and vascular adhesion protein-1. During reperfusion, ROS levels can exceed the antioxidant defense system, resulting in cellular oxidative stress that aggravates MVI. Moreover, increased ROS generation can directly and indirectly lower intracellular nitric oxide (NO) bioavailability. Increased production of the reactive oxygen ion superoxide during reperfusion interacts with NO to form peroxynitrate, in turn inhibiting endothelial-dependent vasorelaxation (O'Donnell et al., 1997). Furthermore, ROS can indirectly lower NO levels by uncoupling eNOS, which modifies eNOS monomers into ROS-producing units (Landmesser et al., 2003). However, clinical trials that administered vasodilating agents did not show a reduction in MVI (Vijayalakshmi et al., 2006; Desmet et al., 2011; Nazir et al., 2016).
Studies that target ROS formation, including antioxidant treatment and various forms of preconditioning, have mainly focussed on cardiomyocyte protection and have led to contradictory results (Rodrigo et al., 2013; Kalogeris et al., 2014; Zhou et al., 2015; Rodrigo et al., 2022). In part, these conflicting results may relate to the observation that, albeit ROS is involved in damage to the microvasculature, at certain dosages ROS has cardioprotective effects. Studies that specifically aim to reduce reperfusion-induced endothelial cell damage are scarce. In a mouse model of cerebral IR injury, transgenic overexpression of antioxidant superoxide dismutase (SOD) showed less vascular endothelial cell death, but did not reduce the rate of hemorrhagic transformation (Maier et al., 2006). In isolated rat hearts, SOD did not preserve coronary vascular reactivity nor improve left ventricular function after IR (Whitman et al., 1997). However, in isolated rat hearts subjected to IR, treatment with Coenzyme Q10 (CoQ10), which is a component of the mitochondrial electron transport chain, preserved endothelium-dependent and -independent vasodilation via free radical scavenger action, and improved left ventricular function (Whitman et al., 1997). Additionally, CoQ10 treatment reduced infarct size in rats, guinea pigs, and Yorkshire pigs (Awad et al., 2022). This means that targeting ROS shows promise in limiting endothelial cell injury in preclinical studies, but no clinical data is available yet. In addition, the ability of ROS (dependent on its dose) to be both harmful and protective against injury, makes ROS a target in this area challenging.
Glycocalyx disruption
A 0.5 µm thick glycocalyx layer covers the vascular endothelium (van den Berg et al., 2003; Nieuwdorp et al., 2008) and consists of mainly proteoglycans (syndecans and glypicans) with long glycosaminoglycan side chains (such as heparan sulfate and hyaluronan), glycoproteins with short carbohydrate side chains, and soluble components such as albumin, thrombomodulin, and extracellular SOD (Reitsma et al., 2007). The endothelial glycocalyx plays a crucial role in vascular permeability (Vink and Duling, 2000; van Haaren et al., 2003; Rehm et al., 2004; Betteridge et al., 2017) and shear stress-induced signalling pathways, including NO production (Pahakis et al., 2007; Green et al., 2017). In animal experiments, enzymatic degradation of the glycocalyx resulted in an increase of the interstitial space, indicative of myocardial edema (van den Berg et al., 2003). In isolated guinea pig hearts subjected to short IR, addition of enzymatic degradation of the glycocalyx significantly increased vascular permeability (Rehm et al., 2004). Ischemia induces only limited damage to the glycocalyx, whereas addition of reperfusion strongly accelerates the damage to the glycocalyx (Beresewicz et al., 1998). Shedding of endothelial glycocalyx has been reported in experimental IR models (Beresewicz et al., 1998; Platts et al., 2003) and in patients undergoing cardiac surgery such as aortic valve replacement (Bruegger et al., 2015; Passov et al., 2021). The consistency of the glycocalyx can be modulated by several factors, including shear stress (Wang et al., 2020), locally produced ROS (Rubio-Gayosso et al., 2006) and cytokines (Wiesinger et al., 2013). Thickness and stiffness of the glycocalyx is reduced by tumor necrosis factor-alfa (Wiesinger et al., 2013) via activation of matrix metalloproteinases (MMPs) (Ramnath et al., 2014). Furthermore, degradation and shedding of the glycocalyx contribute to leukocyte-endothelial cell adhesion (Constantinescu et al., 2003; Schmidt et al., 2012).
Several studies in animals have focussed primarily on the preservation of the glycocalyx after IR and its potential clinical benefit. In isolated guinea pig hearts, NO reduced shedding of the glycocalyx, accompanied by reduced coronary vascular resistance, coronary fluid extravasation, and tissue edema (Bruegger et al., 2008). Also SOD (Beresewicz et al., 1998; ubio-Gayosso et al., 2006), hydrocortisone (Chappell et al., 2007; Chappell et al., 2010), and sevoflurane (Annecke et al., 2010; Chappell et al., 2011; Chen et al., 2016) have shown to preserve the endothelial glycocalyx after IR. In mice subjected to IR, endothelial specific deletion of angiopoietin-2 attenuated endothelial glycocalyx degradation and vascular leakage, and reduced infarct size in the acute phase of myocardial infarction (Lee et al., 2018). Interestingly, simulating a new glycocalyx by a selectin-targeting glycocalyx-mimetic reduced neutrophil extravasation, macrophage accumulation, improved cardiac function, and reduced infarct size (Dehghani et al., 2022). These preclinical studies suggest that preventing glycocalyx degradation in IR can be a promising goal in cardioprotection, although studies in humans are lacking to date.
Endothelial barrier dysfunction
Endothelial cells form the inner lining of the vascular system and maintain vascular homeostasis. An early indicator of loss of vascular integrity is an increase in vascular permeability, which is mainly controlled by endothelial cell calcium homeostasis and cell junctions, importantly contributing to endothelial dysfunction.
Calcium homeostasis
Calcium (Ca2+) is a second messenger involved in signalling pathways that are crucial for regulating endothelial barrier function. Reperfusion can provoke endothelial barrier dysfunction by causing an increase in cytosolic Ca2+ levels and changing endothelial cell shape by activating its cytoskeletal contractile elements. Consequently, the formation of intercellular gaps promotes endothelial permeability and edema formation (Kasseckert et al., 2009; Li et al., 2020). Furthermore, Ca2+ plays a critical role in cell-cell adhesion. Chelation of extracellular Ca2+ promotes the internalization of the cell adhesive cadherin molecules (Le et al., 1999). On the other hand, repletion of extracellular Ca2+ resulted in restoration of vascular endothelial (VE)-cadherin integrity (Gao et al., 2000).
Endothelial cell junction disruption
Endothelial permeability is regulated via transcellular (through the endothelial cell via caveolae-mediated vesicular transport) and paracellular (between the endothelial cells via interendothelial cell junctions) pathways. Here we will focus on the paracellular pathway since this is the dominant mechanism of increased vascular permeability in the heart under pathophysiological conditions (Bazzoni and Dejana, 2004). In endothelial cells, adherens junctions and tight junctions are intermingled along sites of cell-to-cell contacts and have crucial roles in cell-signaling (Bazzoni and Dejana, 2004; Dejana, 2004; Hu et al., 2013).
Adherens junctions
The key component of adherens junctions is the transmembrane adhesion protein VE-cadherin. VE-cadherin interacts with intracellular proteins, including β- and γ-catenins. In turn, these catenins link to α-catenin and connect VE-cadherin to actin filaments, supporting junctional stability (Bazzoni and Dejana, 2004; Dejana, 2004; Hu et al., 2013). The function of VE-cadherin and catenins can be modulated by tyrosine phosphorylation (Esser et al., 1998; Potter et al., 2005; Orsenigo et al., 2012; Wessel et al., 2014). For example, VE-cadherin phosphorylation at the Tyr658 residue was shown to disrupt endothelial barrier function in vitro (Potter et al., 2005). In vivo, besides the VE-cadherin Tyr658 residue, the Tyr685 residue seems to play an important role in endothelial barrier function. In response to mediators such as vascular endothelial growth factor (VEGF), histamine, and bradykinin, phosphorylated VE-cadherin is internalized and ubiquitinated, worsening endothelial barrier function (Orsenigo et al., 2012; Wessel et al., 2014).
Tight junctions
Key components of tight junctions are claudin, occludin, and junction adhesion molecules (JAMs) (Bazzoni and Dejana, 2004; Dejana, 2004; Hu et al., 2013). Of the claudin members, endothelial cells predominantly express claudin-5. In claudin-5 deficient mice, blood-brain barrier permeability was increased for small molecules, suggesting that claudin-5 actively contributes to maintaining endothelial barrier function (Nitta et al., 2003). Claudin, occludin and JAMs link with the actin cytoskeleton via zona occludin-1 protein, thereby stabilizing endothelial barrier function (Bazzoni and Dejana, 2004; Dejana, 2004; Hu et al., 2013). Besides its role in regulating endothelial barrier function, JAMs appear to mediate direct leukocyte/platelet/endothelial cell binding interactions (Garrido-Urbani et al., 2014).
Targeting endothelial cell junctions
Function of both adherens and tight junctions are important in maintaining vascular integrity. Whereas ischemia alone did not reduce the number of endothelial cell junctions in a rat coronary artery model, subsequent reperfusion disrupted almost all cell junctions (Hollander et al., 2016). This further highlights the importance of the reperfusion phase in causing injury to the (coronary) microvasculature. In a mouse model of myocardial IR injury, the peptide hormone relaxin partially restored VE-cadherin protein expression, and subsequently reduced the extent of no-reflow, and improved myocardial function (Gao et al., 2019). Furthermore, losartan, an angiotensin II receptor blocker, inhibited phosphorylation of Src and VE-cadherin. This resulted in increased VEGFR2-Src-VE-cadherin complex formation, a reduction in vascular permeability, hemorrhage, and infarct size (Li et al., 2019). Moreover, in angiopoietin-like 4 (ANGPLT4) knock-out mice, vascular permeability, edema, hemorrhage, no-reflow, and infarct size were increased, accompanied by dissociation of the VEGFR2/VE-cadherin complex (Galaup et al., 2012). Injecting recombinant ANGPTL4 in rabbits reduced the extent of hemorrhage, no-reflow, and infarct size (Galaup et al., 2012). In another study, overexpression of histidine triad nucleotide-binding 2 enhanced VE-cadherin/β-catenin interaction, and subsequently attenuated microvascular leakage, improved myocardial perfusion, and reduced infarct size (Li et al., 2021). Angiopoietin-1, an endothelial-specific angiogenic factor and ligand for the endothelial-specific Tie2 receptor, promoted endothelial barrier function via reduction of tyrosine phosphorylation of VE-cadherin, reduced infarct size, and improved myocardial function (Lee et al., 2011). In addition to these targeted preclinical studies, several studies explored the impact of the tyrosine kinase inhibitor imatinib, and found it to be effective in preserving endothelial barrier function in cultured endothelial cells, mainly by interfering with VE-cadherin and β-catenin (Dejana et al., 2008; Aman et al., 2012; Chislock and Pendergast, 2013). Interestingly, imatinib has been investigated in a wide range of animal models. In a sepsis model (Aman et al., 2012), pulmonary IR model (Tanaka et al., 2016; Magruder et al., 2018), cardiopulmonary bypass model (Koning et al., 2018), and cerebral IR models (Su et al., 2008; Merali et al., 2015), imatinib was able to reduce the extent of microvascular leakage. In addition, in mice with nonreperfused myocardial infarction, treatment with imatinib alleviated cardiac remodeling, including a higher capillary density, less fibrosis, and less inflammation compared to vehicle (Hamid et al., 2022).
Taken together, these preclinical studies provide support for pharmacologically targeting endothelial cell-cell junctions in order to prevent injury to the endothelial barrier and limit reperfusion-induced microvascular leakage. These effects may ultimately translate to smaller infarct sizes, such as demonstrated in preclinical studies, although studies in humans are currently lacking.
Basement membrane degradation
The endothelial basement membrane is a thin, continuous layer of extracellular matrix and has a critical role in vessel stabilization. A basement membrane mainly consists of collagens, laminins, and proteoglycans, and can be degraded by proteinases, such as MMPs (Davis and Senger, 2005). Neutrophils are a major source of MMPs (Romanic et al., 2002). Of those MMPs, MMP-2 and MMP-9 play an important role in different forms of IR injury (Cheung et al., 2000; Peterson et al., 2000; Romanic et al., 2002; Sumii and Lo, 2002; Machado et al., 2006). Therefore, MMPs represent a potential target in IR, and inhibitors of MMPs hold promise as protection against MVI. Indeed, in a cerebral IR model, rats treated with a broad spectrum MMP inhibitor showed significantly reduced hemorrhagic volumes and mortality rate (Sumii and Lo, 2002). In support of these observations specific inhibition of MMP-9 also reduced cerebral hemorrhage, swelling, infarction, and mortality (Saleem et al., 2021). Comparable observations were reported in isolated rat hearts, as treatment with a MMP-inhibitory nitrate hybrid compound increased coronary flow and improved myocardial function after IR (Krzywonos-Zawadzka et al., 2019). In summary, these preclinical studies provide support for using (selective) MMP-inhibitors to attenuate injury to the basement membrane following reperfusion and may translate to (partial) preservation of myocardial function following IR.
Current challenges in translation from bench to bedside
Despite numerous experimental animal studies with promising cardioprotective strategies, the translation into clinical therapy is challenging and has been disappointing. As already extensively reviewed (Heusch, 2017; Kleinbongard et al., 2020), several experimental specific factors, such as study design and reproducibility, and animal specific factors, such as the lack of co-morbidities and the use of co-medication, have significantly contributed to the loss of translation in cardioprotection from bench to bedside. Indeed, in most preclinical studies young and healthy animals have been used, but mainly with the purpose to reveal underlying mechanisms rather than demonstrating translational value. Regarding AMI models, animal gender should be thoroughly reported, as estrogen can be a major driver of microvascular cardioprotective pathways (Querio et al., 2021). These specific translational issues have already been addressed by Bøtker and colleagues, who published practical guidelines to ensure rigor and reproducibility of experiments in cardioprotection, including important remarks on experimental design and predefined in- and exclusion criteria (Bøtker et al., 2018). Furthermore, Lecour and colleagues (2021) published step-by-step criteria to improve the likelihood of translation of cardioprotective interventions into clinical therapy. Both guidelines will facilitate in designing more robust future cardioprotective approaches.
To date, in the clinical setting, no risk factors or clinical predictors have consistently been found for the development of MVI and/or IMH following AMI. Besides the use of additional glycoprotein IIb/IIIa inhibitors (Amier et al., 2017) and the anterior infarct location (Amier et al., 2017; Reinstadler et al., 2019), it remains unclear why some patients show MVI while others do not. Some evidence exists that admission glucose level in STEMI patients is independently related to MVI (Jensen et al., 2011) and IMH (Ota et al., 2022). However, having diabetes (Amier et al., 2017; Ota et al., 2022) or lowering blood glucose concentration with exenatide, a glucagon-like peptide-1 analogue, did not affect MVI (Roos et al., 2016). Interestingly, one day after primary PCI for STEMI, serum syndecans-1 level, a major component of the endothelial glycocalyx, was independently associated with MVO (Huang et al., 2021). Furthermore, local MMP-9 levels in the coronary artery upon reperfusion was higher in patients with IMH (Ota et al., 2022), showing the potential of coronary microvascular degradation products as biomarker for reperfusion injury over clinical risk factors.
In addition to optimizing the preclinical study design according to the consensus guidelines (Bøtker et al., 2018; Lindsey et al., 2018; Lecour et al., 2021), patient selection should be improved. Focusing on MVI, patients with a relatively short ischemic time, a large area at risk (such as can be expected in anterior infarcts), and a completely occluded coronary artery prior to primary PCI will benefit the most from cardioprotective strategies (Heusch, 2017). Importantly, as the first few minutes to hours is the most critical phase in generating reperfusion injury, timing of pharmacological interventions should be prior to or immediately at the onset of reperfusion, in addition to current care.
To the best of our knowledge, no clinical trials primarily targeting components of the coronary microvasculature exist. Changing focus from primarily targeting cardiomyocytes to the coronary microvasculature could pave the way for novel therapeutic approaches in reperfusion injury, starting in rodent models and proceeding to the porcine model, as this most closely resembles the (patho)physiology of the human heart (Heusch et al., 2011).
Conclusion
Microvascular barrier function requires intact glycocalyx, endothelium, and basement membrane. Damage to any of these vascular components, which seems a common feature in IR injury, can result in a leaky microcirculatory environment, edema formation, hemorrhage, and impaired ventricular function. Accordingly, microvascular injury in AMI patients seems a potential target, especially since current medicine does not directly target microvascular injury, but also because microvascular injury predicts clinical outcomes independent of infarct size. While studies in AMI patients on minimizing reperfusion injury have demonstrated limited clinical benefit, preclinical studies suggest that pharmacologically targetting the glycocalyx, the endothelium, endothelial cell junctions and/or the basement membrane may have potential as an adjuvant therapy in AMI patients. Based on these preclinical studies, primarily targeting endothelial cell-junctions seems the most promising approach in limiting microvascular injury and subsequent myocardial damage. Future work is required, especially in translating these observations to humans, to better understand the potential benefits of targeting the microvasculature to further minimize injury following AMI.
Acknowledgements
Figure 1 was created with BioRender.com.
Sources of Funding
None
Conflicts of Interests
The authors declare that they have no conflicts of interest.
References
Lara S.F. Konijnenberg1
1Department of Cardiology, Radboud University Medical Center, Nijmegen, the Netherlands.
Carolien T.A. Kuster1
1Department of Cardiology, Radboud University Medical Center, Nijmegen, the Netherlands.
Tom T.J. Luiken2
2Department of Physiology, Radboud Institute for Health Sciences, Radboud University Medical Center, Nijmegen, the Netherlands.
Nico Sommerdijk3,4
3Department of Biochemistry, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, the Netherlands. 4Electron Microscopy Center, Radboudumc Technology Center Microscopy, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, the Netherlands.
Anat Akiva3
3Department of Biochemistry, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, the Netherlands.
Dick H.J. Thijssen2
2Department of Physiology, Radboud Institute for Health Sciences, Radboud University Medical Center, Nijmegen, the Netherlands.
Robin Nijveldt1
1Department of Cardiology, Radboud University Medical Center, Nijmegen, the Netherlands.
Niels van Royen1
1Department of Cardiology, Radboud University Medical Center, Nijmegen, the Netherlands.
Corresponding author:
Niels van Royen
Email: niels.vanroyen@radboudumc.nl
In a new window | Download PPT
Figure 1: Reperfusion of an occluded coronary artery is often accompanied by microvascular injury. Microvascular injury is reflected by a sequeala of pathophysiological changes, including increased production of reactive oxygen species, disruption of the glycocalyx, endothelial barrier dysfunction with increased cytosolic calcium levels, loss of endothelial cell-cell junctions, degradation of the basement membrane, and in more severe cases formation of edema and intramyocardial hemorrhage. In turn, hemolysis of erythrocytes can expose cardiomyocytes to cytotoxic heme. Preclinical studies suggest that pharmacologically targetting components of the coronary microvasculature may have potential as an adjuvant therapy in acute myocardial infarction patients. MMP, matrix metalloproteinases; PCI, percutaneous coronary intervention; ROS, reactive oxygen species; Tie2, receptor tyrosine kinase; VEGF, vascular endothelial growth factor; VEGFR, VEGF-receptor.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 6487 | 32 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA