Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The role of Pannexin1 channels in cardiac ischemia/reperfusion injury
Time:2022-12-04
Number:8300
Author Affiliations

Conditioning Medicine 2022. 5(4): 123-130.
Abstract
Pannexin1 (Panx1) is a broadly expressed glycoprotein forming heptameric channels in the plasma membrane that allow for the controlled release of ATP from cells. Extracellular ATP is a crucial signaling molecule during inflammation and injury responses, and by stimulating purinergic (P2) receptors it amplifies NLRP3 inflammasome activation, enhances leukocyte infiltration, and provides a positive feedback loop in T lymphocytes, leading to their sustained activation. Extracellular ATP is rapidly converted to adenosine through the action of CD39 and CD73 ectonucleotidases, which dampens inflammation and promotes the resolution of tissue injury. Genetic or pharmacological inhibition of Panx1 channels has been shown to regulate these processes during the initiation and resolution of the inflammatory response. Moreover, by releasing ATP from cells undergoing apoptosis, Panx1 channels provide a “find me” signal for phagocytes to locate and clear these dying cells from injured tissues. Here, we review the current knowledge on the contribution of the Panx1/P2X7/NLRP3 axis to cardiac ischemia/reperfusion injury, a condition that involves accelerated cardiomyocyte death and an excessive inflammatory response, which can lead to heart failure. Similarities with Panx1-mediated signaling during ischemia/reperfusion are examined in other organs like the brain and kidneys. Finally, targeting of Panx1 channels with re-purposed drugs, such as probenecid, carbenoxolone, mefloquine or spironolactone, or with novel specific approaches involving mimetic peptides or mini-antibodies are discussed.
Keywords: Cardiac ischemia, Reperfusion injury, ATP, Inflammasome, NLRP3, Panx1
Abstract
Pannexin1 (Panx1) is a broadly expressed glycoprotein forming heptameric channels in the plasma membrane that allow for the controlled release of ATP from cells. Extracellular ATP is a crucial signaling molecule during inflammation and injury responses, and by stimulating purinergic (P2) receptors it amplifies NLRP3 inflammasome activation, enhances leukocyte infiltration, and provides a positive feedback loop in T lymphocytes, leading to their sustained activation. Extracellular ATP is rapidly converted to adenosine through the action of CD39 and CD73 ectonucleotidases, which dampens inflammation and promotes the resolution of tissue injury. Genetic or pharmacological inhibition of Panx1 channels has been shown to regulate these processes during the initiation and resolution of the inflammatory response. Moreover, by releasing ATP from cells undergoing apoptosis, Panx1 channels provide a “find me” signal for phagocytes to locate and clear these dying cells from injured tissues. Here, we review the current knowledge on the contribution of the Panx1/P2X7/NLRP3 axis to cardiac ischemia/reperfusion injury, a condition that involves accelerated cardiomyocyte death and an excessive inflammatory response, which can lead to heart failure. Similarities with Panx1-mediated signaling during ischemia/reperfusion are examined in other organs like the brain and kidneys. Finally, targeting of Panx1 channels with re-purposed drugs, such as probenecid, carbenoxolone, mefloquine or spironolactone, or with novel specific approaches involving mimetic peptides or mini-antibodies are discussed.
Keywords: Cardiac ischemia, Reperfusion injury, ATP, Inflammasome, NLRP3, Panx1
Introduction
Ischemic heart disease is the leading cause of mortality worldwide (Townsend et al., 2022). Myocardial ischemia occurs when blood flow to the myocardium is restricted, for example after rupture or superficial erosion of an atherosclerotic plaque provoking thrombus formation and occlusion of a coronary artery (Libby, 2021). Prolonged ischemia leads to myocardial infarction, and rapid reperfusion of the ischemic territory is required to prevent cardiomyocyte death. However, reperfusion itself can accelerate cardiomyocyte death and induce an excessive inflammatory response leading to arrhythmias, diminished contractile function, and progression towards heart failure (Heusch, 2020). Additional cardioprotective measures are thus needed on top of timely reperfusion.
As cardioprotective interventions are primarily aimed at reducing infarct size, research has focused for many years on reperfusion signaling involved in cardiomyocyte death such as the reperfusion injury salvage kinase (RISK) and survivor activating factor enhancement (SAFE) pathways (reviewed in: Hausenloy et al., 2016). Cardiac ischemia and reperfusion (I/R) are however also associated with a strong inflammatory response and, although the arrival of inflammatory cells and fibroblasts in the first few days after I/R is needed for efficient cardiac repair, exaggerated responses may further damage the heart (Andreadou et al., 2019).
A crucial role for extracellular ATP and Panx1 channels in inflammation
Inflammation is a complex process involved in a plethora of severe diseases, including I/R injury. One key mediator of inflammation is extracellular ATP, which is released from inflammatory, necrotic, or apoptotic cells as a signaling molecule to attract phagocytic cells (reviewed in: Idzko et al., 2014). Moreover, ATP and its metabolites serve as ligands for purinergic receptors, including P2X or P2Y receptors, present at the plasma membrane of activated immune cells and endothelial cells to trigger NOD-like receptor protein 3 (NLRP3) inflammasome activation (reviewed in: Adamson and Leitinger, 2014). Inflammation is triggered by damage-associated molecular patterns (DAMPs), which act on Toll-like receptors (TLRs) leading to activation of nuclear factor kappa B (NF-κB), thereby controlling the transcription of pro-inflammatory cytokines and the immature forms of interleukin (IL)-1β (pro-IL-1β) and pro-IL-18 (reviewed in: Haneklaus and O'Neill, 2015). After activation of P2X7 receptors by extracellular ATP, ionic fluxes induce an augmentation of intracellular Ca2+ and reduction of intracellular K+ leading to activation of the inflammasome complex. Subsequently, the formation of mature caspase-1 induces the cleavage of pro-ILs resulting in their mature and active forms IL-1β and IL-18 (Figure 1) (Haneklaus and O'Neill, 2015).
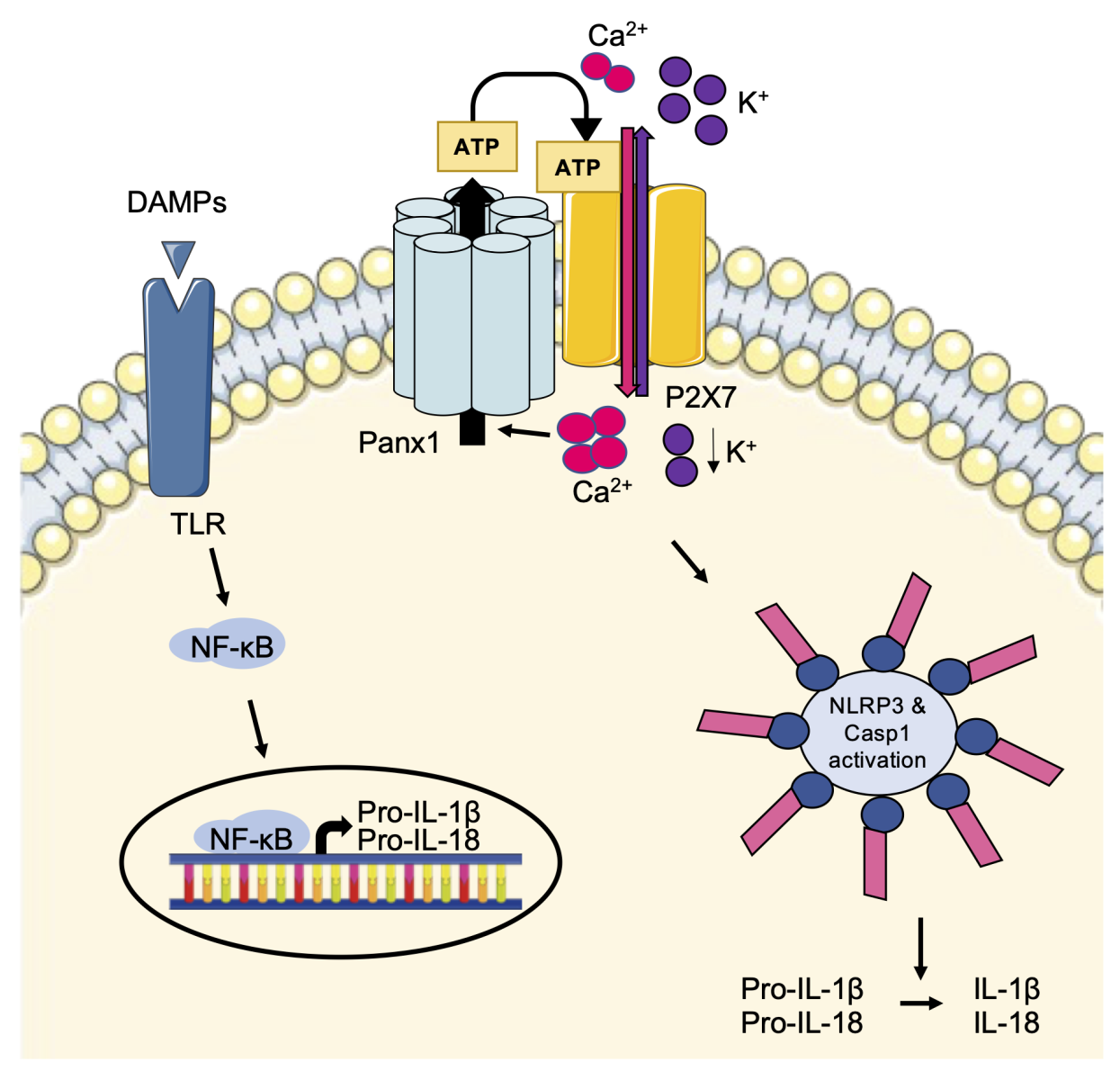
In a new window | Download PPT
Figure 1: The Panx1-P2X7-NLRP3 signaling axis. DAMPs trigger TLR activation leading to the synthesis of immature forms of the pro-inflammatory cytokines pro-IL-1β and pro-IL-18. Extracellular ATP binds to P2X7 receptors resulting in NLRP3 inflammasome activation and caspase 1-mediated cleavage of the pro-ILs into their mature and active forms. By releasing ATP, Panx1 channels can modulate P2X7 receptor function and enhance inflammasome activation.
Given the crucial role of ATP in the inflammatory response, the involvement of ATP release channels in I/R injury is gaining interest (see for a review: Koval et al., 2021). Pannexin1 (Panx1) channels are well known for their role in the regulation of various processes that are essential in cardiovascular physiology, such as vasoconstriction, blood pressure homeostasis, and platelet aggregation (Billaud et al., 2011; Molica et al., 2015; Molica et al., 2019; DeLalio et al., 2020). These channels have also been shown to modulate P2X7 receptor function in a paracrine/autocrine manner and to regulate activation of the NLRP3 inflammasome (Figure 1) (Pelegrin and Surprenant, 2006). Panx1 is a plasma membrane glycoprotein forming heptameric channels (Penuela et al., 2007; Boyce et al., 2014; Michalski et al., 2020). Each of the seven Panx1 subunits is composed of four α-helix transmembrane domains, two extracellular loops (EL1 and EL2), one intracellular loop, and intracellular N-terminal and C-terminal domains (NT and CT) (Michalski et al., 2020). Panx1 is expressed at varying levels in most mammalian cells and tissues, including cardiac and smooth muscle, endothelium of blood and lymphatic vessels, fibroblasts, and multiple types of inflammatory and immune cells (reviewed in: Molica et al., 2018; Kwak and Nielsen, 2021). Panx1 channels allow the passage of ions, small signaling molecules, and metabolites such as ATP, lactate, and glutamate (Nielsen et al., 2020; Narahari et al., 2021). Panx1 channels are normally closed and they open in response to various “stress stimuli” including ischemia, high extracellular potassium, high intracellular calcium, Src-mediated phosphorylation, or caspase-mediated cleavage of the Panx1-CT (see for a review: Chiu et al., 2018). ATP released through Panx1 channels has not only been shown to regulate NLRP3 inflammasome activation but has also been implicated in many aspects of inflammation. By releasing ATP from cells undergoing apoptosis, Panx1 channels provide the necessary “find me” signal for phagocytes to locate and clear these dying cells from injured tissues (Chekeni et al., 2010). Endothelial Panx1 channels also contribute to leukocyte activation and chemoattraction to inflamed or injured areas. In fact, binding of tumor necrosis factor-α (TNF-α) to its receptor leads to Src family kinase-mediated phosphorylation of the tyrosine residue at position 198 (Y198) in the Panx1-CT, resulting in channel opening and ATP release from the endothelial cells. This signal mediates neutrophil and/or monocyte adhesion and emigration through the endothelial layer to the site of injury (Figure 2) (Lohman et al., 2015). Conversely, ATP is rapidly converted to adenosine through the action of CD39 and CD73 ectonucleotidases, which dampens inflammation and promotes the resolution of tissue injury by activating adenosine subtype 2 (A2) receptors expressed on endothelial and inflammatory cells (Figure 2) (reviewed in: Eltzschig and Eckle, 2011). Finally, Panx1-dependent ATP release mediates a positive feedback loop in T lymphocytes, leading to their sustained activation (Schenk et al., 2008). Taken together, Panx1 channels may represent a new player in the regulation of the delicate balance between the initiation of inflammation and tissue repair in I/R.
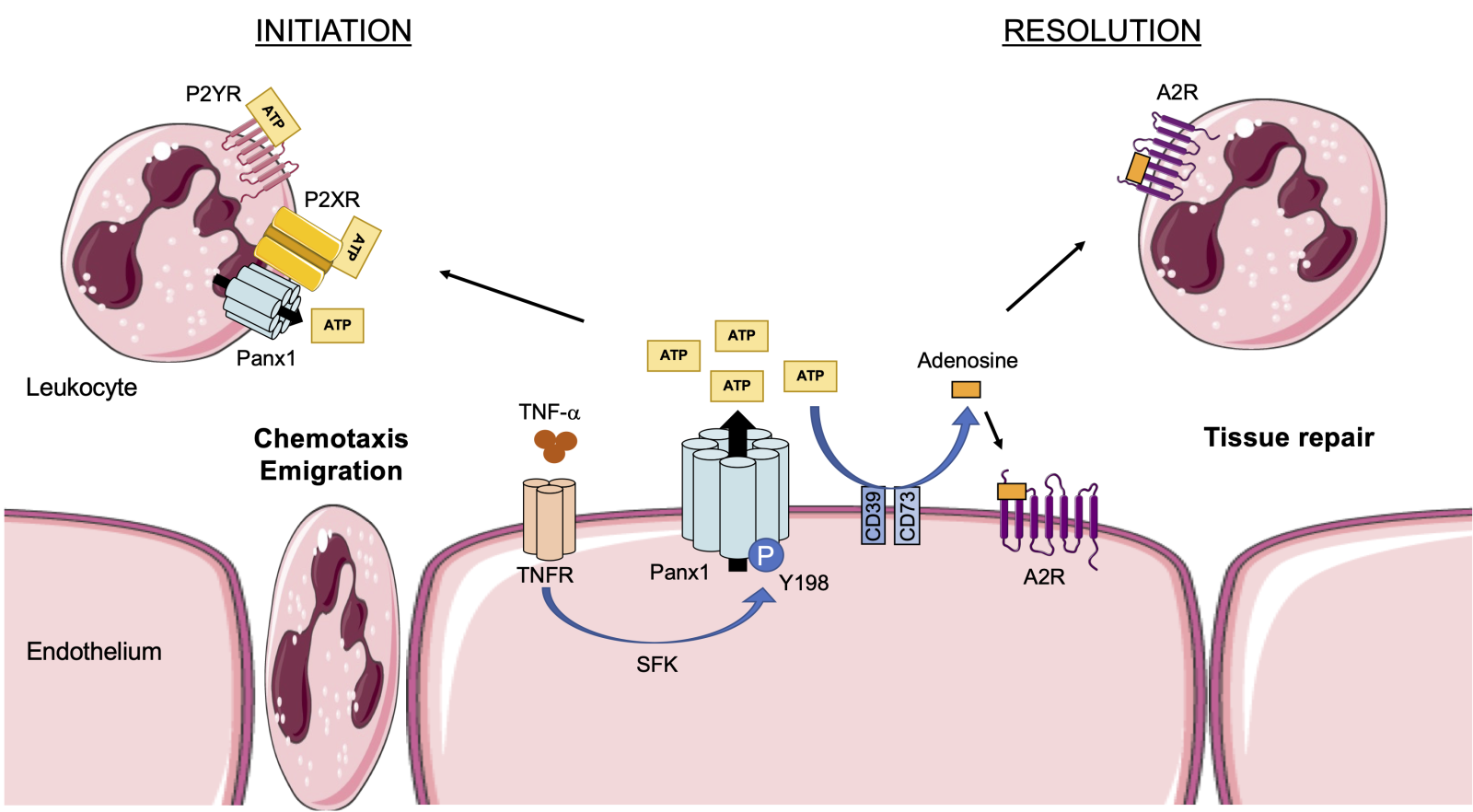
In a new window | Download PPT
Figure 2: Panx1 in the initiation and resolution of inflammation. Binding of TNF-α to the endothelial TNF receptor induces Src family kinase (SFK)-mediated phosphorylation of Panx1 channels on residue Y198. Panx1 channels open and release ATP from the endothelium, which subsequently binds to P2X or P2Y receptors on different types of leukocytes triggering their chemoattraction and emigration through the endothelium to the site of injury. Extracellular ATP is converted to adenosine by the action of CD39 and CD73 ectonucleotidases. Adenosine binds to A2 receptors on leukocytes and endothelial cells causing tissue repair and resolution of inflammation.
In vivo studies examining the contribution of Panx1 channels in the inflammatory response typically involve mouse models with ubiquitous or cell-specific deletion of Panx1 as well as pharmacological inhibition of Panx1 channels. It should be kept in mind however that the presently used “Panx1 channel blockers” are in fact re-purposed drugs, such as probenecid, carbenoxolone, mefloquine, spironolactone, or Brilliant Blue FCF (BB-FCF), a FDA-approved blue colorant for processed foods, dietary supplements, and medications (Wang et al., 2013; Chiu et al., 2018). Although a binding site was identified for carbenoxolone in EL1 containing the highly conserved tryptophan residue at position 74 (W74) at the extracellular entrance of the Panx1 channel (Michalski et al., 2020), these non-selective compounds likely exert off-target effects. The most specific inhibitor so far is 10Panx1, an EL1 mimetic peptide that has been shown to inhibit Panx1 channel-mediated currents in Panx1-transfected cells, ATP release in many Panx1-expressing cell types, and IL-1β secretion from human and mouse macrophages (Pelegrin and Surprenant, 2006; Molica et al., 2018). Linear peptides, however, display poor serum stability limiting the in vivo application of 10Panx1. A so-called “mini-antibody” targeting the same region in EL1 has recently been developed (Rusiecka et al., 2019). Such a single-chain variable fragment (scFv) consists of the variable regions of the heavy and light chains of immunoglobulin connected by a peptide linker and exhibits, in general, better tissue penetration than a full-length antibody. This first Panx1 mini-antibody was shown to reduce ATP release from platelets, inhibit platelet aggregation in vitro, and reduce collagen-induced thrombosis in vivo (Molica et al., 2019). The suitability of this Panx1 mini-antibody for in vivo investigations of longer duration has not been explored yet.
The NLRP3 inflammasome and Panx1 channels in cardiac ischemia/reperfusion
The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial assessing the efficacy of canakinumab, a monoclonal antibody against IL-1β, revealed that inflammasome targeting may be a promising new therapeutic avenue for various cardiovascular diseases. Not only were beneficial outcomes reported for heart failure patients, but the anti-IL-1β therapy was also associated with reduced cardiovascular event rates in stable atherosclerosis patients for the composite primary endpoint of recurrent myocardial infarction, stroke, or cardiovascular death (Ridker et al., 2018; Everett et al., 2019).
The involvement of the NLRP3 inflammasome in cardiac I/R has been extensively studied (see for a review: Frangogiannis, 2014). NLRP3 activation by DAMPs in innate immune cells activates caspase-1, thereby regulating the secretion of bioactive IL-1β and IL-18. These interleukins further amplify the inflammatory response and promote the release of reactive oxygen species (ROS) from neutrophils, which further injures the cardiomyocytes. Obviously, the NLRP3 inflammasome can also be activated in other cell types in the heart. Active caspase-1 in cardiomyocytes leads to pyroptosis (Toldo and Abbate, 2018). NLRP3 inflammasome activation in cardiac microvascular endothelial cells increased adhesion molecule expression, which is required for leukocyte recruitment (Liu et al., 2014). NLRP3 inflammasome activation and release of IL-1β by fibroblasts induce profibrotic changes that contribute to infarct scar enlargement and adverse cardiac remodeling (Kawaguchi et al., 2011; Mezzaroma et al., 2011; Sandanger et al., 2013). Nlrp3-deficient mice subjected to cardiac I/R injury displayed a smaller infarct size and preserved cardiac function as compared to wildtype mice (Sandanger et al., 2013; Liu et al., 2014). Moreover, NLRP3 inflammasome inhibitors administered at the onset of reperfusion reduced infarct size in various animal models (Marchetti et al., 2014; Mastrocola et al., 2016; van Hout et al., 2017; Toldo et al., 2019). As described in the previous paragraph, assembly of the NLRP3 inflammasome and IL-1β release are critically regulated by the concerted action of P2X7 receptors and Panx1 channels (Crespo Yanguas et al., 2017; Chen et al., 2019). In mice, pharmacological inhibition of Panx1 channels with probenecid (a clinically used uricosuric drug) at the onset of reperfusion improved the ejection fraction (EF) 14-days post-I/R compared with saline-treated controls (Good et al., 2021). Infarct sizes were, however, not different between both groups at 14-days post-I/R (Good et al., 2021). By reducing inflammasome activation, probenecid also prevented the deterioration of left ventricular ejection fraction (LVEF) and the development of left ventricular hypertrophy in a rat model of pressure overload-induced chronic heart failure (Onodi et al., 2021). Interestingly, in a large cohort study of 38,888 elderly patients with gouty arthritis, probenecid treatment was associated with a modestly lower risk of cardiovascular events, including myocardial infarction and heart failure exacerbation, than treatment with the alternative uric acid-lowering drug allopurinol (Kim et al., 2018). Together, these studies suggest that reduction of NLRP3 inflammasome activation via inhibition of Panx1 channels may exert cardioprotective effects against long-term complications of I/R. The exact mechanisms and cell type(s) involved are however unknown.
Several studies suggest an additional early involvement of Panx1 channels after ischemic insults in the heart. An increase in Panx1 expression has been observed within 24 hours after I/R in the heart of various animals (Dolmatova et al., 2012; Kristiansen et al., 2018). Moreover, ischemia rapidly increased the glycosylation of Panx1, thereby enhancing the trafficking of these channels to the plasma membrane of cardiomyocytes (Dolmatova et al., 2012). The authors elegantly demonstrated that ischemic stress enhanced ATP release from a cardiac muscle cell line (HL-1), which in turn promoted activation of fibroblasts and their conversion to myofibroblasts (Dolmatova et al., 2012). Thus, ATP release through cardiomyocyte Panx1 channels during ischemia may be an early paracrine event launching a profibrotic response in the ischemic heart. The impact of endothelial Panx1 on cardiac I/R has been studied using mice with a tamoxifen-inducible deletion of Panx1 in endothelial cells (Cdh5-CreERT2+/Panx1fl/fl) and control Panx1fl/fl mice. Genetic deletion of Panx1 from the endothelium appeared to result in improved EF 14 days post-I/R, although no effects on infarct size were seen at this time (Good et al., 2021). The infiltration of leukocytes was studied 2 days post-I/R in these mice. Whereas the total neutrophil and macrophage infiltration into infarcted hearts was not different, the number of proinflammatory lymphocyte antigen 6 complex (Ly6C)high macrophages was reduced in hearts with endothelial deletion of Panx1. Surprisingly, this reduction in Ly6Chigh macrophages was most apparent in the non-infarct region of these hearts (Good et al., 2021).
Protection of the heart from I/R injury can be achieved by ischemic preconditioning (IPC), i.e. brief periods of ischemia followed by reperfusion prior to the prolonged ischemic event (reviewed in: Hausenloy et al., 2016). When administered during IPC, carbenoxolone, mefloquine, and Brilliant Blue G were shown to prevent the beneficial effects of IPC (Vessey et al., 2010), suggesting that the Panx1/P2X7 complex is responsible for the release of endogenous cardioprotectants induced by IPC. Moreover, these Panx1 channel inhibitors also prevented the cardioprotective effects of IPC when administered during reperfusion after a prolonged period of ischemia (Vessey et al., 2011b). Finally, it was shown in the same study that preconditioning of isolated rat hearts with a low dose of ATP minimized the infarct size and augmented the recovery of the left ventricular developed pressure (LVDP) upon subsequent I/R. Each of the Panx1 channel inhibitors impeded these beneficial effects of ATP-induced preconditioning (Vessey et al., 2011a). It remains unknown, however, which of the Panx1-expressing cell types in the heart are involved in the beneficial effects of IPC. It should also be kept in mind that mitochondrial connexin 43 (Cx43) is implicated in IPC-induced cardioprotection, likely by affecting mitochondrial respiration and/or ROS production (reviewed in: Boengler et al., 2022). Non-specific Panx1 channel blocking agents like carbenoxolone also inhibit Cx43 hemichannels and carbenoxolone-induced cardioprotection may thus, in part involve Cx43. Although a picture emerges in which Panx1 channels act on multiple cell types implicated in cardiac I/R injury, further studies are needed to discern the different mechanisms involved. Such mechanisms may be similar to the ones found to be involved in I/R in other organs.
Role of Panx1 in ischemic injury of the brain and kidney
The contribution of Panx1 channels to ischemic injury, with or without reperfusion, has been investigated in vivo in other organs like the brain and kidney (Figure 3). Panx1 is widely expressed in the brain, where it has been found in astrocytes, neurons, and microglia, as well as in endothelial and smooth muscle cells of the cerebral vasculature (reviewed in: Yeung et al., 2020). Since the initial publication of Roger Thompson and colleagues in 2006 (Thompson et al., 2006) in which they found that ischemia-like conditions (oxygen/glucose deprivation (OGD)) activate a large conductance channel in neurons likely involved in ischemic neuronal death, later identified to be Panx1 (Thompson and Macvicar, 2008, a wave of intense research on the role of Panx1 in ischemic stroke commenced. It turned out that anoxia drives activation of the N-methyl-D-aspartate receptor (NMDAR) and subsequent opening of Panx1 channels inducing aberrant depolarization and subsequent neuronal death. This opening of Panx1 channels involved Src family kinase activation and phosphorylation of the tyrosine at position 308 (Y308) in the Panx1 C-terminus (Weilinger et al., 2013). Disruption of the NMDAR-Src-Panx1 signaling complex in a rat model of middle cerebral artery occlusion by administration of a TAT-Panx1308 peptide reduced the ischemic lesion size and improved sensorimotor deficits (Weilinger et al., 2016). Likewise, inhibition of Panx1 channels with probenecid reduced cerebral infarct size after middle cerebral artery occlusion and improved the recovery of memory and motor functions in mice (Xiong et al., 2014).
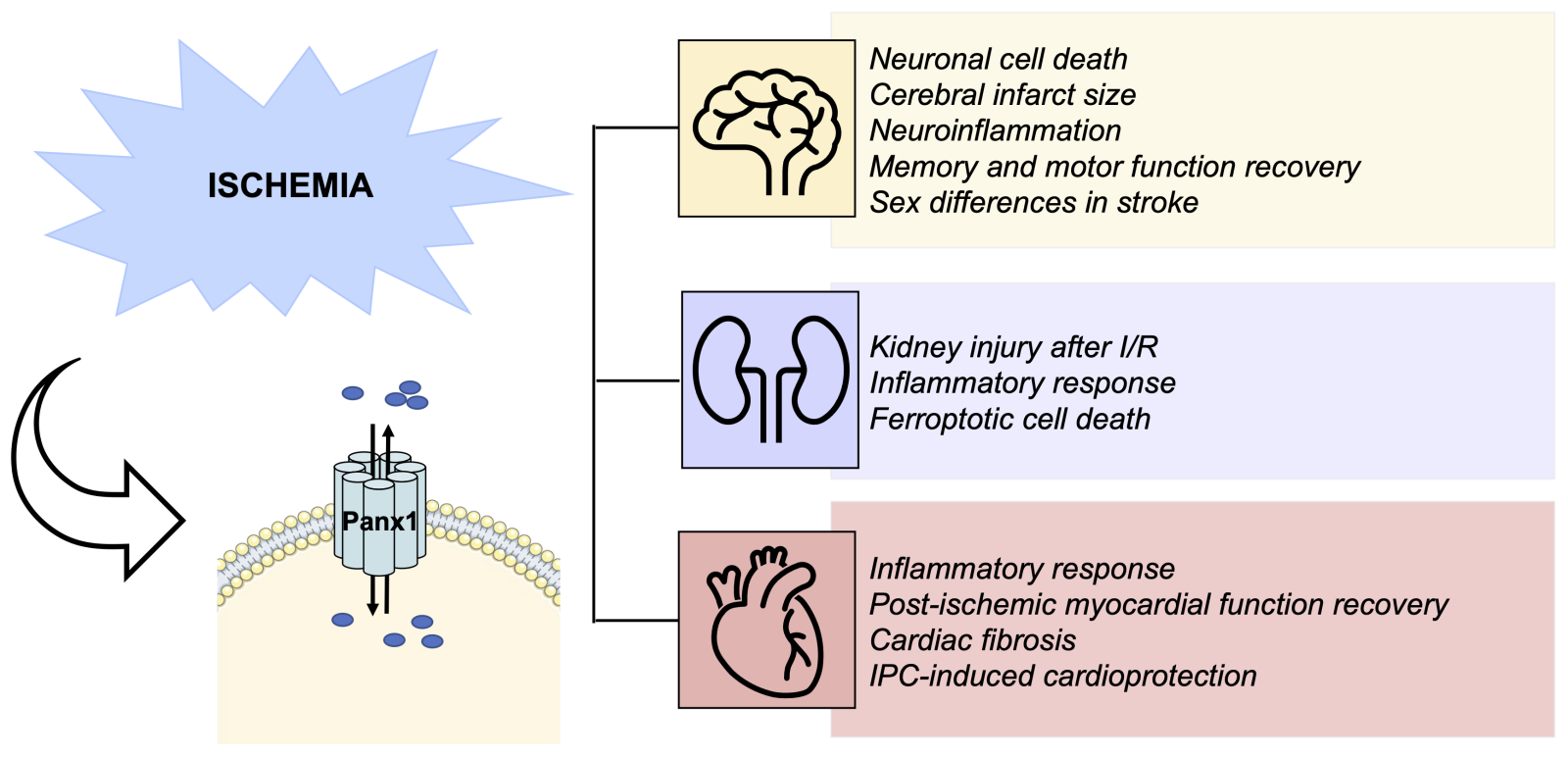
In a new window | Download PPT
Figure 3: Summary of current knowledge on the role of Panx1 in I/R injury. Emerging data reports describe various roles for Panx1 channels in key features of I/R injury in various organs. Thus, Panx1 induces neuronal cell death and neuroinflammation during brain I/R injury, and pharmacological inhibition of Panx1 reduces infarct size and improves memory and motor functions. A sexually dimorphic nature of Panx1 deletion on stroke outcomes has been reported. Panx1 deletion or pharmacological inhibition is also protective against inflammation, cell death, and renal injury. Finally, pharmacological or genetic inhibition of Panx1 channels reduces cardiac inflammation and fibrosis, and it improved myocardial function recovery after 14 days. Furthermore, Panx1 seems involved in IPC-induced cardioprotection.
In addition to the direct involvement of Panx1 channels in neuronal death, the Panx1-P2X7-inflammasome axis has been shown to induce neuroinflammation, further contributing to the ischemic injury (Silverman et al., 2009; Cisneros-Mejorado et al., 2015; Chen et al., 2017; Freitas-Andrade et al., 2017). In a model of permanent middle cerebral artery occlusion, middle-aged Panx1-deficient female mice exhibited a 50% reduction in stroke volume 4-days post-ischemia and reduced signs of neuroinflammation (Freitas-Andrade et al., 2017). Surprisingly, the infarct size and neuroinflammatory response were comparable in Panx1-/- and wildtype male mice. The sexually dimorphic nature of Panx1 deletion was also observed upon pharmacological blocking of Panx1 channels with probenecid (Freitas-Andrade et al., 2017). As mentioned by the authors, women are disproportionally affected by stroke with a higher level of mortality and disability than men (Herson et al., 2013; Gasbarrino et al., 2022). Although the mechanism of the sexually dimorphic nature of Panx1 deletion on stroke was not been explored in this study, the interesting possibility of an estrogen receptor β-mediated regulation of the inflammasome has been evoked (de Rivero Vaccari et al., 2016). A smaller injury size was observed after middle cerebral artery occlusion in an endothelial-specific Panx1 knock-out mouse model, which appeared to be linked to diminished inflammatory cell recruitment and a reduced myogenic tone in cerebral arteries. Importantly, pharmacological Panx1 channel inhibition induced the same responses, which were not observed in mice with smooth muscle cell-specific Panx1 deletion (Good et al., 2018). Finally, a controlled cortical impact (CCI) model was used in mice with a myeloid-specific Panx1 deletion (Cx3cr1-Cre+/Panx1fl/fl). Indeed, these mice showed markedly reduced immune cell infiltration to the brain parenchyma and reduced blood-brain barrier leakage compared to Panx1fl/fl mice. Moreover, motor and memory functions were improved in Cx3cr1-Cre+/Panx1fl/fl mice within a week post-CCI injury (Seo et al., 2020). Altogether, these studies point to an important role for Panx1 in regulating neuroinflammation after cerebral I/R.
Similar to the heart and the brain, the kidney receives a relative high percentage of cardiac output at rest and is vulnerable to ischemic injury. Panx1 expression has been shown in the apical membrane of proximal tubules, thin descending limbs, and collecting ducts, in addition to endothelial and smooth muscle cells of the renal vasculature, including afferent and efferent arterioles (Hanner et al., 2012; Jankowski et al., 2018). Moreover, Panx1 channels in juxtaglomerular cells have been shown to regulate steady-state renin secretion and are thus important in blood pressure homeostasis (DeLalio et al., 2020). Pharmacologic and genetic approaches have been used to assess the contribution of Panx1 to renal I/R in a mouse model representing acute kidney injury (AKI). Administration of the non-specific pharmacological Panx1 channel inhibitor carbenoxolone attenuated renal I/R injury in mice (Jankowski et al., 2018). As carbenoxolone is known to possess off-target effects, including on connexin channels, the authors subsequently exposed a variety of (conditional) Panx1 knockout mice to renal I/R injury. Panx1-/- mice were protected from renal I/R injury and reduced expression of cytokines, chemokines, and adhesion molecules was observed in their kidneys after I/R (Jankowski et al., 2018). Using bone marrow chimeras, it was subsequently revealed that Panx1 deficiency in parenchymal cells (and not in bone marrow-derived cells) was primarily responsible for the protection against renal I/R injury in Panx1-/- mice (Jankowski et al., 2018). Finally, these studies showed that both proximal tubule- and endothelial-specific Panx1 knockout mice were protected against renal I/R injury. In a separate study, Panx1 knockout mice subjected to renal I/R had decreased plasma creatinine, malondialdehyde (MDA) levels in kidney tissues, and tubular cell death compared to wildtype mice. Mechanistically, it was shown that ubiquitous Panx1 deletion protected against renal I/R injury by regulating ferroptotic cell death (Su et al., 2019). Altogether, Panx1 seems to play an important role in acute ischemic kidney disease, which clearly warrants further mechanistic studies.
Concluding remarks
Ischemic heart disease accounts worldwide for nearly nine million deaths each year (Townsend et al., 2022). Given the rising life expectancy and the adverse trends in major cardiovascular risk factors, it is expected that the prevalence of acute myocardial infarction will increase within the next years. This increase will further augment the burden of heart failure, which causes high medical costs for society and reduces the patient’s quality of life. The most effective treatment for limiting infarct size and preventing heart failure is timely myocardial reperfusion using percutaneous coronary intervention, but this procedure itself may also accelerate cardiomyocyte death and augment inflammation. There is thus an unmet clinical need for additional cardioprotective measures on top of timely reperfusion.
The cardiac damage after I/R results from a delicate balance between cardiomyocyte death, inflammation, and tissue repair, and effective cardioprotective therapy is therefore expected to involve multiple targets (Davidson et al., 2019). The P2X7-Panx1-NLRP3 signaling axis has gained attention in recent years, because its involvement in cardiac I/R injury appeared consistent when genetic or pharmacological targeting to either of its three components was performed. The targeting of the Panx1 channel seems particularly attractive as its beneficial effects in I/R injury seems to extend beyond the P2X7-Panx1-NLRP3 signaling axis and also involves effects on neutrophil/macrophage chemotaxis, apoptotic clearance of cells, T cell activation, and fibroblast activation, thus affecting both the initiation and resolution of inflammation.
It is certainly attractive to use re-purposed drugs like probenecid, carbenoxolone, mefloquine, or spironolactone for cardioprotection research. Some of these drugs have already been proven beneficial in rodent cardiac I/R studies in individual laboratories, but validation in other laboratories and larger animal models is necessary. Future research should also include in vitro experiments followed by in vivo studies targeting key regulatory sites of Panx1 channels, genetically or with specific peptides, to enhance our understanding of the molecular mechanisms involved before specific Panx1-targeting therapeutic approaches can be explored in patient populations.
Conflicts of Interest
The authors declare no conflict of interest.
Funding
This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology). This work was supported by the Swiss National Science Foundation (grant numbers 310030_182573, IZCOZ0_189878 and 310030E-176050). In addition, funding was received from the European Union’s Horizon 2020 Future and Emerging Technologies program under grant agreement number 858014.
References
Malaury Tournier1,2
1Department of Pathology and Immunology, University of Geneva, 1211 Geneva, Switzerland. 2Geneva Centre for Inflammation Research, Faculty of Medicine, University of Geneva, 1211 Geneva, Switzerland.
Olga M. Rusiecka1,2
1Department of Pathology and Immunology, University of Geneva, 1211 Geneva, Switzerland. 2Geneva Centre for Inflammation Research, Faculty of Medicine, University of Geneva, 1211 Geneva, Switzerland.
Filippo Molica1,2
1Department of Pathology and Immunology, University of Geneva, 1211 Geneva, Switzerland. 2Geneva Centre for Inflammation Research, Faculty of Medicine, University of Geneva, 1211 Geneva, Switzerland.
Brenda R. Kwak1,2
1Department of Pathology and Immunology, University of Geneva, 1211 Geneva, Switzerland. 2Geneva Centre for Inflammation Research, Faculty of Medicine, University of Geneva, 1211 Geneva, Switzerland.
Corresponding author:
Brenda R. Kwak, PhD
Email: Brenda.KwakChanson@unige.ch
In a new window | Download PPT
Figure 1: The Panx1-P2X7-NLRP3 signaling axis. DAMPs trigger TLR activation leading to the synthesis of immature forms of the pro-inflammatory cytokines pro-IL-1β and pro-IL-18. Extracellular ATP binds to P2X7 receptors resulting in NLRP3 inflammasome activation and caspase 1-mediated cleavage of the pro-ILs into their mature and active forms. By releasing ATP, Panx1 channels can modulate P2X7 receptor function and enhance inflammasome activation.
In a new window | Download PPT
Figure 2: Panx1 in the initiation and resolution of inflammation. Binding of TNF-α to the endothelial TNF receptor induces Src family kinase (SFK)-mediated phosphorylation of Panx1 channels on residue Y198. Panx1 channels open and release ATP from the endothelium, which subsequently binds to P2X or P2Y receptors on different types of leukocytes triggering their chemoattraction and emigration through the endothelium to the site of injury. Extracellular ATP is converted to adenosine by the action of CD39 and CD73 ectonucleotidases. Adenosine binds to A2 receptors on leukocytes and endothelial cells causing tissue repair and resolution of inflammation.
In a new window | Download PPT
Figure 3: Summary of current knowledge on the role of Panx1 in I/R injury. Emerging data reports describe various roles for Panx1 channels in key features of I/R injury in various organs. Thus, Panx1 induces neuronal cell death and neuroinflammation during brain I/R injury, and pharmacological inhibition of Panx1 reduces infarct size and improves memory and motor functions. A sexually dimorphic nature of Panx1 deletion on stroke outcomes has been reported. Panx1 deletion or pharmacological inhibition is also protective against inflammation, cell death, and renal injury. Finally, pharmacological or genetic inhibition of Panx1 channels reduces cardiac inflammation and fibrosis, and it improved myocardial function recovery after 14 days. Furthermore, Panx1 seems involved in IPC-induced cardioprotection.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 8300 | 39 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA