Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Mechanism-driven therapies for hypertrophic cardiomyopathy
Time:2023-08-28
Number:8667
Author Affiliations

Conditioning Medicine 2023. 6(1): 15-24.
Abstract
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease that manifests with left ventricular hypertrophy in the absence of an identifiable cause. HCM was initially viewed as a disease of the sarcomere, as several mutations in various contractile proteins have been identified in familial cases. However, this ideology is being contested as many cases do not carry sarcomere mutations, and not all sarcomere-mutation carriers develop HCM. The clinical spectrum is vast, ranging from individuals who are completely asymptomatic to those who manifest chest pain, exercise intolerance, heart failure, and arrhythmia. Current management with β-blockers, calcium channel antagonist, and antiarrhythmics is mainly aimed at relieving symptoms and do not address the underlying pathophysiology. Crucially, while mortality remains low, a significant number of patients experience death or hospitalization due to ventricular tachycardia, heart failure, or stroke. Recently, several targeted approaches have been undertaken to attenuate pathological features, including myocardial fibrosis, cardiomyocyte hypercontractility, perturbed metabolism, and oxidative stress. While most of these targeted therapies have shown promise in preclinical animal models, their translation into clinical cohorts has been disappointing, with the exception of cardiac myosin inhibitor, mavacamten. Considering conventional management strategies do not improve long-term prognosis, there is an unmet need to develop mechanism-driven treatments for HCM. While this would require in-depth understanding of the causal effects of each mutation, models based on human physiology may provide unprecedented opportunity to interrogate the pathogenicity of novel mutations and to develop mechanism-driven therapies for improving health outcomes in HCM patients.
Keywords: Hypertrophic cardiomyopathy, Clinical management, Diastolic dysfunction, Heart failure, Mavacamten, Induced pluripotent stem cells
Abstract
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease that manifests with left ventricular hypertrophy in the absence of an identifiable cause. HCM was initially viewed as a disease of the sarcomere, as several mutations in various contractile proteins have been identified in familial cases. However, this ideology is being contested as many cases do not carry sarcomere mutations, and not all sarcomere-mutation carriers develop HCM. The clinical spectrum is vast, ranging from individuals who are completely asymptomatic to those who manifest chest pain, exercise intolerance, heart failure, and arrhythmia. Current management with β-blockers, calcium channel antagonist, and antiarrhythmics is mainly aimed at relieving symptoms and do not address the underlying pathophysiology. Crucially, while mortality remains low, a significant number of patients experience death or hospitalization due to ventricular tachycardia, heart failure, or stroke. Recently, several targeted approaches have been undertaken to attenuate pathological features, including myocardial fibrosis, cardiomyocyte hypercontractility, perturbed metabolism, and oxidative stress. While most of these targeted therapies have shown promise in preclinical animal models, their translation into clinical cohorts has been disappointing, with the exception of cardiac myosin inhibitor, mavacamten. Considering conventional management strategies do not improve long-term prognosis, there is an unmet need to develop mechanism-driven treatments for HCM. While this would require in-depth understanding of the causal effects of each mutation, models based on human physiology may provide unprecedented opportunity to interrogate the pathogenicity of novel mutations and to develop mechanism-driven therapies for improving health outcomes in HCM patients.
Keywords: Hypertrophic cardiomyopathy, Clinical management, Diastolic dysfunction, Heart failure, Mavacamten, Induced pluripotent stem cells
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease with a prevalence of 1:200-1:500, that manifests as left ventricular hypertrophy (LVH) in the absence of an identifiable cause (Maron, 2018). Approximately 60% of HCM cases have a clear familial link with missense variants in MYH7 and truncating variants in MYBPC3 accounting for most familial cases (Marian and Braunwald, 2017; Sabater-Molina et al., 2018). While this may lend support that HCM is a disease of the sarcomere, the absence of sarcomere mutations in a vast number of cases contests this ideology and may explain the heterogeneous nature of the disease (Maron et al., 2019). Indeed, the clinical spectrum of HCM is vast, ranging from cases who are completely asymptomatic to those who present with chest pain, shortness of breath, fatigue, and syncope, with a small subset experiencing sudden cardiac death, in particular children and athletes (Ostman-Smith et al., 2008; Maron et al., 2009). HCM patients are grouped into two subsets; those with LV outflow tract (LVOT) obstruction and those without. LVH can precipitate LVOT obstruction at rest or with provocation, reduce LV compliance, and induce diastolic dysfunction. As expected, patients with obstructive HCM (oHCM) are associated with more severe symptoms and adverse outcomes, and though nonobstructive HCM (nHCM) was initially viewed as a benign condition with minimal symptoms, recent findings support high rates of adverse clinical events, comparable to those with oHCM (Lu et al., 2018; Maron et al., 2018a).
The presence or absence of obstruction guides clinical management. For patients with oHCM, measures that increase cardiac preload and afterload, and those that reduce cardiac contractility are favored. Pharmacotherapies like β-blockers, calcium channel antagonists, and antiarrhythmics represent the latter, where the LVOT obstruction is attenuated by reducing cardiac inotropy and chronotropy, reducing myocardial oxygen demand and enhancing diastolic filling. Invasive septal reduction strategies (either by surgical myectomy or alcohol septal ablation) may be considered for patients whose symptoms are refractory to pharmacotherapy (Rovner et al., 2003), although this can increase the risk of complete heart blockage due to damage of bundle branches (Talreja et al., 2004). Interventions for patients with nHCM mainly attempt to manage arrhythmias and improve diastolic filling; however, these approaches are less effective than for those with oHCM (Matsubara et al., 1995; Olivotto et al., 2018). Ultimately, these conventional management strategies aim to relieve symptoms and are unable to reverse pathological features that accompany LVH, including cardiomyocyte hypertrophy and disarray, or interstitial fibrosis (Cui et al., 2021). Crucially, despite low incidence of mortality, long-term follow-up studies reveal poor prognosis in HCM patients with ~50% experiencing death or hospitalization due to ventricular tachycardia, heart failure, or stroke (Sugiura et al., 2022), which is compounded by the modest efficacy and tolerability of conventional pharmacotherapies (Spoladore et al., 2012). As such, there is an unmet need to develop new treatments to improve health outcomes in HCM patients.
Proteomic analysis of myocardial tissue from HCM patients has revealed alterations in various cellular pathways, including metabolism, muscle contraction, calcium regulation, and oxidative stress (Coats et al., 2018), and recent attempts to target some of these pathways have been promising. In this review, we discuss conventional pharmacotherapies for managing HCM and draw special attention to new treatment strategies that can attenuate pathological features, and highlight the use of human models for interrogating the pathogenicity of novel mutations with the aim of developing mechanism-driven therapies for improving health outcomes in HCM patients.
Conventional pharmacotherapies
Since it was first described over 60 years ago, the clinical management of HCM has centered on relieving symptoms and improving quality of life. This section focuses on non-selective pharmacological modalities that are used in line with existing treatment guidelines.
β-blockers
The use of non-vasodilating β-adrenergic blockers is one of the oldest strategies for treating symptomatic cases and represents the mainstay of therapy (Figure 1). The beneficial effects of β-blockers are mediated by sympathetic modulation of heart rate, contractility, and stiffness, which in turn improve LV relaxation and increases time for diastolic filling. Several β-blockers have been evaluated over time with some found to be more effective than others (Cohen and Braunwald, 1968; Hubner et al., 1973). For HCM presenting in childhood (which is associated with a higher mortality than when presented in adulthood), high-dose propranolol treatment drastically reduced the risk of death compared to those who were managed conventionally (Ostman-Smith et al., 1999). Due to their negative chronotropic effects, β-blockers are unable to promote improvements in exercise capacity (Ikram and Fitzpatrick, 1981; Gilligan et al., 1993); however, recent studies with metoprolol have shown that this agent can reduce obstruction at rest and during exercise (Dybro et al., 2021) as well as improve exercise hemodynamics (Dybro et al., 2022) and LV global longitudinal strain (Dybro et al., 2023), which imply improvements in LV systolic function. While these studies suggest β-blocker treatment is effective, it is unclear whether these agents can improve long-term prognosis of HCM patients. Moreover, it is unlikely these agents can exert meaningful benefits in patients with more severe gradients and heart failure status (Monda et al., 2022) or attenuate adverse ventricular remodeling (Dybro et al., 2021).
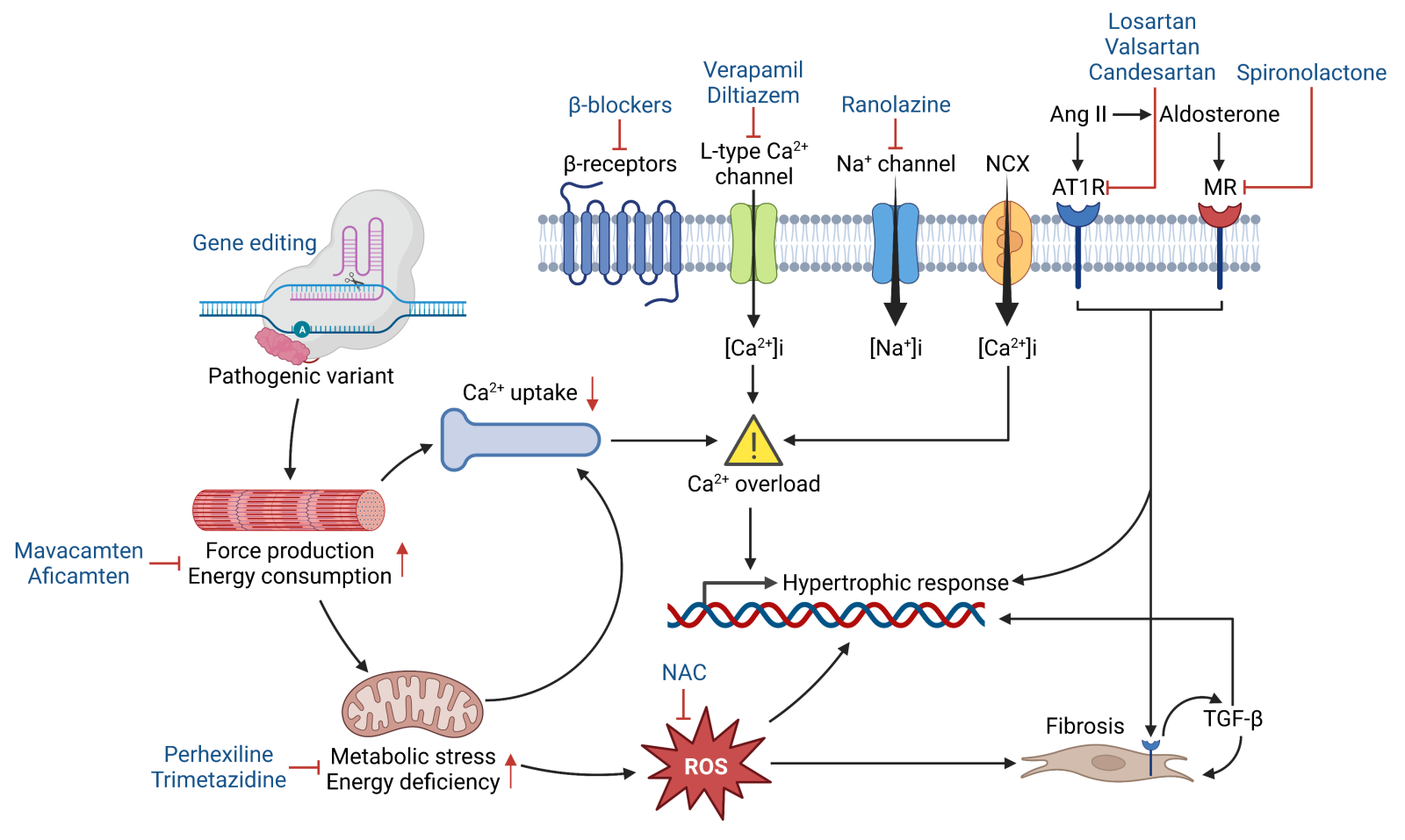
In a new window | Download PPT
Figure 1. Schematic illustrating the pathophysiology of HCM, whereby pathogenic variants potentiate hypertrophic and fibrotic responses through abnormal sarcomere mechanics, impaired energetics, calcium mishandling, and oxidative stress. Several therapeutic modalities (blue) have been developed to target these maladaptive processes. Abbreviations: NCX- Na+/Ca2+ exchanger; Ang II- angiotensin II; AT1R- angiotensin II receptor type 1; MR- mineralocorticoid receptor; NAC- N-acetylcysteine; ROS- reactive oxygen species; TGF-β- transforming growth factor beta.
Calcium channel antagonists
An alternative to β-blocker therapy is the use of the non-dihydropyridine calcium channel antagonist, verapamil, which has been shown to relieve symptoms in patients with different degrees of disease severity (Rosing et al., 1980; Gilligan et al., 1993). Due to its negative inotropic and chronotropic effects, verapamil mediates its action by improving diastolic filling, which has been attributed to reduction in diastolic asynchrony, rather than changes in diastolic velocities of the myocardial fibers (Rosing et al., 1985; Pacileo et al., 2000). Whether verapamil can improve exercise capacity remains controversial (Rosing et al., 1980; Rosing et al., 1985; Gilligan et al., 1993). The rationale for using calcium channel antagonists to treat the disease is supported by several studies that report intracellular calcium handling abnormalities in the setting of HCM (Figure 1). However, despite promoting better therapeutic outcomes than β-blockers (as suggested by comparative studies), verapamil is contraindicated for patients at high risk for adverse electrophysiological events, atrioventricular block, and sinus arrest (Rosing et al., 1985). Diltiazem is another calcium channel antagonist that has been shown to improve LV relaxation and diastolic filling without altering systolic function in HCM patients (Suwa et al., 1984; Iwase et al., 1987). Though it is considered to have a more favorable safety profile than verapamil, diltiazem can increase pulmonary artery wedge pressure, which is likely due to the combined action of a negative inotropic effect with no improvements in LV compliance, and this may increase risk of pulmonary congestion (Betocchi et al., 1996). Another strategy to prevent calcium overload in cardiomyocytes is by inhibiting the late sodium current (INa,late), which is enhanced in the setting of HCM and facilitates the exchange of intracellular sodium for extracellular calcium (Figure 1). However, the INa,late inhibitor, ranolazine, was unable to improve diastolic function, exercise capacity, biomarker levels, or quality of life, despite demonstrating acceptable safety profiles and reducing premature ventricular complex burden (Olivotto et al., 2018).
Antiarrhythmics
Antiarrhythmics, either alone or in combination with other agents, have been shown to decrease pressure gradients through their negative inotropic and lusitropic properties (Pollick, 1988; Cokkinos et al., 1989; Hamada et al., 1997). In one study, the Class 1a antiarrhythmic, disopyramide, relieved symptoms in patients with oHCM by improving diastolic function through afterload reduction but had minimal effect on patients with nHCM (Matsubara et al., 1995). Similarly, cibenzoline was shown to reduce pressure gradients in patients with oHCM (Hamada et al., 1997) by reducing diastolic pressures and attenuating diastolic dysfunction (Hamada et al., 2001; Hamada et al., 2005; Hamada et al., 2007). Although an increase in exercise capacity has been reported following disopyramide treatment (Pollick, 1988), whether improvements in diastolic function truly translate to better exercise tolerance requires validation in larger cohorts. Despite promoting substantial functional improvements in most symptomatic cohorts, antiarrhythmics may promote early incidence of sudden cardiac death by triggering malignant arrhythmias or conduction abnormalities, especially when diastolic filling is reduced post-treatment (Fananapazir et al., 1991).
In summary, these conventional pharmacotherapies have been shown to relieve symptoms in several small cohorts. However, many of these studies were underpowered and employed surrogate endpoints, such as change in LVOT gradients or exercise tolerance; and hence, it has been difficult to assess the true impact of these agents on long-term health outcomes in patients. Strikingly, recent findings imply these agents may be ineffective in the long-term as a significant number of patients who were on these medications experienced HCM-related adverse events (Sugiura et al., 2022). While this calls for adequately designed clinical trials, the inclusion of functional imaging techniques, genetic information, and hard endpoints should be considered when determining the efficacy of novel treatments in future.
Mechanism-driven therapies
Several approaches have been undertaken to target various pathological features of HCM. This section focuses on therapies designed to attenuate myocardial fibrosis, oxidative stress, impaired myocardial energetics, and hypercontractility.
Angiotensin receptor blockers
Myocardial fibrosis is an independent predictor of adverse outcomes in HCM patients (O'Hanlon et al., 2010; Raman et al., 2019); and hence, it is not surprising that attempts have been made to prevent the development of fibrosis in the setting of HCM. Elevated levels of transforming growth factor beta-1 (TGF-β1) and its receptor were first identified in ventricular biopsies from HCM patients, suggestive of its involvement in disease pathogenesis (Li et al., 1997; Li et al., 1998). Indeed, the activation of TGF-β signaling in non-myocytes mediates an increase in fibrosis, which may contribute to diastolic dysfunction and heart failure in mice with HCM (Teekakirikul et al., 2010) (Figure 1). Interestingly, treatment with the angiotensin II type 1 receptor antagonist, losartan, prevented the development of LVH and fibrosis in these mice, but only if administered prior to the onset of LVH. These findings support the use of angiotensin receptor blockers (ARBs) to treat patients with less established HCM, and consistently, valsartan improved cardiac structure and function in patients with early-stage HCM (Ho et al., 2021). In another study, losartan attenuated progression of LVH and fibrosis in patients who were mostly asymptomatic (Shimada et al., 2013). On the contrary, losartan was unable to attenuate LVH (Axelsson et al., 2015) or improve cardiac function and exercise capacity in patients with overt HCM (Axelsson et al., 2016).
The patient genotype could be a major determinant of the response to ARBs, as though candesartan was able to promote LVH regression, and improve LV function and exercise tolerance in carriers with MYH7 mutations, it had moderate effects in carriers of MYBPC3 mutations and no effect in those with TNNI3 (troponin I) mutations (Penicka et al., 2009). It can be speculated the differential ARB response observed here is governed by TGF-β signaling, as mice with different HCM-causing mutations differed in several properties, with only those with troponin T mutations demonstrating a biosignature consistent with elevated TGF-β signaling (Vakrou et al., 2018). While this may imply elevated TGF-β signaling underpins the pathophysiology in specific genetic subgroups, plasma analysis confirmed this pathway is generally upregulated in HCM patients when compared to those presenting with hypertensive LVH (Shimada et al., 2021) and can also serve as a predictor of major adverse cardiovascular events (Shimada et al., 2022).
Mineralocorticoid receptor antagonist
In certain cases, myocardial fibrosis has been found to be an early manifestation in patients who are yet to develop LVH, where substantial scar formation precipitates arrhythmia or progressive heart failure in the absence of LVOT obstruction. Aldosterone is thought to be a critical mediator of cardiac hypertrophy and fibrosis as it is elevated in patient myocardium, evokes a hypertrophic response in rat cardiomyocytes, and promotes collagen and TGF-β expression in rat fibroblasts (Tsybouleva et al., 2004) (Figure 1). Interestingly, the mineralocorticoid receptor (MR) antagonist, spironolactone, attenuated fibrosis, cardiomyocyte disarray, and diastolic dysfunction in mice (Tsybouleva et al., 2004), supporting the need for clinical trials to evaluate the potential beneficial effects of MR blockade in the setting of HCM. However, spironolactone was unable to attenuate fibrosis, nor improve LV remodeling and functional capacity in a small cohort of patients with or without LVOT obstruction (Maron et al., 2018b).
Antioxidants
The occurrence of LVH and fibrosis are considered secondary to the activation of numerous mitotic pathways, including those mediated by oxidative stress (Figure 1). Consistent with being a critical mediator of cardiac hypertrophy and fibrosis (Ramachandra et al., 2021a), oxidative stress is elevated in HCM patient myocardium and correlates with LV dilation and systolic dysfunction (Nakamura et al., 2005), while inhibition of these pathways through direct or indirect measures attenuates the disease phenotype in animal models of HCM (Senthil et al., 2005; Marian et al., 2006; Lombardi et al., 2009; Wilder et al., 2015). N-acetylcysteine (NAC), a precursor of glutathione (the most abundant intracellular defense mechanism against oxidative damage), is one of the most studied compounds for attenuating oxidative stress, and it has been shown to reverse established LVH and fibrosis, and reduce susceptibility to ventricular arrhythmia (Marian et al., 2006; Lombardi et al., 2009). NAC can also attenuate diastolic dysfunction that may arise through oxidative modifications of myofilament proteins (Wilder et al., 2015; Ryba et al., 2019; Ramachandra et al., 2022). Despite these beneficial effects, NAC had no major impact on indices of LVH and fibrosis in HCM patients (Marian et al., 2018). It is important to note most of these patients presented with minimal symptoms and did not have exercise intolerance; and hence, whether NAC can relieve symptoms or prevent disease progression despite the lack of reduction in fibrosis remains to be determined. Interestingly, fingolimod, a sphingosine-1-phosphate receptor modulator used for treating multiple sclerosis, decreased oxidative modifications of myofilament proteins, which in turn attenuated diastolic dysfunction, but with no reduction in fibrosis (Ryba et al., 2019). These findings may suggest that restoring myofilament function is sufficient for improving cardiac function in the setting of HCM.
Metabolic modulators
Elucidating common pathophysiological mechanisms across diverse HCM genotypes is crucial for developing therapies that can promote maximum response across the entire clinical spectrum. Impaired myocardial energetics is considered an early and common driver of HCM (Ramachandra et al., 2019; Ramachandra et al., 2021b) independent of family history (Jung et al., 2000), clinical status (Jung et al., 1998), and patient genotype (Crilley et al., 2003). In support, reduction in myocardial phosphocreatine/adenosine triphosphate (PCr/ATP) ratios and increases in inorganic phosphate/phosphocreatine (Pi/PCr) ratios are found in asymptomatic patients (Jung et al., 1998), and in carriers with different sarcomere mutations, including those who are yet to develop LVH (Crilley et al., 2003). This energy deficit is exacerbated during exercise and may explain the development of diastolic dysfunction in HCM patients during peak exercise (Dass et al., 2015). Impaired myocardial energetics was initially viewed as a result of ischemia mediated decreases in oxygen supply (Sieverding et al., 1997); however, findings from several animal models suggest the induction of energetic abnormalities and diastolic dysfunction are more likely to be primary effects of sarcomere mutations and are unlikely to be secondary consequences of LVH (Spindler et al., 1998) (Figure 1). Indeed, sarcomere mutations do facilitate chronic mismatch between ATP synthesis and ATP consumption by overall crossbridge activity; and hence, it is not surprising that inefficient use of ATP at the myofilament level could provoke cardiac dysfunction with the severity dependent on the type of mutation and the amount of mutant protein in the sarcomere (Montgomery et al., 2001; Javadpour et al., 2003). While these studies imply energetic impairment is a direct consequence of alterations of the myocardium, other studies, albeit controversial, suggest the low myocardial PCr/ATP ratios are a result of fibrosis (Esposito et al., 2009; Petersen et al., 2009). However, an argument can be made that some regions of the myocardium are more energy deprived than others, which is supported by findings whereby increased myofilament calcium sensitivity (due to sarcomere mutations) rapidly precipitated energy deprived regions during stress, which in turn decreased intercellular coupling and increased arrhythmia susceptibility (Huke et al., 2013).
Energy and metabolism are tightly coupled cellular processes; and hence, energetic impairment is usually associated with perturbed metabolism. Indeed, comprehensive multiomics profiling of LV septal myectomy samples revealed substantial dysregulation in fatty acid metabolism, reduction of acylcarnitines, and accumulation of free fatty acids that coincided with elevated levels of oxidative stress, mitochondrial damage, and reduced mitochondrial clearance (Ranjbarvaziri et al., 2021). Other studies also report a reduced capacity for fatty acid oxidation, but an increase in ketone bodies and branched chain amino acids may suggest the use of alternate fuels in HCM hearts (Previs et al., 2022). Since these metabolic derangements were observed across diverse genotypes, therapeutic strategies aimed at normalizing myocardial energetics and metabolism could have immediate clinical implications. In support, the carnitine palmitoyl transferase-1 inhibitor, perhexiline, attenuated myocardial energy impairment, improved diastolic function, and increased exercise capacity in patients with symptomatic nHCM (Abozguia et al., 2010), and is being evaluated for its ability to promote LVH regression (Ananthakrishna et al., 2021). Despite these promising outcomes, modalities that suppress fatty acid oxidation could be potentially ineffective or harmful given that fatty acid metabolism is perturbed in HCM hearts (Ranjbarvaziri et al., 2021; Previs et al., 2022). In support, trimetazidine, an inhibitor of fatty acid β-oxidation decreased exercise capacity in patients with nHCM (Coats et al., 2019).
Cardiac myosin inhibitors
Hypercontractility is another pathological feature of HCM, which is attributed to the destabilization of the myosin super-relaxed (SRX) state (Toepfer et al., 2019; Vander Roest et al., 2021). In healthy hearts, myosin undergoes physiological shifts between the SRX conformation (energy conserving) and the disordered relaxed state (DRX) conformation (energy consuming), while in hypertrophied hearts, pathogenic mutations increase the proportion of myosins in the DRX state evoking hypercontractility, impaired relaxation, and increased energy consumption that precipitates higher rates of heart failure and arrhythmias (Toepfer et al., 2020) (Figure 1). Destabilization of the SRX state is not restricted to carriers with myosin mutations and is found to occur in animal models with mutations in cardiac myosin-binding protein C (McNamara et al., 2016; Toepfer et al., 2019), cardiac myosin essential light chain (Sitbon et al., 2020), and ventricular regulatory light chain (Yadav et al., 2019; Yuan et al., 2022).
Given the DRX state facilitates crossbridge formation with greater ATP consumption, novel cardiac myosin inhibitors have been developed to reduce myosin ATPase activity. Since its discovery, where it attenuated the development of LVH, cardiomyocyte disarray, and fibrosis in mice (Green et al., 2016), MYK-461 (mavacamten) is now a viable therapeutic modality for HCM. Like previous therapies, mavacamten demonstrated a more pronounced therapeutic effect in mice only when administered prior to the onset of overt LVH (pre-hypertrophic state), which may imply maximum benefit could be achieved in patients with less established HCM. Strikingly, mavacamten has been associated with favorable outcomes in patients with and without obstruction (Heitner et al., 2019; Ho et al., 2020; Olivotto et al., 2020). In the EXPLORER-HCM clinical trial, which evaluated the effect of mavacamten in symptomatic patients with obstruction, this agent was associated with improved New York Heart Association functional class, better exercise capacity, less LVOT obstruction, and improved health status (Olivotto et al., 2020; Spertus et al., 2021). Interestingly, sub-study analysis revealed mavacamten treatment reduced LV mass and LV wall thickness, as well as left atrial volume index (Saberi et al., 2021), with accompanying improvements in diastolic function and systolic anterior motion of the mitral valve (Hegde et al., 2021; Cremer et al., 2022). Consistently, mavacamten treatment in patients without obstruction was associated with improvements in myocardial wall stress as evidenced by reductions in NT-proBNP and troponin I levels (Ho et al., 2020).
Mechanistically, mavacamten is thought to decrease force production, inhibit myosin ATPase activity, and accelerate cross-bridge detachment rate (Anderson et al., 2018; Mamidi et al., 2018; Rohde et al., 2018; Awinda et al., 2021), but as to how this agent attenuates LVH, fibrosis, and diastolic dysfunction warrants further investigation. Mavacamten treatment may also reduce the need for septal reduction therapy in highly symptomatic patients with obstruction (Desai et al., 2023). Finally, when evaluated in patients on β-blockers, mavacamten improved heart-rate independent measures, but not indices that were heart-rate dependent, including peak exercise capacity (Wheeler et al., 2023). This finding may argue for dose reduction or withdrawal of β-blockers to gain maximum effect of this agent, however such decisions should be considered carefully with respect to patient history.
In summary, several mechanism-driven therapeutic modalities have been identified in animal models of HCM. The poor clinical translation of these modalities highlights the large gap between preclinical target discovery and clinical implementation. Mavacamten seems to be an exception, providing symptom relief and reversing established pathological features in clinical cohorts. However, if this agent is to be considered a mainstay therapy, careful dosing will be required to prevent reductions in LV ejection fraction and atrial fibrillation (Heitner et al., 2019).
Future Perspectives
HCM is a heterogeneous disease with differences in genetic etiology underpinning symptom severity and patient response to pharmacotherapies. Indeed, an association between position of mutation and disease severity has been described in early studies, whereby different mutations in myosin heavy chain beta (β-MHC) revealed variability in the nature and extent of functional impairment in contractile properties (Lankford et al., 1995). Similarly, different mutations in troponin T precipitate different alterations in muscle fibers (Hernandez et al., 2005). Moreover, when the impact of mutations on cardiac structure and function were evaluated in pre-hypertrophic individuals, the R92W troponin T mutation was associated with increased systolic function, the A797T β-MHC mutation with reduced diastolic function, and the R403W β-MHC mutation with both reduced systolic and diastolic function (Revera et al., 2008). These findings support the need for interrogating the causal effect of novel mutations, especially those prevalent in specific ethnic populations (Viswanathan et al., 2018; Pua et al., 2020; Wu et al., 2020), and which are of non-sarcomere origin, as this will facilitate the development of mechanism-driven therapies.
Treatment with mavacamten, or next-in-class, aficamten (Maron et al., 2023) have provided substantial benefits in symptomatic patients; however, the exclusion of individuals with severe heart failure symptoms and the low representation of non-Caucasian participants and children in these studies argues whether these agents can provide universal benefits across the entire clinical spectrum. Such concerns are derived from past findings whereby several therapeutic compounds that improved cardiac structure and function in animal models of HCM were unable to promote meaningful benefits in humans. For several decades, animal models with various pathogenic mutations (predominantly in contractile proteins) have helped to increase our understanding of HCM, and while these translational disparities could be attributed to shortcomings of the respective clinical trials, it must be acknowledged that animal models may not completely represent human diseases. For instance, most mouse models of HCM fail to develop overt LVH and/or fibrosis amongst other clinical features (Gannon and Link, 2021); and hence, it may be prudent to place greater emphasis on studying human cells or tissue to elucidate the causal effects of novel mutations and to discover new therapeutic targets.
Human induced pluripotent stem cells (iPSCs) offer an unprecedented opportunity to study human physiology and disease at the cellular and organ level. Human iPSCs are pluripotent, which means they can be differentiated into any cell type of the human body, and being stem cells, they can be expanded into millions of cell progeny, circumventing the limitations associated with primary tissues. Being derived from patients, iPSCs allow for the identification and validation of therapeutic targets across diverse genotypes, thereby accounting for clinical heterogeneity and maximizing patient response to novel therapeutics, while mitigating adverse effects. We and others have derived iPSCs from HCM patients with different mutations, and remarkably, cardiomyocytes differentiated from these iPSCs show clinical hallmarks, including cellular hypertrophy, arrhythmia, myofibril disarray, calcium mishandling, and impaired relaxation (Lan et al., 2013; Viswanathan et al., 2018; Prondzynski et al., 2019; Wu et al., 2019; Zhou et al., 2019; Ramachandra et al., 2022). As expected, different mutations evoke different pathological features in these human models, reaffirming the need for mechanism-driven therapies (Prajapati et al., 2018; Smith et al., 2018; Bhagwan et al., 2020). Importantly, these iPSC-derived HCM models have provided new mechanistic insight into disease pathogenesis (Qiu et al., 2021; Vander Roest et al., 2021; Ramachandra et al., 2022), and functionally characterized the pathogenicity of novel mutations, both in sarcomere and non-sarcomere encoding genes (Liu et al., 2014; Pua et al., 2020; Kondo et al., 2022), positioning these human cellular models as effective tools for developing patient-tailored therapies. Establishing patient-specific iPSC models for every uncharacterized HCM genotype will be extremely challenging, and so for now priority could be given for novel mutations with high risk of pathogenicity as determined by combined assessment of in silico modelling, population prevalence, and clinical severity.
Gene therapy as a potential future treatment modality for HCM
Recently, the effect of genomic base editing to correct the pathogenic MYH7 R403Q mutation has been evaluated in two independent studies, where this modality was able to correct and rescue pathological phenotypes in HCM patient-derived cardiomyocytes and in mice with HCM (Chai et al., 2023; Reichart et al., 2023) (Figure 1). These studies mark the first demonstration of efficient single nucleotide gene correction in postnatal mammalian cardiomyocytes in vivo, and highlight permanent genomic correction of all cardiomyocytes is not compulsory to prevent the onset of HCM. Gene editing could potentially be a one-time treatment modality for HCM patients and it would be interesting to see whether this approach can also attenuate pathological features in more established cases, although dosing will require rigorous calculations to minimize bystander editing and prevent adverse effects (Reichart et al., 2023). In summary, gene editing could serve as a very specific therapeutic modality for individuals with known pathogenic mutations, and as we interrogate the causality of other mutations in models relevant to human physiology there is much opportunity to develop mechanism-driven therapies for HCM.
Conclusions
Recent findings highlight conventional pharmacotherapies as being ineffective in improving long-term prognosis in HCM patients, which can be attributed to their non-selective mode of action and their inability to target the underlying pathophysiology. Much of our mechanistic understanding of HCM is centered on studies interrogating the genotype-phenotype relationship of a few specific pathogenic sarcomere variants. However, it must be acknowledged, we still lack substantial understanding of many other pathogenic sarcomere variants, non-sarcomere variants, and variants prevalent in non-Caucasian populations. To improve health outcomes in HCM patients, we should strongly consider moving away from the one-size fits all approach of using non-selective pharmacotherapies and focus on developing mechanism-driven therapies based on the genetic background of each patient. A workflow combining genetic data, in silico modeling, and patient-specific iPSC models could (1) give impetus for unravelling the complexity of HCM, (2) help to risk stratify genotype-positive/phenotype-negative carriers, and (3) facilitate the development of more effective therapies that can potentially prevent disease progression and improve health outcomes in patients with established HCM.
Conflicts of interest
The authors declare that they have no conflicts of interest. Chrishan Ramachandra and Derek Hausenloy did not participated at any level in the editorial review of this manuscript.
Acknowledgements
Chrishan Ramachandra is supported by the Goh Cardiovascular Research Award (Duke-NUS-GCR/2022/0027). Iswaree Balakrishnan is supported by the SingHealth Duke-NUS Academic Medicine Research Grant (AM/TP073/2023 (SRDUKAMR2373)). Weng Khong Lim is supported by the National Precision Medicine Programme (NPM) PHASE II FUNDING (MOH-000588). Derek Hausenloy is supported by the Duke-NUS Signature Research Programme funded by the Ministry of Health, Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research Investigator Award (MOH-STaR21jun-0003), Centre Grant scheme (NMRC CG21APR1006), and Collaborative Centre Grant scheme (NMRC/CG21APRC006).
References
Chrishan J. Ramachandra1,2
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore.
Iswaree Devi Balakrishnan3,4
3Department of Cardiology, National Heart Centre Singapore, Singapore. 4Yong Loo Lin School of Medicine, National University Singapore, Singapore.
Weng Khong Lim5-8
5SingHealth Duke-NUS Institute of Precision Medicine, Singapore. 6SingHealth Duke-NUS Genomic Medicine Centre, Singapore. 7Cancer & Stem Cell Biology Program, Duke-National University of Singapore Medical School, Singapore. 8Laboratory of Genome Variation Analytics, Genome Institute of Singapore, Agency for Science, Technology and Research, Singapore.
Derek J. Hausenloy1,2,4,9
1National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 2Cardiovascular & Metabolic Disorders Program, Duke-National University of Singapore Medical School, Singapore. 4Yong Loo Lin School of Medicine, National University Singapore, Singapore. 9The Hatter Cardiovascular Institute, University College London, London, UK.
Corresponding author:
Chrishan J. Ramachandra
Email: chrishan.ramachandra@nhcs.com.sg
In a new window | Download PPT
Figure 1. Schematic illustrating the pathophysiology of HCM, whereby pathogenic variants potentiate hypertrophic and fibrotic responses through abnormal sarcomere mechanics, impaired energetics, calcium mishandling, and oxidative stress. Several therapeutic modalities (blue) have been developed to target these maladaptive processes. Abbreviations: NCX- Na+/Ca2+ exchanger; Ang II- angiotensin II; AT1R- angiotensin II receptor type 1; MR- mineralocorticoid receptor; NAC- N-acetylcysteine; ROS- reactive oxygen species; TGF-β- transforming growth factor beta.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 8667 | 24 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA