Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
The reciprocal correlation between oxidative stress and diabetes and antioxidant therapeutic approaches
Time:2024-03-08
Number:7318
Author Affiliations

Conditioning Medicine 2023. 6(4): 98-119.
Abstract
The establishment of diabetes mellitus (DM) is multifactorial, depending on both genetic and environmental factors, including oxidative stress. Oxidative stress could be an early event in the pathology of DM and not just a consequence of chronic hyperglycemia. Oxidative stress leads to the onset of DM through insulin resistance induction, insulin biosynthesis suppression, and β-cell apoptosis. However, the correlation between oxidative stress and DM is found to be reciprocal since the diabetic state itself contributes to further deterioration of the oxidative state in diabetic patients. Hyperglycemia, elevated free fatty acids, and inflammation result in exacerbated reactive oxygen species generation, which not only affects DM establishment but is also responsible for a plethora of diabetic complications. Even though hyperglycemia seems to be the main mediator of diabetic complications, there is strong evidence that the initial event of oxidative damage could be by either elevated free fatty acids or inflammation that already exists in the prediabetic state. Insulin resistance also precedes many years before the onset of type 2 DM and the subsequent establishment of the diabetic state, which is characterized by hyperglycemia. Therefore, antioxidant therapeutic approaches could prevent diabetic onset, improve hyperglycemia and other parameters of the diabetic state, such as inflammation, and subsequently ameliorate diabetic complications. This review focuses on the pathophysiological reciprocal regulation of oxidative stress and diabetes and therapeutic antioxidant approaches, namely non-enzymatic, enzymatic, and mechanism-based antioxidants.
Keywords: Oxidative stress, Hyperglycemia, Diabetes mellitus, Insulin resistance, New therapeutic approaches, Antioxidant therapy
Abstract
The establishment of diabetes mellitus (DM) is multifactorial, depending on both genetic and environmental factors, including oxidative stress. Oxidative stress could be an early event in the pathology of DM and not just a consequence of chronic hyperglycemia. Oxidative stress leads to the onset of DM through insulin resistance induction, insulin biosynthesis suppression, and β-cell apoptosis. However, the correlation between oxidative stress and DM is found to be reciprocal since the diabetic state itself contributes to further deterioration of the oxidative state in diabetic patients. Hyperglycemia, elevated free fatty acids, and inflammation result in exacerbated reactive oxygen species generation, which not only affects DM establishment but is also responsible for a plethora of diabetic complications. Even though hyperglycemia seems to be the main mediator of diabetic complications, there is strong evidence that the initial event of oxidative damage could be by either elevated free fatty acids or inflammation that already exists in the prediabetic state. Insulin resistance also precedes many years before the onset of type 2 DM and the subsequent establishment of the diabetic state, which is characterized by hyperglycemia. Therefore, antioxidant therapeutic approaches could prevent diabetic onset, improve hyperglycemia and other parameters of the diabetic state, such as inflammation, and subsequently ameliorate diabetic complications. This review focuses on the pathophysiological reciprocal regulation of oxidative stress and diabetes and therapeutic antioxidant approaches, namely non-enzymatic, enzymatic, and mechanism-based antioxidants.
Keywords: Oxidative stress, Hyperglycemia, Diabetes mellitus, Insulin resistance, New therapeutic approaches, Antioxidant therapy
Highlights
We focus on the cellular mechanisms involved in the reciprocal correlation between diabetes mellitus and oxidative stress. We aim to define the sequence of events that lead to the onset of diabetes mellitus and complications of oxidative damage. In addition, we analyze the molecular pathways involved in diabetes establishment, including the significant role of oxidative stress. Moreover, we explore promising therapeutic antioxidant approaches, namely non-enzymatic, enzymatic, and mechanism-based antioxidants. Significantly, targeting oxidative stress offers a paradigm of how a conditioning strategy could influence the therapeutic approach for diabetes mellitus, leading to new clinically efficient treatments.
Introduction
The prevalence of diabetes mellitus (DM) is growing exponentially worldwide at an epidemic proportion (Yaribeygi et al. 2020). Over the past three decades, the global burden of DM has swelled from 30 million in 1985 to 382 million in 2014, with current trends indicating that these rates will only continue to rise (Leon, 2015). At present, there are approximately 463 million diabetic patients in the world, and on average, one in 11 adults (20-79 years old) has diabetes (Wei et al., 2022). The latest estimates by the international diabetes federation project are that 592 million (1 in 10 persons) worldwide will have DM by 2035 (Leon, 2015).
According to the current classification, there are two major types of diabetes, type 1 diabetes (T1DM) and type 2 diabetes (T2DM) (Adler et al., 2021), which account for more than 90% of diabetes cases, making them the most common types of diabetes (Wei et al., 2022). The distinction between the two types has historically been based on age at onset, degree of loss of β-cell function, degree of insulin resistance, presence of diabetes-associated autoantibodies, and requirement for insulin treatment for survival. However, none of these characteristics unequivocally distinguishes one type of diabetes from the other, nor account for the entire spectrum of diabetes phenotypes (Adler et al., 2021). A relatively small proportion (10%) of patients suffering from diabetes mellitus have type 1 or insulin-dependent diabetes. However, most diabetes patients are not insulin-dependent and are able, at least initially, to produce the hormone (Rochette et al., 2014). It is now generally agreed that the underlying characteristic common to all forms of diabetes is the dysfunction or destruction of pancreatic β-cells. Many mechanisms can lead to a decline in function or the complete destruction of β-cells. These mechanisms include genetic predisposition and abnormalities, epigenetic processes, insulin resistance, autoimmunity, concurrent illnesses, inflammation, and environmental factors (Adler et al., 2021).
Although there are several studies to elucidate the molecular mechanisms underlying the development of diabetes complications, their precise pathophysiology is not completely understood. One of the primary mechanisms for the development of diabetes complications is through oxidative stress (Yaribeygi et al. 2020). Oxidative stress is when ROS overproduction in vivo exceeds the buffering capacity of antioxidant enzymes and antioxidants, resulting in a local imbalance between ROS production and destruction (Kayama et al., 2015). When generated in appropriate amounts, ROS can be produced as part of the inflammatory response, providing tissue protection (Chatterjee, 2016). In contrast, when available in significant excess, they are known to modify/degenerate biological macromolecules, e.g., nucleic acid (DNA degeneration), lipids (lipid oxidation), and proteins (membrane protein degeneration), thus inducing cell dysfunction or death (Kayama et al., 2015). However, the concept of oxidative stress as a global imbalance of pro-oxidants and antioxidants has been questioned by several researchers. Recognition of the existence of multiple, discrete redox signaling pathways suggests that a more suitable definition for oxidative stress is a condition that disrupts redox signaling and control (Jones, 2006). Therefore, ROS play a dual role as both beneficial or protective signaling molecules and damaging ones (Rochette et al., 2014; Chatterjee, 2016; Hurrle and Hsu, 2017).
Mitochondria are the primary sources of ROS production, mainly through the electron transport chain (Rains and Jain 2011; Andreadou et al. 2021a). Other sources of oxidative stress are ROS-generating enzymes, mainly NAD(PH) oxidases (NOXs), xanthine oxidase (XO), endothelial nitric oxide synthase (eNOS), lipoxygenases (LOX), CYP450 enzymes, and myeloperoxidase (MPO) (Rochette et al., 2014; Kayama et al., 2015; Yaribeygi et al., 2019; Gonzalez, 2005; Łuczak et al. 2020; Desikan et al. 2017). ROS may also be derived from exogenous sources such as excessive dietary nutrients or tobacco smoking (Newsholme et al., 2019). Regardless of ROS levels, their ultimate effects are determined by the balance between the amount of ROS produced, the ROS-scavenging capacity, and the additional defense mechanisms of individual cells (Rochette et al., 2014; Kayama et al., 2015). Defense mechanisms against free radical-induced oxidative stress involve (a) preventive mechanisms, (b) repair mechanisms, and (c) antioxidant defenses. An antioxidant can be defined as any substance that, when present in very low concentrations compared to that of an oxidizable substrate, significantly delays or inhibits the oxidation of that substrate. Antioxidants can be classified as enzymatic and non-enzymatic (Rochette et al., 2014). The primary enzymatic antioxidants are superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), and thioredoxin peroxidase. The primary non-enzymatic antioxidants are vitamins, such as vitamins A, C, and E, coenzyme Q10, glutathione, and uncoupling protein (UCP) (Rochette et al., 2014; Kayama et al., 2015; Fisler and Warden 2006; Andreadou et al. 2021b).
This review aims to clarify the correlation between DM and oxidative stress and to define the sequence of events that lead to the onset of DM and complications of oxidative damage. Therefore, in light of published literature reviews surrounding DM and oxidative stress, our review focuses on new potential targets in antidiabetic treatment. Thus, we suggest the antioxidant approach as a potential mechanism-based key element to treat DM rather than another anti-hyperglycemic approach.
Oxidative stress leading to DM establishment
Oxidative stress plays a significant role in the development of both T1DM and T2DM. Despite the fact there are some different aspects of the pathogenesis of these two types of DM, oxidative stress is a principal mechanism in their progression since it actively affects not only insulin resistance but also glucose tolerance, insulin secretion, dyslipidemia, apoptosis, and autoimmune destruction of β-cells, ultimately leading to both types of DM and the onset of many diabetic complications (Kayama et al., 2015; Yang et al. 2011; Tangvarasittichai, 2015; Kaneto et al., 2007).
Failure of pancreatic β cells is a common characteristic of type 1 and type 2 DM. T1DM is induced by the destruction of pancreatic β cells, mediated by an autoimmune mechanism and the consequent inflammatory process. The biological dialogue between the immune cells and the β-cells results in an autoimmune assault against the β-cells. Various inflammatory cytokines (such as tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-1β) and oxidative stress are produced during this process, which has been proposed to play an important role in mediating β-cell destruction. ROS release from phagocytes can damage adjacent cells, leading to failure to resolve inflammation (Kaneto et al., 2007; Newsholme et al., 2019). β-cell destruction by ROS, whether induced by oxidants given exogenously or elicited by cytokines, occurs through both apoptotic and necrotic mechanisms. There are also indications that oxidative stress is involved in other aspects of T1DM, including macrophage-mediated clearance of apoptotic cells and complications that develop in the vasculature of diabetic patients (Ηaskins et al., 2003).
On the other hand, T2DM is the most prevalent and severe metabolic disease, and β-cell dysfunction and insulin resistance are the hallmark of T2DM (Kaneto et al., 2007). Individuals with T2DM display impaired insulin-stimulated glucose uptake into muscle and adipocytes, defective insulin suppression of hepatic glucose output, and are referred to as insulin resistant. Although insulin resistance could play a protective role when key metabolic tissues are exposed to potentially injurious levels of nutrients repressing tissue nutrient overload, this comes at a cost - concomitant hyperinsulinemia, which could lead to the inability of the pancreas to compensate for the large increase in glucose levels (James et al., 2021). This situation is described as the severest pathophysiological consequence of insulin resistance, which occurs many years before the onset of T2DM as a result of a plethora of factors, including genetic and environmental factors (e.g., nutrition, physical inactivity, environmental pollutants) (Evans et al., 2002; Tangvarasittichai, 2015; James et al., 2021). In the case of T2DM, the role of oxidative stress and its downstream effects is crucial since these factors could lead to insulin resistance in the whole body, targeting insulin-sensitive tissues through the mechanisms discussed in the following chapters (Henriksen et al., 2011). Recent findings are consistent with the idea that oxidative stress could be an early event in the pathology of DM and not simply a consequence of chronic hyperglycemia (Evans et al., 2005).
Oxidative stress as a mechanism in insulin resistance
Oxidative stress has been recently recognized as a key mechanism in insulin resistance (Hurrle and Hsu, 2017). Insulin resistance occurs when normal insulin levels are inadequate to produce a normal insulin response from fat, liver, or muscle cells. ROS have been shown to affect the insulin signaling cascade, creating the possibility for multiple disruptions in the ability of insulin to maintain its normal functions, resulting in insulin resistance of the cell, which is the most common outcome of this disruption (Rains and Jain 2011). The development of insulin resistance can also lead to β-cell failure due to defective β-cell insulin response (Wang and Jin, 2009).
There is a strong correlation between the state of oxidative stress, the incidence of insulin resistance, and even late-stage DM cases (Hurrle and Hsu, 2017). The exact link between oxidative stress and impaired insulin signaling has yet to be fully understood, but several well-accepted mechanisms have been proposed and are presented below (Rains and Jain 2011). Glucose transporter 4 (GLUT4) is the main transporter in peripheral insulin-sensitive tissues, namely skeletal muscle, adipose tissue, liver, and heart. The cell responds to insulin by increasing GLUT4 expression in the plasma membrane, thereby increasing cellular uptake of glucose from the bloodstream. However, high levels of insulin signaling negatively affect GLUT4 presence in the membrane (Hurrle and Hsu, 2017). GLUT1 is ubiquitously expressed and primarily responsible for basal glucose uptake, but it is not insulin-responsive and is generally expressed in low levels in adipose tissue. Gene transcription in response to oxidative stress affects insulin signaling by altering the availability of both GLUT4 and GLUT1 (Rains and Jain 2011). Exposing adipocytes to prolonged oxidative stress results in increased expression of GLUT1 due to the enhanced binding activity of activating protein-1 (AP-1) to DNA, which in turn leads to an increase in basal glucose uptake and metabolism in various cell types and, eventually, mitochondrial ROS are also elevated. On the other hand, exposure to oxidative stress results in a marked reduction of GLUT-4 expression in adipocytes due to decreased DNA binding of nuclear proteins to the insulin-responsive element (IRE) sequence in the GLUT4 promoter. Therefore, a decrease in glucose uptake in peripheral insulin-sensitive tissues is observed, and blood glucose levels remain high, leading to a positive feedback loop that drives up intravascular insulin levels, desensitizing peripheral tissues to the insulin (Hurrle and Hsu, 2017; Rains and Jain 2011). The biomolecular consequence of this cycle is the continued downregulated expression of GLUT4 in the cell membrane, exacerbating insulin resistance (Hurrle and Hsu, 2017).
Phosphatidylinositol-3-kinase (PI3K)/protein kinase B (PKB, or Akt) pathway regulates the translocation of GLUT glucose transporters from the cytoplasmic vesicles onto the cell membrane surface and thereby increases the insulin-dependent transport of glucose into the cell. Under normal conditions, appropriate tyrosine phosphorylation of insulin receptor substrate (IRS) proteins leads to the activation of the PI3K/Akt pathway and, subsequently, through Akt phosphorylation, the translocation of GLUT-4 onto the cell membrane (Świderska et al. 2020). However, oxidative stress activates several major stress-sensitive kinases attenuating insulin signaling (Evans et al., 2005). A large family of serine/threonine stress-sensitive kinases are mitogen-activated protein kinases (MAPK), which are also involved in cellular responses to proinflammatory cytokines. Three major MAPK activating cascades have been identified: c-jun N-terminal kinases (JNK), extracellular signal-regulated kinases (ERKs), and p38 kinase (Pereda et al. 2006). JNK is involved in the reduction of insulin signaling through IRS proteins, and ERKs seem to contribute to insulin resistance by suppressing the activity of the transcription factor nuclear factor erythroid-2 (E2)-related factor 2 (Nrf2), which is the primary regulator of the antioxidant response (Evans et al., 2005; Tan et al., 2011). In addition, other stress-sensitive kinases play a role in insulin resistance, such as Iκ kinase B, which controls the activation of NF-kB, which subsequently increases IRS-1 serine phosphorylation inhibiting insulin’s action, or mammalian target of rapamycin (mTOR) and glycogen synthase kinase-3, which acts synergistically to desensitize insulin signaling by phosphorylating insulin receptors or IRSs on selective serine/threonine residues (Rains and Jain 2011; Evans et al., 2005).
Concerning effects on IRS proteins, stress-sensitive kinases increase serine phosphorylation and, therefore, decrease the extent of tyrosine phosphorylation. The serine/threonine phosphorylated forms of IRS molecules are less able to associate with the insulin receptor and downstream target molecules, especially PI3K, resulting in impaired insulin action and glucose transport. In addition, the serine-threonine phosphorylated forms of IRS molecules are more susceptible to proteasome-mediated degradation (Evans et al., 2005). Therefore, when PI3K is not activated, GLUT4 translocation from its intracellular pool to the plasma membrane is not achieved, resulting in reduced activation of glucose transport and other downstream events, leading to insulin resistance (Evans et al., 2005; Rochette et al., 2014).
In summary, oxidative stress dysregulates glucose uptake from insulin-sensitive (GLUT-4) and other (GLUT-1) tissues, altering GLUT activity by activating stress-sensitive kinases or affecting transcription factors. This situation, combined with other effects of oxidative stress-sensitive kinases, leads to the development of insulin resistance and further deterioration of ROS production.
Oxidative stress suppresses insulin biosynthesis and secretion
Induction of oxidative stress leads to suppression of insulin biosynthesis and secretion. A decrease in the number of functioning pancreatic β-cells and insufficient insulin biosynthesis and/or secretion are the hallmarks of DM (Kaneto et al., 2009). One molecular mechanism of action through which chronic hyperglycemia can impair β cell function is via decreased protein expression of two important transcription factors, pancreatic and duodenal homeobox-1 (PDX-1) and MAF basic leucine zipper (bZIP) transcription factor A (MafA). Both proteins are critical for normal insulin gene expression, as the absence or mutation of their DNA binding sites on the insulin promoter leads to decreased insulin levels, insulin content, and insulin secretion (Robertson, 2006). These factors directly bind to the insulin gene promoter and are important transcription factors in pancreatic β-cell differentiation and mature β-cell function (Kaneto et al., 2009). Oxidative stress can lead to deterioration of insulin biosynthesis through multiple effects on PDX-1 and MafA pathways, as shown in Figure 1 (Kaneto, 2013; Kaneto and Matsuoka, 2012; Zhu et al., 2013; Vallée and Lecarpentier, 2018).
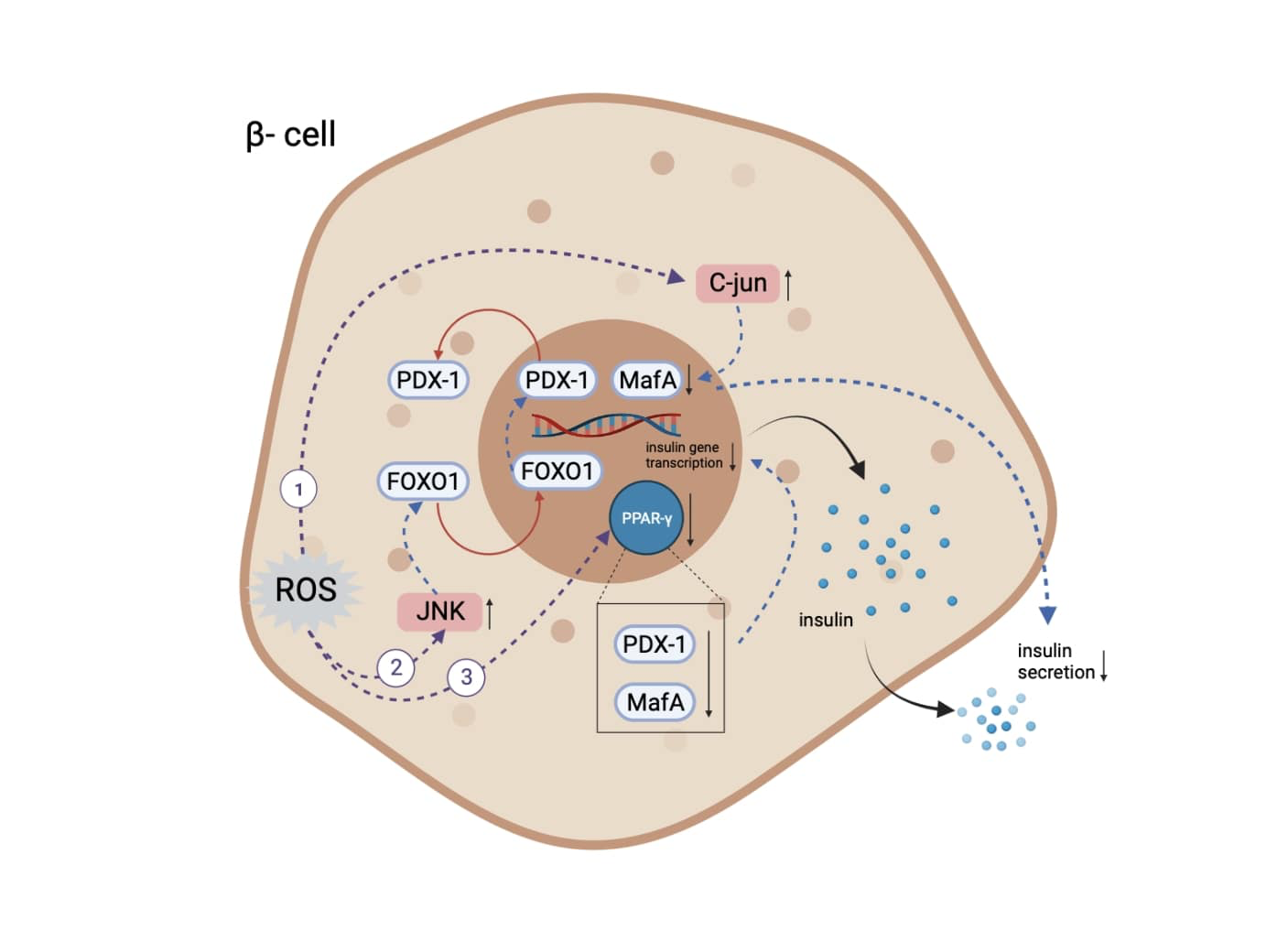
In a new window | Download PPT
Figure 1. The mechanisms through which oxidative stress leads to insulin expression and secretory deficiencies. (1) Increase of level and activity of c-Jun protein, leading to suppressed expression of MafA and subsequent regulation of insulin expression and secretion. (2) Activation of JNK kinase pathway, leading to translocation of FOXO-1 and subsequent opposite translocation of PDX-1. (3) Inactivation of PPARγ, resulting in PDX-1 and MafA degradation. Abbreviations: MafA, MAF bZIP transcription factor A; JNK, c-Jun N-terminal kinase; FOXO-1, Forkhead/winged helix box protein O1; PDX-1, pancreatic and duodenal homeobox -1; PPARγ, peroxisome proliferator activated receptor γ (Created with BioRender.com.)
Oxidative stress activates the JNK kinase pathway, leading to translocation of the transcription factor Forkhead/winged helix box gene, group O-1 (FOXO-1) from the cytoplasm to the nucleus, which results in the opposite translocation of PDX-1 from the nucleus to the cytoplasm and subsequent suppression of insulin biosynthesis (Kaneto, 2013; Kaneto and Matsuoka, 2012). Oxidative stress also increases the level and activity of c-Jun protein. C-Jun suppresses the expression of MafA, which regulates insulin expression and is involved in insulin secretory defects (Kaneto, 2013). Furthermore, ROS production leads to the inactivation of peroxisome proliferator-activated receptor-γ (PPARγ), whose activation prevents PDX-1 and MafA proteins from degrading and eventually maintains the insulin-producing and secreting phenotype (Zhu et al., 2013; Vallée and Lecarpentier, 2018).
In addition to the classical paradigms for insulin regulation by glucose, reports suggest a role for UCP in insulin secretion and utilization (Bordone et al., 2005). Through UCP’s function, a faster flow of electrons is achieved, thus reducing the membrane potential, resulting in a reduction in ROS production (Bordone et al., 2005; Fisler and Warden 2006). Despite this beneficial effect of UCPs, UCP-2 may contribute to the development of insulin resistance. Pancreatic β-cells secrete insulin in response to a meal by perceiving the cell's ATP/ADP ratio resulting from glucose metabolism. UCP-2 mediates a slight increase in proton leakage, thus reducing the ATP/ADP ratio of the cell and, consequently, the effect of glucose on insulin secretion. Therefore, there is strong evidence that UCP-2 is a negative glucose regulator (Fisler and Warden 2006). Additionally, up-regulation of UCP-2 can impair proteolytic processing of proinsulin to mature insulin, which results in increased secretion of proinsulin (Chan and Kashemsant, 2006).
In conclusion, the augmented expression of c-Jun in diabetic islets decreases MafA activity, followed by reduced insulin biosynthesis and secretion, thereby explaining, at least in part, the molecular mechanism for β-cell glucose toxicity often observed in T2DM (Kaneto, 2013; Kaneto and Matsuoka, 2012).
Oxidative stress induces β-cell apoptosis
Oxidative stress can induce pathways of β-cell apoptosis through mechanisms associated with cytokine mobilization and mitochondrial dysfunction (Anuradha et al., 2014; Drews et al., 2010). Oxidative stress in β-cells indirectly leads to the expression of Fas (cluster of differentiation-95 or apoptosis antigen-1) and TNF-α receptor-1, which are responsible for the extrinsic pathway of apoptosis (Anuradha et al., 2014). Another pathway sensitive to oxidative stress is the JNK1/2 pathway, which responds specifically to stress-induced signals that drive apoptosis. Among other specific molecular targets of JNK1/2 are β-cell lymphoma (Bcl-2) proteins, which are closely related to apoptotic cell death factors (Li et al., 2012). Additionally, mTOR complexes play important roles in regulating β-cell proliferation, apoptosis, and insulin biosynthesis and secretion, partially via ameliorating oxidative or endoplasmic reticulum (ER) stress. Oxidative stress inhibits mTOR complex-1, which usually inhibits autophagy in mitochondria and strengthens mitochondrial capacity and function. Thus, dysregulated mTOR signaling triggers cell death, autophagy, and apoptosis, resulting in failed protein synthesis in vital organs, including pancreatic β-cells. mTOR complexes have been consistently identified as key modulators of β-cell mass (Wang et al., 2016). Mitochondria are also targets of ROS and reactive nitrogen species (RNS). Free radicals mediate the opening of the mitochondrial permeability transition pore (PTP), a multi-protein complex that functions as a mitochondrial Ca2+ release channel. Free radicals trigger the opening of the mitochondrial PTP to an extent that collapses the mitochondrial membrane potential, leading to ATP depletion and the release of proapoptotic factors. This results in the formation of a multimeric complex known as the apoptosome, which initiates the caspase cascade, culminating in β-cell death (Drews et al., 2010).
In summary, oxidative stress deteriorates insulin resistance and biosynthesis and directly damages β-cells, inducing apoptosis by affecting mainly stress-sensitive kinases and mitochondrial function.
DM promotes oxidative stress and oxidative stress promotes diabetic complications
In the early stage, before the establishment of T2DM, β-cells increase their secretory capacity to compensate for and control hyperglycemia (Tangvarasittichai, 2015). However, continued exposure to elevated glucose has detrimental effects on pancreatic β-cell insulin secretion and is correlated with the onset of insulin resistance in the periphery. Elevated glucose (or other elevated plasma substrates) can initially result in enhanced metabolism of β-cells and subsequent hyperinsulinemia, but chronic exposure induces oxidative stress that may cause β-cell dysfunction and even β-cell death (Keane et al., 2015). Above optimal insulin concentrations, the insulin signaling pathway amplifies the activity of NOX4, which increases ROS levels, leading to further deterioration and β-cell dysfunction (Hurrle and Hsu, 2017). Thus, β-cell dysfunction, in which oxidative stress plays a significant role, is essential for T2DM development and progression (Tangvarasittichai, 2015). On the other hand, hyperglycemia in a patient with T1DM results from genetic, environmental, and immunologic factors. These factors lead to the destruction of pancreatic β-cells and insulin deficiency, resulting in hyperglycemia (Mouri and Badireddy, 2023). Elevated cell glucose levels result in an increased production of ROS, contributing to the evolution of DM and further deterioration of pancreatic β-cells (Maiese et al., 2007; Yang et al. 2011; Tangvarasittichai, 2015).
Hyperglycemia as the major factor of ROS generation in DM
Based on in vivo and in vitro studies, free radical generation during chronic hyperglycemia occurs via the following molecular mechanisms: a) Electron transport chain (ETC)-dependent mechanisms, b) ROS-independent activation of free radical generators, and c) Disabling of cellular antioxidant systems (Maritim et al., 2003; Luevano-Contreras and Chapman-Novakofski, 2010; Yaribeygi et al., 2019).
Electron transport chain (ETC) dependent mechanisms
The overproduction of superoxide anion by the mitochondrial ETC plays a major role in hyperglycemia-induced pathogenetic mechanisms. In diabetic cells, more glucose passes through the glycolytic pathway, producing more pyruvate and acetyl coenzyme A (CoA), pushing more electron donors (NADH, FADH2) into the ETC (Brownlee, 2005; Yan, 2014). As a result, the voltage gradient across the mitochondrial membrane increases until a critical threshold is reached, and therefore, the normal process through the ETC is impaired (Brownlee, 2005; Yan, 2014; Kayama et al., 2015). Electron transfer inside complex III is blocked, causing the electrons to back up to coenzyme Q, generating superoxide (Brownlee, 2005).
Another pathway that leads to increased levels of ROS is hyperglycemia-induced activation of p66Shc. p66Shc is a redox enzyme that acts specifically in the mitochondrion, reducing oxygen and generating H2O2 (Rochette et al., 2014). p66Shc directly oxidizes cytochrome c instead of inducing a non-enzymatic increase of superoxide anion through a higher metabolic rate (Brownlee, 2005; Rochette et al., 2014). Additionally, p66Shc decreases the expression of antioxidant enzymes by inhibiting FOXO transcription factors (Rochette et al., 2014).
Although it appears that mitochondria are required to initiate hyperglycemia-induced superoxide production, some evidence indicates that this, in turn, can activate several other superoxide production pathways that may amplify the original damaging effect of hyperglycemia. These include redox changes, NOXs, and uncoupled eNOS (Giacco and Brownlee, 2010).
ROS originating from the ETC, along with high levels of NADH, can impair the activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which is modified through the action of the enzyme poly (ADP-ribose) polymerase (PARP). PARP is activated by DNA damage caused by glucose-generated free radicals in the mitochondria and, therefore, splits the NAD+ molecule into its two component parts: nicotinic acid and ADP-ribose. PARP then proceeds to make polymers of ADP-ribose, which accumulate on GAPDH, resulting in its inhibition. As a result, all the glycolytic intermediates upstream of GAPDH are increased (Brownlee, 2005; Giacco and Brownlee, 2010; Yan, 2014). This increase consequently activates five pathways that can branch off the glycolytic pathway under chronic hyperglycemic conditions (Yan, 2014). These pathways include: (1) increased glucose flux through the polyol pathway, (2) increased formation of advanced glycation end products (AGEs), (3) activation of protein kinase C (PKC), (4) increased glucose flux through the hexosamine pathway, and (5) glyceraldehyde autoxidation pathway (Rochette et al., 2014; Yan, 2014). These pathways are minor and insignificant in glucose metabolism under normoglycemic conditions but can become significant pathways to pass high glucose levels. As discussed below, all these pathways have been linked to ROS production, oxidative stress, the pathogenesis of DM, and diabetic complications (Yan, 2014). In summary, hyperglycemia and oxidative stress form a vicious circle, leading to further ROS generation and exacerbation of hyperglycemia, mainly via ETC-dependent pathogenetic pathways, as represented in Figure 2.
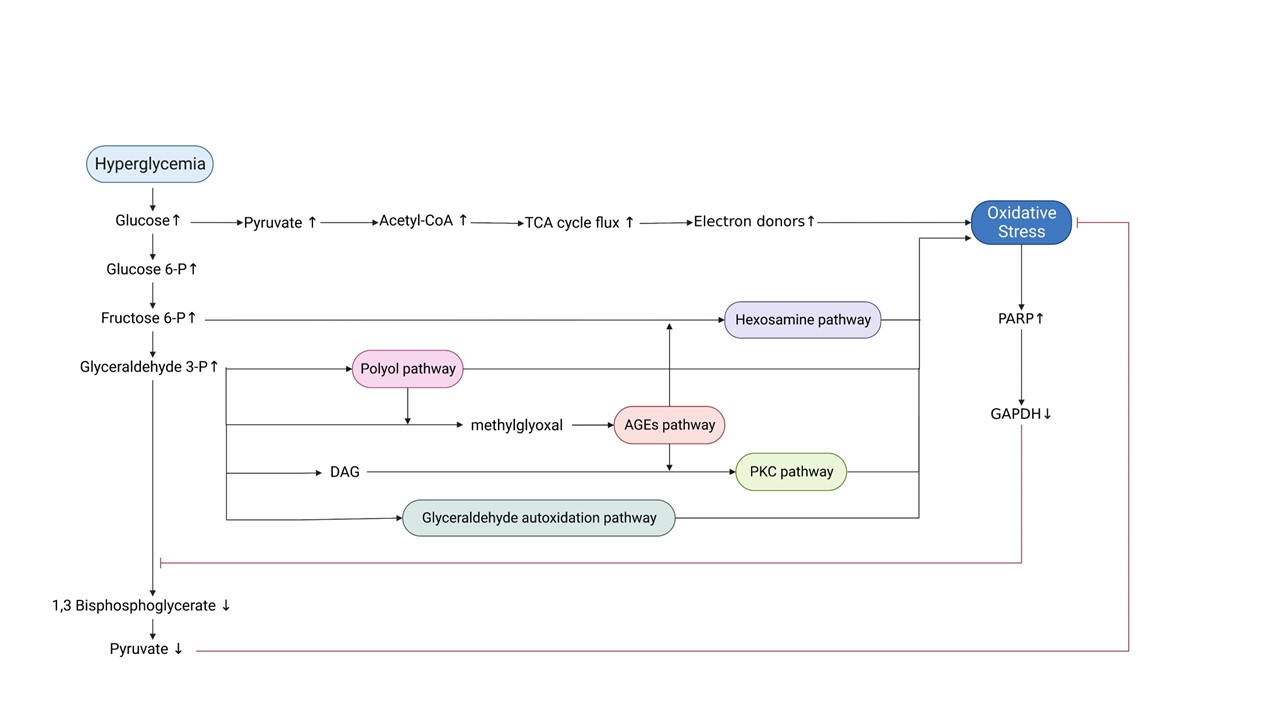
In a new window | Download PPT
Figure 2. Schematic representation of the mechanisms through which hyperglycemia leads to oxidative stress and diabetic complications. Hyperglycemia induces oxidative stress through increased TCA cycle flux. Oxidative stress-mediated inactivation of GAPDH: a) leads to accumulation of glycolytic intermediates, which in turn trigger the five major pathogenetic pathways responsible for diabetic complications and further promotion of oxidative stress, and b) counteracts the accumulation of pyruvate to prevent excessive TCA cycle flux. Abbreviations: TCA, tricarboxylic Acid; PARP, poly-ADP ribose polymerase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; DAG, diacylglycerol; AGEs, advanced glycation end products; PKC, protein kinase C (Created with BioRender.com.)
Increased glucose flux through the polyol pathway
The polyol pathway comprises the rate-limiting enzyme aldose reductase (AR) and sorbitol dehydrogenase. Under normoglycemic conditions, AR reduces toxic aldehydes to inactive alcohols, thereby protecting cells. AR has a low affinity for glucose and typically does not metabolize significant amounts of glucose (Brownlee, 2005; Gleissner et al., 2007). However, inhibiting GAPDH increases the flow through the polyol pathway because of increased glucose levels and an increase in glycolytic metabolites such as glyceraldehyde 3-phosphate, for which AR has much higher affinity (Tahrani et al., 2011; Yan, 2014).
Thus, increased flux of the polyol pathway leads to increased oxidative stress through three different molecular mechanisms: (1) AR reduces glucose to sorbitol, consuming NADPH, and then sorbitol is oxidized to fructose by sorbitol dehydrogenase, with NAD+ as a cofactor, leading to redox imbalance between NADH and NAD+. As the ratio NAD+/NADH decreases, oxidative stress occurs due to NADH being the substrate for NOX to generate ROS (Chung et al., 2003; Brownlee, 2005; Gleissner et al., 2007; Giacco and Brownlee, 2010; Yan, 2014). (2) Depletion of NADPH leads to a significant decrease in the reduced form of glutathione (GSH) levels, which is an important endogenous scavenger of ROS (Chung et al., 2003; Giacco and Brownlee, 2010; Yan, 2014; Andreadou et al. 2021b). (3) Through the polyol pathway, glucose is metabolized into fructose. Being more potent nonenzymatic glycation agents, fructose and its metabolites are converted into α-oxaldehydes (glyoxal, methylglyoxal, 3-deoxyglucosone), which can eventually lead to increased AGEs (Chung et al., 2003; Gleissner et al., 2007; Luevano-Contreras and Chapman-Novakofski, 2010). AGEs formation is another important pathway of oxidative stress, as discussed below.
Activation of PKC
PKCs are a family of kinases with many isoforms whose activity is greatly enhanced by diacylglycerol (DAG) (Giacco and Brownlee, 2010). Intracellular hyperglycemia increases DAG levels by increasing intracellular levels of glyceraldehyde 3-phosphate, a DAG precursor molecule, resulting from GAPDH inhibition (Gleissner et al., 2007; Giacco and Brownlee, 2010). Additionally, indirectly activating PKC may occur because of crosslinking with other pathways, such as the AGEs pathway (Gleissner et al., 2007). Activating PKC can induce ROS formation through several pathways. It has been shown that isoforms of PKC can activate NOX or p66ShcA, resulting in H2O2 production and decreased ROS-scavenging enzymes (Rains and Jain 2011; Rochette et al., 2014; Yan, 2014). Furthermore, PKC activation may lead to further deterioration of insulin resistance due to decreased insulin-stimulated IRS-1 tyrosine phosphorylation, decreased PI3K activation, and inhibited Akt-dependent eNOS activation, therefore, plays a significant role in DM establishment (Greene et al., 2004; Yan, 2014).
PKC activates transcription factors, such as NF-κB, which plays a crucial role in inflammation, immunity, cell proliferation, development, survival, and apoptosis (Brownlee, 2001; Rochette et al., 2014; Chatterjee, 2016). This activation, in turn, triggers the generation of pro-inflammatory cytokines and chemokines. Once triggered, these cytokines and chemokines bind to their respective receptors, such as platelet-derived growth factor receptor, vascular endothelial growth factor (VEGF) receptor, and epidermal growth factor receptor (EGFR) that are known to generate ROS (Chatterjee, 2016). Apart from triggering ROS production, PKC activation can mediate a plethora of diabetic complications, such as vessel permeability changes (through VEGF), increased vasoconstriction (through eNOS inhibition and endothelin-1 activation), increased microvascular matrix, and overexpression of fibrinolytic inhibitor plasminogen activator inhibitor -1, which leads to vascular occlusion etc. (Brownlee, 2005; Aronson, 2008; Rains and Jain 2011).
Increased flux through the hexosamine pathway
Fructose 6-phosphate is diverted from glycolysis to provide substrates for the rate-limiting enzyme of the hexosamine pathway, namely glutamine-fructose 6-phosphate amidotransferase (Giacco and Brownlee, 2010). In the first step of the pathway, glutamine-fructose 6-phosphate amidotransferase converts the accumulated fructose-6-phosphates to glucosamine-6-phosphate, which in turn is converted to uridine diphosphate N-acetylglucosamine (James et al., 2002; Giacco and Brownlee, 2010). Uridine diphosphate N-acetylglucosamine is the substrate for specific O-linked β-N-acetylglucosamine transferases that catalyze post-translational modifications of proteins on serine (Ser)/threonine (Thr) residues (James et al., 2002; Giacco and Brownlee, 2010; Yan, 2014). Under hyperglycemia, GAPDH inhibition significantly increases levels of fructose-6-phosphate, the proximal glycolytic intermediate, diverting it to the hexosamine pathway (Du et al., 2000; Giacco and Brownlee, 2010).
It has been suggested that glucosamine increases H2O2 levels, indicating that glucosamine in β-cells could produce ROS. Moreover, O-linked β-N-acetylglucosamine can affect transcription factors related to oxidative stress and inflammation, such as NF-κB, p53, FOXO1, and specificity protein 1 (Issad and Kuo, 2008). Of relevance to diabetic establishment is the inhibition of eNOS activity in arterial endothelial cells (Giacco and Brownlee, 2010). In addition, activation of the JNK pathway has been linked with hyperactivity of the hexosamine pathway (Ighodaro, 2018).
Increased formation of AGEs
AGEs are a heterogeneous group of molecules formed from glycation, which differs from the enzymatic reaction of glycosylation (Gkogkolou and Böhm, 2012; Prabhakar, 2021). In addition to the polyol pathway, high glucose levels can induce the formation of methylglyoxal from glyceraldehyde 3-phosphate when GAPDH function is impaired (Yan, 2014). Methylglyoxal and other intracellular dicarbonyls react with amino groups of intracellular and extracellular proteins to form AGEs (Brownlee, 2001). AGEs and their precursors can cause damage at intracellular and extracellular levels, leading to increases in oxidative stress and inflammation (Goldin et al., 2006; Giacco and Brownlee, 2010; Ighodaro, 2018).
AGEs can bind to different AGE receptors (AGE-R1, AGE-R2, AGE-R3, and RAGE) or interact abnormally with components of the extracellular matrix (ECM) to promote ROS generation (Ighodaro, 2018). Apart from AGEs, plasma proteins modified by AGE precursors may also bind to AGE receptors (Giacco and Brownlee, 2010). The AGE-receptor system could be broadly divided into two arms: one is associated with ROS production and inflammatory effects, best represented by RAGE, whereas the other is involved in AGE detoxification and suppression of proinflammatory processes represented by AGE-R1, -R2, and -R3 (Lu et al., 2004; Nowotny et al., 2015). One of the main consequences of RAGE–ligand interaction is the production of intracellular ROS via the activation of a NOX system, while AGE-R1 inhibits its activity (Rains and Jain 2011; Nowotny et al., 2015). RAGE mediates signal transduction by generating ROS, activating NFκB, while AGE-R1 suppresses NF-kB-mediated actions. Long-term exposure to AGEs depletes AGE-R1 while RAGE expression is elevated, which favors inflammatory and oxidative stress phenomena (Lu et al., 2004; Giacco and Brownlee, 2010; Nowotny et al., 2015).
Additionally, high levels of glycated proteins activate PKC, enhance the hexosamine pathway through the induction of increased recruitment of O-GlcNAc-transferase by methylglyoxal, and alter the structure and function of antioxidant enzymes, further exacerbating oxidative stress (Maritim et al., 2003; Giacco and Brownlee, 2010; Rains and Jain 2011; Yan 2014). AGEs, particularly modified proteins, occur in the intracellular compartments and ECM (Ighodaro, 2018). Intracellular proteins modified by AGEs have altered function, leading to changes in cellular properties (Goldin et al., 2006; Giacco and Brownlee, 2010). Extracellular matrix components modified by AGE precursors interact abnormally with other matrix components and matrix receptors (integrins) that are expressed on the surface of cells (Giacco and Brownlee, 2010). Accumulation of AGEs on proteins in the ECM can cause cross-links to form, which can “trap” other local macromolecules. Thus, AGEs can alter the properties of large matrix proteins such as collagen (Goldin et al., 2006).
Apart from the GAPDH pathway, other sources can generate AGEs during hyperglycemia, such as glucose autoxidation or the Maillard reaction (Negre-Salvayre et al., 2009; Luevano-Contreras and Chapman-Novakofski, 2010; Gkogkolou and Böhm, 2012; Yaribeygi et al., 2019). There are also exogenous sources of AGEs, such as tobacco smoke and food intake (Peppa and Vlassara, 2005). AGEs ingested through food might be one of the major contributing factors to the development of diabetic complications (Rhee and Kim, 2018).
The glyceraldehyde autoxidation pathway
One alternative to the classic pathway of glucose metabolism is the less familiar pathway of enediol, a by-product originating from the autoxidation of glyceraldehyde-3-phosphate in the glycolytic pathway. Autoxidation of α-hydroxyaldehydes generates ROS, especially hydrogen peroxide and α-ketoaldehydes. Although glyceraldehyde is characteristically thought of as an insulin secretagogue, when present in excess, it may also inhibit insulin secretion (Robertson, 2004; Yan, 2014; Luo et al., 2016).
ETC-independent activation of free radical generating enzymes
Recent evidence indicates that free radical generating enzymes can be directly induced by hyperglycemia (Giacco and Brownlee, 2010; Yaribeygi et al., 2019). Non-mitochondrial sources of ROS include NAD(P)H oxidase, xanthine oxidase, uncoupled eNOS, lipoxygenase, cyclooxygenase, cytochrome P450 enzymes, and other hemoproteins (Bajaj et al., 2012).
NADPH oxidases are also strongly activated by hyperglycemia either directly or by inhibiting adenosine monophosphate (AMP)‐-activated protein kinase (Bajaj et al., 2012; Yaribeygi et al., 2019). It also has been demonstrated that hyperglycemia induces NOX2 activation, the major isoform of NOX in the heart, leading to ROS production independently of glucose metabolism (Van Steenbergen et al., 2017).
Xanthine oxidase is induced by chronic hyperglycemia, leading to β-cell superoxide anion production in the cytosol and potential deterioration of oxidative stress (Bajaj et al., 2012; Newsholme et al., 2019; Yaribeygi et al., 2019).
eNOS also produces ROS. Evidence has shown that red blood cells (RBCs) from patients with T2DM exhibit endothelial dysfunction in healthy vascular tissues, suggesting potential crosstalk between RBCs and the endothelium in T2DM. The fact that arginase shares a common substrate (L-arginine) with eNOS may limit L-arginine availability and lead to eNOS uncoupling, generating ROS rather than NO (Vilahur 2018). eNOS also needs co-factors (tetrahydrobiopterin: BH4) to convert O2 to nitric oxide (NO) (Rochette et al., 2014). Otherwise, “uncoupled” eNOS produces superoxide (Bajaj et al., 2012). DM is characterized by diminished BH4 bioavailability, and therefore, through uncoupling of eNOS, oxidative stress is exacerbated (Yaribeygi et al., 2019).
CYP450 enzymes are activated by hyperglycemia, and their catalytic and hydroxylation effects on ketone bodies and fatty acids are upregulated in DM, generating higher levels of ROS. Moreover, altering CYP activity leads to mitochondrial respiratory chain damage that further induces oxidative stress (Yaribeygi et al., 2019).
Lipoxygenase (LOX) enzymes are upregulated by hyperglycemia, and their catalytic activity is enhanced, resulting in further conversion of arachidonic acid (AA) to a broad range of signaling molecules (Othman et al. 2013; Yaribeygi et al., 2019). LOX-derived metabolites act upstream of NOX pathways, suggesting the existence of interconnected signaling between LOXs and NOXs (Othman et al. 2013; Wang et al., 2020). Moreover, a LOX-derived metabolite, 12-hydroxyeicosatetraenoic acid (12-HETE), increases ROS as a result of its function as a proinflammatory lipid mediator through activation of ERK1/2 and NFκB, through the increase in VEGF and mitochondrial NO, and possibly via inhibition of nuclear translocation of Nrf2 (Evans et al., 2002; Dobrian et al., 2011; Guo et al., 2011; Chatterjee, 2016; Hernandez-Perez et al., 2017).
In summary, hyperglycemia affects the activity of the aforementioned key enzymes, leading to further ROS production in a mitochondrion-independent manner. Interestingly, the diabetes‐induced free radicals that result from enzymatic reactions and activation of oxidative enzymes like NOX, COX, LOX, and XO contribute as much as those derived from the mitochondria (Yaribeygi et al., 2019).
The initial event that leads to ROS production in DM
Although hyperglycemia is the major driver of oxidative stress in diabetes, insulin resistance occurs many years before the onset of T2DM and subsequent hyperglycemia, implying that hyperglycemia is not the initial event that leads to insulin resistance. Considering that oxidative stress is the major factor that causes insulin resistance, another pathological state may lead to excessive ROS generation as an initial event (Evans et al., 2002; Kayama et al., 2015; Tangvarasittichai, 2015). Studies with first-degree relatives of individuals with T2DM proved there are individuals with significant insulin resistance in muscle and liver (and probably fat) with mild hyperinsulinemia at a time when they are neither obese nor glucose intolerant (James et al., 2021). Therefore, even with excessive nutrient intake and weight gain, the initial event that produces insulin resistance seems to be increased oxidative stress (Boden et al. 2015). The strong association between obesity and insulin resistance suggests that a major mediator of oxidative stress-induced insulin resistance at the prediabetic stage might be inflammation due to circulating factors secreted by adipocytes, such as TNF-α, leptin, or resistin, or free fatty acids (FFAs) (Evans et al., 2002). However, there is stronger evidence that FFAs play a primary role in the pathogenesis of insulin resistance and impaired β-cell function. For instance, a study has shown that first-degree relatives of individuals with T2DM have higher levels of circulating FFAs and intramuscular lipids than healthy control individuals, suggesting that inappropriate lipid accumulation might be more important for promoting insulin resistance than total adiposity (James et al., 2021).
Link between elevated FFAs and oxidative stress
FFAs represent a causative link between obesity, insulin resistance, and the development of T2DM (Evans et al., 2002). Obesity is linked to high plasma levels of FFAs usually observed in DM because the enlarged adipose tissue mass releases more FFAs while FFAs clearance may be reduced. Once plasma FFA levels are elevated, they inhibit insulin’s anti-lipolytic action, further increasing the rate of FFA release into the circulation (Boden, 2008). Increased FFA flux from adipocytes into arterial endothelial cells increases FFA oxidation by mitochondria (Negre-Salvayre et al., 2009).
The increase in β-oxidation and the accumulation of FFA and their metabolites, especially long-chain acyl-CoAs and acylcarnitines, are associated with increased ROS production and subsequent oxidative stress (Dambrova et al., 2021). Since β-oxidation of fatty acids and oxidation of FFA-derived acetyl CoA by the tricarboxylic acid (TCA) cycle generate the same electron donors (NADH and FADH2) generated by glucose oxidation, increased FFA oxidation causes mitochondrial overproduction of ROS by precisely the exact mechanism described above for hyperglycemia (Brownlee 2005; Newsholme et al., 2019). Similar to hyperglycemia, FA-linked ROS production is associated with mitochondrial dysfunction, which includes inhibition of the ETC and activation of the same damaging pathways, namely AGEs, PKC, and the hexosamine pathway (N-acetylglucosamine) (Brownlee, 2005; Dambrova et al., 2021). ROS is exacerbated in the latter pathway because FFAs induce a state of oxidative stress by stimulating ROS-producing enzymes and impairing endogenous antioxidant defenses by decreasing intracellular glutathione (Newsholme et al., 2019; Evans et al., 2002).
In addition, diabetic dyslipidemia is characterized by an increase in n-6 polyunsaturated fatty acids (PUFAs), such as arachidonic acid (AA) (Othman et al. 2013). 12/15 LOX acts on free arachidonate, increasing AA levels and producing oxygenized PUFAs and, subsequently, more ROS (Hernandez-Perez et al., 2017; Hanna and Hafez, 2018). Moreover, the activity of 12/15 LOX is also induced by elevated FFAs in a diabetic state, enhancing the aforementioned process (Dobrian et al., 2011).
Moreover, as shown for the heart, enhanced protein acetylation caused by fatty acids can be considered an early event in FA-induced inhibition of glucose transport. It has been reported that cardiac substrates impair glucose uptake by increasing global protein acetylation from acetyl-CoA. Both palmitate and oleate promote a rapid increase in protein acetylation in vivo, which is correlated with an inhibition of insulin-stimulated glucose uptake. This glucose absorption deficit was caused by an impairment in the translocation of vesicles containing the glucose transporter GLUT4 to the plasma membrane, although insulin signaling remained unaffected (De Loof et al., 2023).
Palmitate may mediate another mechanism proposed to lead to insulin resistance. Upon palmitate exposure, endosomal alkalinization was observed in cardiomyocytes and appears to be an early lipid-induced event preceding the onset of insulin resistance. Alkalinization of endosomes occurs due to the inhibition of the proton pumping activity of vacuolar-type H+-ATPase (v-ATPase) (Liu et al., 2017). The mechanism of palmitate-induced v-ATPase inhibition involved its dissociation into two parts: the cytosolic V1 and the integral membrane V0 sub-complex, followed by endosomal de-acidification/dysfunction (Wang et al., 2022; Liu et al., 2017). Interestingly, oleate also inhibits v-ATPase function, yielding triacylglycerol accumulation but not insulin resistance (Liu et al., 2017). Endosomal alkalization could lead to translocation of the fatty acid transporter CD36 from its endosomal storage compartment to the sarcolemma since endosomes can no longer serve as a storage compartment for lipid transporter CD36, a fact that further increases lipid uptake (Wang et al., 2022; Liu et al., 2017).
Increased oxidative stress might also provide a mechanistic basis for the observed FFA-induced effect on the activity of the PI3K/Akt pathway, as discussed in the previous section (Evans et al., 2002; Liu and Cao, 2009). Moreover, several long-chain FFAs decrease levels of GLUT4 by decreasing GLUT4 gene transcription (Anderwald and Roden, 2004). There is ample evidence that fatty acids become toxic at elevated concentrations for prolonged periods. Adverse effects of chronic exposure of the β-cell to elevated FFA concentrations include decreased glucose-induced insulin secretion, impaired insulin gene expression, and increased cell death (Robertson et al., 2004). However, it is suggested that lipotoxicity only occurs in chronic hyperglycemia due to the observation that most individuals with increased circulating lipid levels have normal β-cell function (Robertson et al., 2004). These combined deleterious effects of elevated glucose and FFA levels on pancreatic β-cell function and survival can be referred to as glucolipotoxicity (Poitout et al., 2010). Because the same signaling pathways are implicated, it is suggested that FFA- and hyperglycemia-induced oxidative stress causing both insulin resistance and further ROS generation eventually results in late diabetic complications (Evans et al., 2002). Glucolipotoxicity induces not only oxidative stress but also inflammation, affecting pancreatic β-cells and peripheral tissues. Various pro-inflammatory mediators lead to impaired insulin secretion and recruitment of macrophages, enhancing local islet inflammation in β-cells. Additionally, it has been shown that glucolipotoxicity is one of the major contributors to the development of tissue-specific insulin resistance (Rehman and Akash, 2016; Galicia-Garcia et al., 2020). Therefore, a link between elevated FFAs and the inflammatory state is observed in DM.
Link between inflammation and oxidative stress
Chronic inflammation and oxidative stress have been implicated in the pathophysiology of DM. During inflammation, immune cells, such as polymorphonuclear white blood cells, generate ROS via the enzyme NOX2. Although inflammation induces oxidant injury, the reverse sequence of events is also evident; therefore, complex interactions between the oxidative stress and inflammatory pathways involve mechanisms for both their mutual amplification (positive feedback or a “vicious cycle”) (Chatterjee, 2016; Oguntibeju, 2019). It has become apparent that increased ROS production leads to increased production of inflammatory cytokines, and reciprocally, an increase in inflammatory cytokines can stimulate ROS production (Sharma et al., 2018).
ROS-induced activation of kinases, such as PKC, JNK, and p38 MAPK, leads to the activation of redox-sensitive transcription factors, such as NF-κB, AP-1, and Nrf-2 that can alter hundreds of genes that induce expression of growth factors (such as VEGF), cytokines, and chemokines. Overall cytokines and chemokines, such as IL-6 and TNF-α, can, in turn, induce further oxidative stress and mitochondrial dysfunction (Chatterjee, 2016; Pillarisetti and Saxena, 2004). Therefore, ROS act as both upstream and downstream mediators of inflammation and hyperglycemia (Chatterjee, 2016).
Proinflammatory cytokines also play an important role in β-cell pathophysiology. The involvement of TNF-α, interferon-γ, and IL-1β released by infiltrating mononuclear cells in T1DM-associated insulitis and β-cell apoptosis has been documented. Several studies have shown that IL-1β alone, or in combination with TNFα and interferon-γ, promotes β-cell dysfunction and death (Keane et al., 2015).
Obesity, which is commonly present in patients with T2DM, is associated with a proinflammatory state (Aronson 2008). The available evidence strongly suggests that T2DM is an inflammatory disease and that inflammation is a primary cause of obesity-linked insulin resistance hyperglycemia and hyperlipidemia rather than merely a consequence, despite the fact that the aforementioned conditions may also trigger inflammation. It seems likely that the inflammatory response is initiated in the adipocytes as they are the first cells affected by the development of obesity (Wellen and Hotamisligil, 2005). A mechanism that may be relevant in this initiation is oxidative stress, which inflicts oxidative damage and activates inflammatory signaling cascades (Dobrian et al., 2011). In adipose cells, ROS exposure increases the expression of pro-inflammatory adipokines such as TNF-α, IL-6, plasminogen activator inhibitor-1, and macrophage chemotactic protein-1, leading to reduced insulin signaling and activation of transcription factors such as NF-kB and MAPKs, both also leading to insulin resistance (Gleissner et al., 2007; Matsuda and Shimomura, 2014). TNF-α and IL-6 could be the most critical cytokines that induce insulin resistance because they can directly inhibit insulin signaling by stimulating inhibitory phosphorylation of serine residues of IRS-1 and also by inducing indirect anti-insulin effects, such as attenuation of the inhibitory effect of insulin on lipolysis and FFA release (Arner, 2005; Wellen and Hotamisligil, 2005; Burhans et al., 2018).
Also, TNF-α-mediated changes in leptin and adiponectin secretion may stimulate macrophage transport into adipose tissue. Once more macrophages are introduced into adipose tissue, they enhance the local production of inflammatory cytokines, which alters adipocyte function, including adipokine production (Arner, 2005). In addition, ROS exposure suppresses adiponectin expression and secretion. Thus, adiponectin-mediated antagonism of insulin resistance through enhancement of hepatic IRS-2 expression is suppressed (Hurrle and Hsu, 2017). These data show that oxidative stress plays a role in reducing adiponectin levels and increasing the levels of proinflammatory adipokines, which in turn contributes to the pathogenesis of the disease (Matsuda and Shimomura, 2014).
In conclusion, apart from exogenous sources, FFAs and inflammation could be the sources of the initial ROS generation, leading subsequently to hyperglycemia and the establishment of DM (Maiese et al., 2007; Yang et al. 2011; Tangvarasittichai, 2015).
Antioxidant therapeutic approaches
Given that oxidative stress is implicated in the pathophysiology of DM, it has been suggested that antioxidant therapy could represent a potential co-adjuvant to antidiabetic pharmacological treatment to improve glucose metabolism (Zhang et al., 2020; Martini et al., 2010).
Conventional/non-enzymatic antioxidant therapy
Nonenzymatic antioxidants possess scavenging abilities and stimulatory effects on endogenous antioxidant enzymes. In this case, the molecules involved are vitamins, GSH, coenzyme Q10, α-lipoic acid, carotenoids, flavonoids, and trace elements (Cu, Zn, Mg, and Se) (Rochette et al., 2014; Tonin et al., 2020).
Vitamins
Vitamins have antioxidant properties such as (1) directly scavenging ROS/RNS, (2) regulating genes that trigger a biological response to oxidative stress, such as the inhibition of iNOS or the increase of GSH, (3) terminating lipid peroxidation chain reaction, (4) regenerating oxidized forms of other antioxidants, (5) stabilizing NOS cofactors (BH4), and (6) participating in the upregulation of antioxidant enzymes such as CAT, SOD, and GST, although many of these mechanisms are still not completely elucidated (Tonin et al., 2020).
Observational studies indicate a link between deficiencies in vitamins C and D and the prevalence of T2DM, while other studies demonstrate decreased plasma concentrations of vitamin A in individuals with T1DM (Martini et al., 2010; Christie-David et al., 2015). A possible explanation is that antioxidant molecules are extensively consumed due to high levels of oxidative stress (Tonin et al., 2020)
Several animal studies, observational studies, and clinical trials have been performed to evaluate the effects of vitamin supplementation in DM. The most evaluated are vitamins C and E, while only a few studies have assessed vitamin A or B complex (Martini et al., 2010; Christie-David et al., 2015). Current clinical evidence suggests that vitamins C and E significantly improve plasma antioxidant capacity, while little evidence exists regarding the clinical effects of other vitamins (Tonin et al., 2020). Clinical trials of vitamin C supplementation have yielded conflicting results, so thus far, they have not demonstrated sufficient benefit to support a routine supplementation in T2DM patients (Martini et al., 2010; Christie-David et al., 2015). Moreover, when the antioxidant capabilities of vitamin E were examined clinically in non-diabetic, overweight subjects, it was shown that vitamin E supplementation lowered glucose concentrations, insulin levels, and peroxide concentrations in the plasma of participants at a short time point (Manning et al., 2004). However, these effects were not observed at a longer time point (Pazdro and Burgess, 2010). Therefore, it has not yet been confirmed whether vitamin E is beneficial for preventing or treating T2DM since results also remain controversial (Martini et al., 2010). Moreover, randomized controlled trials have not clearly demonstrated the effects of vitamin D either (Christie-David et al., 2015).
The poor performance of antioxidant supplements, such as vitamin C, may result from poor solubility, permeability, stability, and specificity. Thus, novel delivery systems for antioxidants are essential to enhance the effects of these supplements in treating DM (Zhang et al., 2020). However, this poor performance may be justified by the fact that they neutralize reactive oxygen molecules on a one-for-one basis, whereas hyperglycemia-induced overproduction of superoxide is a continuous process (Giacco and Brownlee, 2010; Rochette et al., 2014). Further studies are needed to determine if routine supplementation with these vitamins in DM is beneficial (Martini et al., 2010; Christie-David et al., 2015).
Coenzyme Q10
Coenzyme Q10, also known as ubiquinone, CoQ, or vitamin Q10, is a lipid-soluble benzoquinone endogenously synthesized in the body from phenylalanine and mevalonic acid, but it is also derived from dietary sources (Golbidi et al., 2011; Rochette et al., 2014). Coenzyme Q10 is an effective antioxidant that plays a fundamental role in mitochondrial bioenergetics (Rochette et al., 2014; Zhang et al., 2018). Coenzyme Q10 accepts electrons from electron donors and transfers them to the cytochrome system. This induces a proton gradient (chemiosmosis) essential to produce ATP (Golbidi et al., 2011). A small increase in the concentration of coenzyme Q10 in mitochondrial membranes can lead to an increase in mitochondrial respiration. Coenzyme Q10 may also improve endothelial dysfunction by ‘recoupling’ eNOS (Rochette et al., 2014).
Recent studies demonstrate that T2DM patients have significantly lower levels of coenzyme Q10, which indicates that coenzyme Q10 deficiency may reduce the organism’s ability to counter hyperglycemia-induced oxidative stress (Zhang et al., 2018). These findings suggest the possibility of improving glycemic control with coenzyme Q10 in diabetic patients through various mechanisms, including a decrease in oxidative stress, and therefore improve the oxidative stress-induced abnormalities in mitochondrial functions (Golbidi et al., 2011; Zhang et al., 2018).
Additionally, it has been shown that Coenzyme Q10 exerts beneficial effects on DM-induced oxidative stress by upregulating the expression of Sirt1 and Nrf2 genes, consequently inducing antioxidant enzymes. Hence, it has been found to protect pancreatic β cells, liver cells, and endothelial cells against oxidative stress-induced DM (Samimi et al., 2019).
In clinical trials, it was recently observed that coenzyme Q10 may have long-term benefits in T2DM patients and its complications (Kayama et al., 2015; Al-Taie et al., 2021). However, more randomized and large-sample-size trials for T2DM are needed, and it is worth mentioning that new formulations have been developed that improve the absorption of coenzyme Q10 (Rochette et al., 2014; Al-Tai et al., 2021).
A-lipoic acid
A-lipoic acid (ALA), also known as 1,2-dithiolane-3-pentanoic acid or thioctic acid, is a compound commonly found in mitochondria that is necessary for different enzymatic functions (Salehi et al., 2019). Although it is synthesized by the human body in low amounts, it is mainly obtained from the diet (Golbidi et al., 2011; Salehi et al., 2019). When ALA is administered in the diet, a substantial part is converted to its reduced dithiol form, dihydrolipoic acid (DHLA) (Golbidi et al., 2011; Rochette et al., 2015). Functionally, ALA acts as a cofactor for TCA cycle enzymes and is required for the oxidative decarboxylation of pyruvate to acetyl-CoA, a critical step bridging glycolysis and the TCA cycle (Golbidi et al., 2011). Preclinical studies suggested that improvements in mitochondrial function observed with ALA treatment were achieved by reducing ROS production and attenuating decreased expression of TCA cycle enzymes (Flemming et al., 2016).
In addition, ALA and DHLA are potent antioxidants (Golbidi et al., 2011). Their functions include: 1) quenching ROS and RNS, 2) regeneration of exogenous and endogenous antioxidants such as vitamins C and E and glutathione, 3) chelation of metal ions, 4) reparation of oxidized proteins, 5) regulation of gene transcription factors (GSH, Nrf-2, etc.), 6) inhibition of the activation of NF-kB and AP-1, and 7) decreasing asymmetric dimethylarginine levels (Folli et al., 2011; Golbidi et al., 2011; Rochette et al., 2015).
The clinical properties of ALA are also related to its molecular actions on the insulin signaling pathway (Rochette et al., 2015). ALA stimulates glucose uptake by rapid translocation and regulation of glucose transporters (GLUT1 and GLUT4) to the plasma membrane and tyrosine phosphorylation of IRS-1 (Rochette et al., 2015; Salehi et al., 2019). This effect might be mediated by p38MAPK (Rochette et al., 2015). Moreover, ALA-mediated improvement in endothelial function is partially ascribed to eNOS recoupling and increased NO bioavailability (Salehi et al., 2019).
Dietary supplementation with ALA has been successfully tested in various clinical situations, including DM (Rochette et al., 2015). Given its potential antioxidant properties, ALA may possess several beneficial effects in preventing and treating DM or reducing the risk of various diabetic complications (Golbidi et al., 2011; Rochette et al., 2014). As such, the benefits of ALA treatment have been noted in the treatment of symptomatic diabetic neuropathy, but the results in patients remain contradictory (Rochette et al., 2015). ALA is still under investigation in clinical trials, and there are some promising results, such as decreased markers of oxidative stress, increased insulin sensitivity, and improved endothelial dysfunction during acute hyperglycemia after ALA supplementation (Golbidi et al., 2011).
Mitochondria-targeted antioxidants
Preclinical studies showed that mitochondria-targeted antioxidants have therapeutic potential in physiological aging and several diseases, such as DM, by improving mitochondrial function by increasing mitochondrial biogenesis and preventing their fragmentation, reinforcing antioxidant defense systems, and inhibiting cellular apoptosis (Carvalho and Cardoso 2021). Several studies evaluated the effects of several mitochondria-targeted antioxidants, such as mitochondria-targeted coenzyme Q (MitoQ), MitoTEMPOL, SkQ1, and Szeto‐Schiller (SS) peptides. (Carvalho and Cardoso 2021; Ding et al., 2021).
MitoQ and SKQ1 are derivatives of ubiquinone and plastoquinone, respectively, which attach to the triphenylphosphonium cation (Zinovkin and Zamyatnin, 2019). Both SkQ1 and MitoQ are lipophilic cations. Therefore, they rely on the mitochondrial transmembrane potential (ΔΨm) to penetrate the mitochondria and accumulate at the inner mitochondrial membrane (Zinovkin and Zamyatnin, 2019; Ding et al., 2021). Furthermore, ubiquinone, serving as an electron carrier and an antioxidant, has been shown to efficiently prevent lipid peroxidation (Ding et al., 2021). Contrary to other mitochondria-targeted antioxidants, both compounds are “rechargeable” by virtue of being reduced by the ETC after their oxidation by ROS (Zinovkin and Zamyatnin, 2019). MitoQ is the most studied and widely used mitochondrial antioxidant (Rochette et al., 2014). Data from in vitro and in vivo studies demonstrated that MitoQ is effective in fighting oxidative stress in a safe manner, making it a perfect candidate for investigation in clinical trials (Carvalho and Cardoso 2021). MitoQ and MitoTEMPOL have been shown to protect β-cells and, therefore, improve insulin secretion and the survival of said cells. Additionally, protection against complications associated with DM, such as diabetic nephropathy and retinopathy, has been reported (Rocha et al., 2014). However, compared to MitoQ, SkQ1 exerts antioxidant effects at relatively lower concentrations and also has a larger range of ‘antioxidant’ activity, providing a potential advantage for clinical SkQ1 applications (Zinovkin and Zamyatnin, 2019; Ding et al., 2021).
Mitochondria-targeted antioxidant peptides such as compounds XJB-5–131 or SS31 could be a potential new treatment for diabetic diseases (Rochette et al., 2014). XJB peptides consist of an electron and ROS scavenger (4‐NH2‐TEMPO), and studies have revealed promising results due to their capacity to improve mitochondrial function (Carvalho and Cardoso 2021). SS peptides are a novel family of small cell-permeable mitochondria-targeted peptides that include the water-soluble tetrapeptide SS-31 (H-D-Arg-Dmt-Lys-Phe-NH2), also known as Elamipretide, MTP-131, and Bendavia. SS-31 concentrates in the inner mitochondrial membrane without relying on mitochondrial membrane potential that is altered in most diseases (Ding et al., 2021). SS-31 peptide may restore mitochondrial function and alleviate symptoms caused by DM, and more importantly, may lead to normalization of the mitochondrial potential, preventing insulin resistance development (Fisher-Wellman and Neufer, 2012; Ding et al., 2021).
These agents have thus far achieved positive results in preclinical models of DM, but translation into the clinic warrants further study to better define their efficacy and safety in the context of DM complications (Rochette et al., 2014; Kayama et al., 2015; Flemming et al., 2016; Ding et al., 2021).
Enzymatic antioxidants
Conventional antioxidants act on a “one-to-one” basis, while the hyperglycemia-induced production of superoxide radicals is continuous. Therefore, enzymatic antioxidants may provide an effective antioxidant therapeutic strategy since they may counteract this continuous ROS overproduction (Rochette et al., 2014). The most efficient enzymatic antioxidants involve SOD and GPx mimetics.
SOD mimetics
Many SOD mimetics have been synthesized that can be used as pharmaceutical agents in many diseases in which the native SOD is ineffective (Younus, 2018). These mimetics include the metalloporphyrins, manganese (Mn)-based SOD mimetics cyclic polyamines, nitroxides, Mn–salen complexes, and fullerenes (Bonetta, 2018; Forman and Zhang, 2021). Although SOD mimics have been developed to remove O2•− specifically, most can also reduce other reactive oxygen or nitrogen species such as ONOO− and H2O2. In addition, other protective effects of SOD mimics involve redox-sensitive signaling that might be attributable to activities other than mimicking SOD. The best-studied class of SOD mimics is the Mn porphyrins. Some of these mimetics, such as MnTM-2-pYp5+ and MnTE-2-pYp5+, showed very high SOD activity (Forman and Zhang, 2021). In addition, metalloporphyrin-based SOD mimics have demonstrated protective effects against the progression of T-cell-mediated autoimmune DM (Rochette et al., 2014). MnTM-2-PyP reduced DM-induced oxidative stress and extended the lifespan of streptozotocin diabetic rats (Bonetta, 2018). Another Mn-porphyrin, MnBuOE (or BMX-001), has been shown to reduce downstream apoptotic events associated with oxidative stress, thus preserving viability and function in T1DM (Bruni et al., 2018). A Mn-cyclic polyamine named M40403 has been shown to prevent vascular and neural complications in experimental DM (Di Naso et al., 2011; Bonetta, 2018). M40403 is selective in preventing superoxide formation and does not directly influence the generation of other ROS. (Coppey et al., 2001).
Another class of weaker SOD mimetics is the nitroxides tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), which restores the redox imbalance and prevents renal abnormalities and kidney damage seen in DM. The beneficial effects of tempol in DM may be due to its SOD mimetic activity, its superoxide scavenging activity, inhibition of superoxide-dependent generation of other ROS, inhibition of gene expression of NOX isoforms, limitation of ER stress and inflammation, or indirect modulation of NO levels (Nassar et al., 2002; Peixoto et al., 2009; De Blasio et al., 2017; Bonetta, 2018).
Although SOD and SOD mimetics have displayed efficacy in experimental models of DM, their role in improving the vascular complications of DM remains controversial in patients (Rochette et al., 2014). Due to the instability, high immunogenicity, low cellular uptake, and short in vivo half-life, the clinical applications of SOD mimetics as therapeutic agents are very limited. To overcome these difficulties, SOD can be incorporated either in highly loaded conventional liposomes or long-circulating liposomes (PEG-liposomes) (Younus, 2018).
GPx mimetics
GPx mimetics are a promising antioxidant and anti-inflammatory approach and alternative adjunct therapy to reduce diabetic complications. A variety of mimics have been developed, among them, the selenoorganic compound ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one) has been extensively studied for its therapeutic potential in various experimental models of DM (Rochette et al., 2014; Forman and Zhang, 2021). Ebselen rapidly reacts with peroxynitrite and free radicals, thus protecting against the deleterious effects of ROS and RNS. Furthermore, in T1DM, ebselen antagonizes iNOS activity through NO scavenging, decreasing NO accumulation, which is harmful to insulin-producing cells. Ebselen has exhibited beneficial effects on β-cell mass and function and protects from vascular complications in patients with DM (Azad and Tomar, 2014). Ebselen may induce phase II detoxification enzymes (Forman and Zhang, 2021). However, some trials have questioned its efficacy as an antioxidative agent; thus, further research is needed (Beckman et al., 2016).
ALT-2074 (BXT-51072) is a newer analog of ebselen, displaying increased GPx activity and potency. In vitro, ALT-2074 inhibited the inflammatory response in endothelial cells and reduced oxidative damage. A phase II clinical trial of ALT-2074 in DM has been completed, but data are not yet available (Forman and Zhang, 2021).
Mechanism-based therapies
Currently, mechanistic insights are leading to new therapeutic concepts for DM. ROS-related drug development will have to focus on the delicate task of identifying the main disease-relevant sources of deleterious ROS while at the same time leaving essential physiological sources of ROS and their signaling pathways intact. The use of Nrf-2 agonists and selective inhibition of specific NOX isoforms are characteristic examples of this approach (Elbatreek et al., 2019).
Nrf-2 activators
Nrf-2 is a transcription factor that orchestrates the expression of more than 100 genes related to oxidative stress and cell survival, including those that control antioxidant enzymes and genes that control immune and inflammatory responses (Rochette et al., 2014; Li et al., 2017; Baumel-Alterzon et al., 2021). Nrf-2 initiates a program of gene expression in response to oxidative stress, which confers protection on β-cells against experimentally induced supraphysiological concentrations of ROS (Elbatreek et al., 2019; Baumel-Alterzon et al., 2021). Therefore, Nrf-2 is referred to as the “master regulator” of the antioxidant response (Rochette et al., 2014).
Mechanistically, Nrf-2 is held in an inactive complex in the cytosol through interaction with its negative regulator, Keap1. In response to oxidative stress, Keap1 undergoes oxidative modification, and Nrf-2 is released, enters the nucleus, and binds to the antioxidant response element (ARE), thereby initiating the transcription of key antioxidant genes (Sharma et al., 2017; Pickering et al., 2018). In addition, Nrf-2 has been shown to play a role in decreasing inflammation through interaction with NF-κB and direct inhibition of IL-1β and IL-18 cytokine transcription, as well as positively regulating NO bioavailability (Pereira et al., 2017; Pickering et al., 2018).
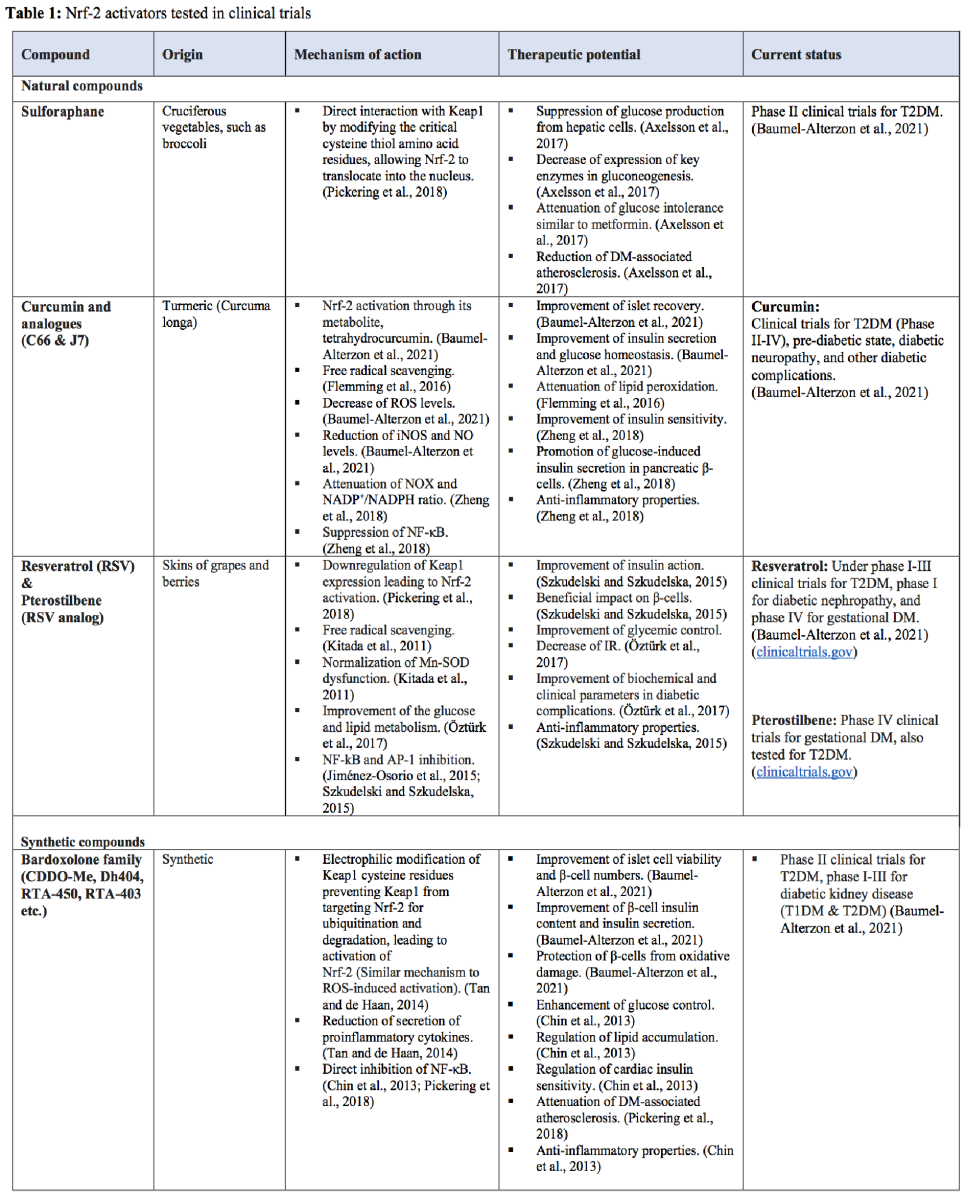
Dysregulation of Nrf-2 signaling is implicated in DM because chronic hyperglycemia and prolonged ROS production inhibit Nrf-2 activation. Moreover, Nrf-2 activators enhance the expression of antioxidant enzymes at their physiological sites, contrary to exogenous scavenging antioxidants that act broadly. Therefore, Nrf-2 activators are regarded as potential therapeutic agents to induce antioxidant capacity and alleviate pathology (Elbatreek et al., 2019; Forman and Zhang, 2021). Furthermore, given that both oxidative stress and inflammation have been interlinked as the underlying etiology in the pathogenesis of DM-associated endothelial dysfunction, strategies to ameliorate both may be beneficial in improving DM-associated endothelial dysfunction (Sharma et al., 2017). Nrf-2 is therefore recognized as an important therapeutic target, which is made more attractive given its responsiveness to activation by pseudo-stressors. These pseudo-stressors are primarily small molecules derived from natural plants, but several synthetic derivatives have been developed with improved bioavailability and Nrf-2-activating ability (Pickering et al., 2018).
The most studied natural compounds are sulforaphane (Axelsson et al., 2017; Pickering et al., 2018; Forman and Zhang, 2021), curcumin and its analogs (Flemming et al., 2016; Zheng et al., 2018; Pivari et al., 2019; Baumel-Alterzon et al., 2021), and resveratrol (Kitada et al., 2011; Jiménez-Osorio et al., 2015; Szkudelski and Szkudelska, 2015; Öztürk et al., 2017; Pickering et al., 2018; Baumel-Alterzon et al., 2021) (see Table 1). Additionally, synthetic Nrf-2 activators have been developed, and they are currently being tested in several clinical trials of DM and its complications. Compounds of the bardoxolone family are synthetic compounds of greater significance (Table 1) (Chin et al., 2013; Tan and de Haan, 2014; Pickering et al., 2018; Baumel-Alterzon et al., 2021). Results from clinical trials have demonstrated that sulforaphane has a favorable impact on the lipid profile and inflammatory markers, significantly improves insulin sensitivity, and lowers fasting glucose levels in T2DM patients. Moreover, orally administered resveratrol significantly improved insulin sensitivity in diabetic patients within one month. Clinical trials with bardoxolone-methyl (CDDO-Me) showed an improved estimated glomerular filtration rate in 90% of the patients, with a parallel reduction in serum creatinine and blood urea nitrogen and increased creatinine clearance. Markers of vascular injury and inflammation were also improved after treatment with CDDO-Me without any life-threatening or other drug-related adverse events. Patients receiving CDDO-Me also had significantly increased glomerular filtration rates compared to placebo. The subsequent phase 3 BEACON study with a larger cohort of patients with stage 4 chronic kidney disease and T2DM had to be prematurely terminated as adverse cardiovascular events and mortality were noticed in the CDDO-Me arm (Matzinger et al., 2018). The Nrf-2 activators tested in clinical trials are presented in Table 1.
NOX-inhibitors
Several studies have suggested a pivotal role of different NOX isoforms in the pathogenesis of diabetic complications and have clearly demonstrated that certain available natural or synthetic NOX inhibitors show beneficial effects against different diabetic complications (Urner et al., 2020). There are two types of agents that inhibit NOXs, those that inhibit enzymatic activity and those that prevent the assembly of the NOX2 enzyme, which is a multiprotein complex (Forman and Zhang, 2021). Many of the molecules described to date are not direct NOX inhibitors but inhibit NOX pathway activity (Teixeira et al., 2017).
Many phytoconstituents, such as apocynin, puerarin, and berberine, are potent inhibitors of cytosolic components of NOXs, such as p40phox, p47phox, and p67phox, and are reported to inhibit the progression of diabetic complications (Laddha and Kulkarni, 2020). Apocynin inhibits downstream events of all isoforms, ameliorating oxidative stress (Teixeira et al., 2017; Laddha and Kulkarni, 2020). Importantly, these compounds have off-target effects and are not considered selective NOX inhibitors because their specificity of action has been questioned in previous reports. However, two nontoxic, pyrazolopyridine dione-based, structurally related compounds, GKT136901 and GKT137831, demonstrating a preferential inhibition of NOX1 and NOX4, have been identified and proposed as a novel therapy for NOX inhibition (Elumalai et al., 2021). These inhibitors slow or prevent disease progression in a range of chronic inflammatory diseases, such as DM, by modulating common signal transduction pathways, and they have shown beneficial effects against diabetic complications in several preclinical studies (Teixeira et al., 2017; Elbatreek et al., 2019). GKT137831 is the first-in-class specific NOX inhibitor reported as a potent inhibitor to treat T2DM that was studied in a phase II clinical trial to alleviate diabetic nephropathy in both T1DM and T2DM patients (Elbatreek et al., 2019; Charlton et al., 2020; Laddha and Kulkarni, 2020; Zhang et al., 2020). Although results remain controversial, there were positive effects in reducing systemic inflammatory markers (Elbatreek et al., 2019).
Other novel NOX inhibitors showed promising results in preclinical studies, and they are currently being tested in clinical trials against diabetic complications. The novel pan‐NOX inhibitor APX‐115 is being tested for efficacy against diabetic nephropathy, and APX 1004 is being tested against diabetic retinopathy (Elbatreek et al., 2019; Urner et al., 2020). In addition, APX-115 protects against nephropathy in a mouse T2DM model and was superior to GKT137831 in preserving renal function. VAS2870, another pan-NOX inhibitor, reduces aortic contractility in diabetic rats by improving endothelial function, providing neuroprotection and improving neurological scores in mice, and inhibiting reperfusion-induced hemorrhagic transformation in hyperglycemic rats. Clearly, isoform-specific NOX inhibitors and further clinical trials focusing on diabetic complications are needed (Elbatreek et al., 2019).
There are other promising strategies, including specifically designing small peptides, such as NOXA1ds, that inhibit the assembly of NOX complexes by blocking NOX1 or interfering with the synthesis of the components of the NOX complexes. However, agents related to these approaches have not yet reached clinical trials (Charlton et al., 2020; Forman and Zhang, 2021).
PKC inhibitors
The involvement of PKC in activating several signaling molecules, such as the pro-oxidant enzyme NOX and NF-κB, makes PKC one of the most important therapeutic targets for controlling oxidative stress and inflammation involved in diabetic complications (Liu et al.; 2012; Volpe et al., 2018). Several potential targets for the pharmacological development of PKC inhibitors include the ATP-binding site in the catalytic region and the DAG-binding site (Lien et al., 2021). Some PKC inhibitors have been tested for diabetic complications. Among them, Ruboxistaurin (RBX), PKC-412 (staurosporine), and PKC-412A (N-benzoyl-staurosporine) were all tested in clinical trials to assess their efficacy to inhibit PKC-β (Volpe et al., 2018).
RBX mesylate (also known as LY333531) is a macrocyclic bisindolylmaleimide compound that specifically and competitively inhibits PKC-β (Deissler and Lang, 2015). Thus, RBX prevents hyperglycemia-induced oxidative stress by inhibiting NOX activation in animal studies (Liu et al.; 2012). Some clinical trials examining RBX in the treatment of diabetic complications revealed that inhibiting PKCβ significantly reduces diabetic retinopathy, nephropathy, neuropathy, and other angiopathies (Lien et al., 2021). The encouraging results of RBX against diabetic retinopathy have yet to receive FDA approval (Golbidi et al., 2011). It has also been demonstrated that RBX is nephroprotective in experimental diabetic models (Volpe et al., 2018). Although the efficacy of RBX against peripheral neuropathy has not been successfully demonstrated in Phase III trials, a pilot study reported a beneficial effect of RBX in treating diabetic nephropathy, but this has not been confirmed in a controlled large-scale trial (Golbidi et al., 2011). Moreover, chronic inhibition of PKC significantly improved the recovery of coronary endothelial function in patients with DM following cardioplegic ischemia and reperfusion (Zhang et al., 2020). Therefore, RBX may represent a promising therapy to prevent diabetic cardiovascular complications (Liu et al.; 2012). To date, clinically significant adverse effects associated with this drug have not been observed, and currently, more than a hundred new PKC inhibitor drugs are under evaluation (Deissler and Lang, 2015; Volpe et al., 2018).
Anti-AGEs therapeutic agents
Currently, there are six therapeutic options to reduce AGEs and antagonize RAGE, including AGE cross-link breakers, AGE inhibitors, and RAGE antagonists. However, only a few have been clinically evaluated (Jud and Sourij, 2019).
AGE-crosslink breakers can break up cross-links between AGEs and extracellular molecules. Members of this class include thiazolium derivatives, like alagebrium (ALT-711), and pyridinium derivatives, like TRC4186 (Jud and Sourij, 2019). AGE cross-link breakers can cleave the protein cross-linking structure in AGEs, breaking diketone compounds up and, therefore, reducing the accumulation of AGEs in tissues (Jud and Sourij, 2019; Yang et al., 2019). So far, clinical data are only available for alagebrium and TRC4186. These data showed a significant improvement in arterial compliance and beneficial effects on diastolic heart failure by reducing the left ventricular mass and improving diastolic filling. In a phase I trial investigating the pharmacokinetic, safety, and tolerability of TRC4186, good tolerance and safety were observed (Jud and Sourij, 2019). Specific information about alagebrium and TRC4186 (Coughlan et al., 2007; Joshi et al., 2009; Byun et al., 2017; Jud and Sourij, 2019; Toprak and Yigitaslan, 2019) is presented in Table 2.
On the other hand, AGE inhibitors prevent the formation of AGE cross-links or even the development of AGEs. The individual substances feature different mechanisms to act as an AGE inhibitor, but all have shown initial beneficial AGE-lowering effects. (Jud and Sourij, 2019; Sourris et al., 2021).
Inhibitors of AGE formation include scavengers of free reactive carbonyl groups, such as aminoguanidine (Byun et al., 2017; Spasov et al., 2017) and OPB-9195 (Spasov et al., 2017), Vitamin B derivatives, such as pyridoxamine (Voziyan and Hudson, 2005; Yang et al., 2019; Ramis et al., 2019; Sourris et al., 2021; Telen, 2020; Jiang et al., 2021), thiamine, benfotiamine, and carnosine (Jud and Sourij, 2019; Prokopieva et al., 2016; Sourris et al., 2021; Matthews et al., 2021; www.clinicaltrials.gov) are presented also in Table 2. Clinical trials for aminoguanidine and OPB-9195 were halted because numerous side effects were observed (Byun et al., 2017; Spasov et al., 2017). On the other hand, pyridoxamine has shown initial beneficial AGE-lowering effects in clinical trials. In a phase II study in patients with diabetic renal disease, pyridoxamine was renoprotective. (Sourris et al., 2021). Moreover, a pilot intervention study with carnosine showed an increase in fasting insulin, and insulin resistance was hampered (de Courten et al., 2016).
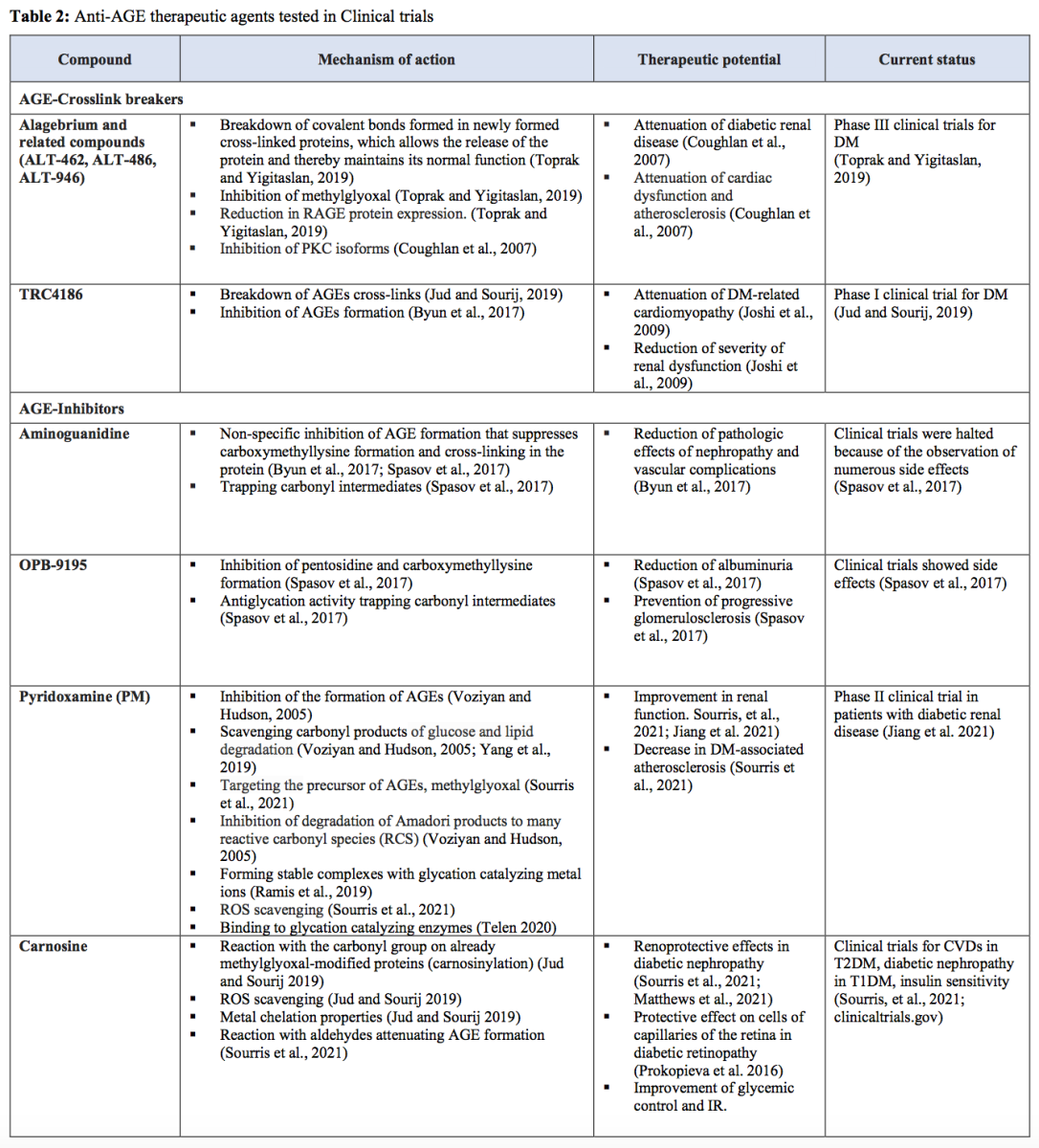
Transketolase activators
Transketolase activators, including thiamine and benfotiamine, are considered potential therapeutic agents. When increased superoxide inhibits GAPDH activity, glycolytic intermediates preceding the enzyme accumulate and are then shunted into the five pathways of hyperglycemic damage. Among the glycolytic intermediates, fructose 6-phosphate and glyceraldehyde 3-phosphate are also final products of the transketolase reaction, which is the rate-limiting enzyme of the non-oxidative branch of the pentose phosphate pathway (Hammes et al., 2003; Wu et al., 2014). Although the function of this enzyme is commonly thought to be the conversion of pentose phosphates to glycolytic intermediates, it can also convert glycolytic intermediates to pentose phosphates (Giacco and Brownlee, 2010). This conversion leads to a reduction in the concentration of these two glycolytic intermediates, inhibiting their flux into several damage-inducing pathways activated by hyperglycemia, such as AGEs, PKC, and hexosamine pathways (Hammes et al., 2003; Wu et al., 2014). Thus, thiamine and benfotiamine normalize all these pathways in the presence of excess glucose, leading to reduced synthesis of ROS. Apart from inhibiting hyperglycemia-induced pathways, thiamine also increases the oxidation of pyruvate to acetyl-CoA, thus counteracting the accumulation of pyruvate and lactate in the cytoplasm. (Beltramo et al., 2021) Moreover, it has been found that benfotiamine can inhibit the hyperglycemia-induced activation of NF-κB and the von Willebrand factor (Hammes et al., 2003). It has also been shown that benfotiamine prevents cardiac dysfunction in DM by modulating the PI3K/Akt pathway (Chakrabarti et al., 2011).
Transketolase requires thiamine (vitamin B1) as a cofactor. Although thiamine only activated transketolase by ~ 25%, the thiamine derivative benfotiamine activated transketolase by 250% in arterial endothelial cells. Based on those findings, several groups have demonstrated the ability of benfotiamine to counteract glucose-mediated toxicity in several experimental models of DM. DM has also been found to cause intracellular thiamine depletion, which provides further evidence that thiamine and benfotiamine are potential therapeutic agents (Giacco and Brownlee, 2010).
A small trial in patients with T1DM showed normalization of increased AGE formation, a 40% reduction in monocyte hexosamine-modified proteins, and normalized prostacyclin synthase activity by using a combination of alpha lipoic acid and benfotiamine (Folli et al., 2011; Ziegler et al., 2021). Moreover, a few pilot studies in humans reported beneficial effects of thiamine administration on diabetic nephropathy. T2DM patients with early-stage nephropathy experienced reduced urinary albumin excretion after three months of thiamine supplementation, while benfotiamine had fewer promising effects (Beltramo et al., 2021). Additionally, benfotiamine prevented experimental retinopathy in diabetic rats. Subsequently, related clinical trials were performed (Beltramo et al., 2021; www.clinicaltrials.gov). However, benfotiamine did not show promising efficacy in phase II and IV trials in treating diabetic kidney disease or peripheral nerve function. Therefore, other transketolase activators await further investigation (Ge et al., 2020).
Xanthine oxidase inhibitors
Selective XO inhibitors such as allopurinol and febuxostat, which are used to lower uric acid levels and reduce the risk of a gout flare in gout patients, may represent new drug candidates for the treatment of DM-related organ injury because of their ROS-lowering effects (Iacobini et al., 2021). As revealed by clinical trials, XO inhibitors also have anti-inflammatory properties (Charlton et al., 2020).
Allopurinol prevented hyperglycemia-induced oxidative stress and attenuated cardiac dysfunction, cardiomyocyte hypertrophy, and cardiac fibrosis in diabetic rats. In all these cases, prevention of ROS-induced oxidative stress may represent one of the major protective mechanisms (Yang et al. 2008). The attenuation of oxidative stress by allopurinol may be mediated by activation of the Nrf-2/heme oxygenase-1 pathway, apart from XO inhibition. Given that allopurinol also prevented an increase in lipid hydroperoxides in the plasma of diabetic patients and that it mediates the abovementioned cardiovascular benefits, allopurinol treatment may be a promising therapy to attenuate cardiovascular disease, particularly in DM, and deserves to be evaluated in clinical studies (Byrne et al., 2021).
Febuxostat reduces arterial ROS levels and endothelial dysfunction, thereby decreasing pro-inflammatory markers and attenuating histological features of atherosclerosis in ApoE−/− mice (Charlton et al., 2020). Febuxostat also attenuated diabetic nephropathy by modulating oxidative stress and endothelial function through the VEGF–NADPH oxidase signaling pathway (Hong et al., 2021).
Topiroxostat, a non-purine selective XO inhibitor, reduces albuminuria by attenuating endothelial damage induced by glomerular oxidative stress derived by XO activation (Itano et al., 2020). XO inhibitors, particularly high doses of topiroxostat, also protect against diabetic neuropathy development by inhibiting ROS generation (Takahashi et al., 2021).
Antioxidant action of drugs clinically used in the management of T2DM
Numerous studies have demonstrated that several widely used antidiabetic agents, such as sulfonylureas (Al-Omary, 2017), thiazolidinediones (Reynolds and Goldberg, 2006; Choi and Ho, 2018), metformin (Esteghamati et al., 2013; Al-Omary, 2017), dipeptidyl peptidase-4 (DPP-4) inhibitors (Kim et al., 2014; Pujadas et al., 2017; Kong et al. 2021), and especially the newest antidiabetic drugs, sodium-glucose cotransporter 2 (SGLT-2) inhibitors possess antioxidant properties.
SGLT-2 inhibitors (SGLT-2is), also called gliflozins, were developed to inhibit sodium-glucose transport protein 2 in the kidneys, reducing renal tubular glucose reabsorption (Tsai et al., 2021). Their beneficial effects are likely due to glucose lowering effects and improved glucose utilization by restoring insulin sensitivity and signaling, all of which prevent downstream glucotoxicity (Steven et al., 2017). Other research supports that SGLT-2is do not directly prevent hyperglycemia-mediated enhanced glucotoxicity in these cells by inhibiting glucose uptake (Semo et al., 2023). Accumulating evidence from experimental and clinical studies supports the antioxidant roles of SGLT-2is in T2DM, with mechanisms that involve both inhibition of free radical generation and regulation of antioxidant systems (Tsai et al., 2021).
SGLT-2is decreased oxidative stress and lipid peroxidation biomarkers in preclinical studies (Andreadou et al., 2017; Bray et al., 2020). Chronic administration of empagliflozin (EMPA) significantly attenuates biomarkers of cardiac oxidative stress in the myocardium after ischemia/reperfusion stimuli, even in the absence of diabetes, in an in vivo model (Nikolaou et al., 2021). Additionally, clinical studies have shown decreased oxidative stress markers after 24 weeks of dapagliflozin treatment in patients with newly diagnosed T2DM (Balogh et al., 2023). Patients treated with SGLT-2is for 12 months achieved remarkable decreases in oxidants and a remarkable increase in antioxidants compared with those who received insulin, albeit all patients achieved adequate glycemic control. This great increase in antioxidant activity, indicating improved radical scavenging capacity, counteracts the potential oxidative effects of hyperglycemia-induced ketogenesis. Therefore, the net effect of SGLT-2is was toward a beneficial equilibrium between oxidant and antioxidant mechanisms (Lambadiari et al., 2021). Some of the putative mechanisms include reduction of AGEs, inactivation of NADPH oxidases, improvement of NOS activity, TNF-α suppression, improvement in antioxidant defenses, improved mitochondrial function, and activation of STAT3, owing to glycemic control (Oelze et al., 2014; Andreadou et al., 2017; Li et al., 2019; Lambadiari et al., 2021; Uthman et al., 2022).
SGLT-2is are highly efficient in preventing glucotoxicity by reducing the formation of AGE and induction of RAGE, as shown in experimental studies (Oelze et al., 2014). However, EMPA may exert some protective effects, e.g., on myocardial function, independent of AGE (Andreadou et al., 2017). Other studies also indicated that SGLT-2is affects NADPH oxidase activity by suppressing NOX1, NOX2, and NOX4 in vivo (Oelze et al., 2014; Terami et al., 2014). In addition, in a pilot clinical study, blood levels of a marker of NOX2 activation and hydrogen peroxide production decreased in patients with T2DM after treatment with SGLT-2is (Balogh et al., 2023).
As shown in cardiac muscle cells, EMPA also decreases oxidative stress by improving NOS activity and increasing NO production. These effects are partly due to the activation of the Nrf-2/ARE signaling pathways since EMPA was proposed to promote the translocation of Nrf-2 to the nucleus and suppression of the TGF-β/Smad pathway (Li et al., 2019; Balogh et al., 2023). Additionally, the antioxidant effects of SGLT-2is mediated by proinflammatory cytokines have been described in animal and in vitro studies (Tsai et al., 2021). EMPA reduces TNF-α-induced ROS by inhibiting the Na+/H+ exchanger and lowering intracellular sodium concentrations (Uthman et al., 2022).
Other studies also indicated that SGLT-2is improve antioxidant defenses (Lambadiari et al., 2021; Nabrdalik-Leśniak et al., 2021). EMPA increases the endogenous cardiac antioxidant capacity and renders the myocardium less susceptible to ROS-induced cell death (Nikolaou et al., 2021). In addition, chronic treatment with EMPA upregulates SOD2 mRNA levels in murine non-diabetic hearts (Nikolaou et al., 2021).
While diabetes is a redox disease, the redox regulation by SGLT-2is, in parallel with their glucose-lowering effects, can be pivotal in managing diabetes and in ameliorating its cardiovascular complications (Andreadou et al., 2017; Ikonomidis et al., 2020).
Conclusions -future perspective
DM remains a pathological condition of great importance and a significant cause of premature mortality. DM prevalence is usually underestimated and is likely to worsen as its prevalence keeps increasing at a high rate, obtaining pandemic proportions. Different factors contribute to the pathogenesis and progression of DM. A deep understanding of these factors and the sequence of the events leading to the establishment of this disease may provide important insight for improving antidiabetic therapeutic strategies. Although current therapeutic strategies can help diabetic patients and alleviate the symptoms, new efficient treatments are required since DM remains an unsolved health problem that affects millions of people worldwide.
Oxidative stress is a hallmark event of DM, and it is found to be one of the major mechanisms for the onset and progression of this disease. Herein, we reviewed the reciprocal correlation between diabetes and oxidative stress since not only does excessive ROS production lead to the onset of diabetes, but also the diabetic state leads to exaggerated generation of free radicals that play a significant role in diabetic complications. On the one hand, oxidative stress seems to play a significant role in initiating this pathology since solid evidence indicates that it leads to insulin resistance, which pre-exists hyperglycemia, and simultaneously affects insulin signaling and contributes to β-cell dysfunction. On the other hand, hyperglycemia, the main characteristic of the diabetic state, results in excessive free radical generation through various pathways analytically presented in our review. Thus, a vicious circle between oxidative stress and hyperglycemia is observed. Given that hyperglycemia is subsequent to both insulin resistance and excessive ROS production, our review suggests that other pre-existing factors may provide a reasonable explanation for the mechanism through which oxidative stress leads to insulin resistance. Interestingly, these pre-existing factors, namely increased circulating FFAs and inflammation, are not only possible initial events but also develop a reciprocal interplay with oxidative stress. The exact correlation between diabetes and oxidative stress is represented in Figure 3.
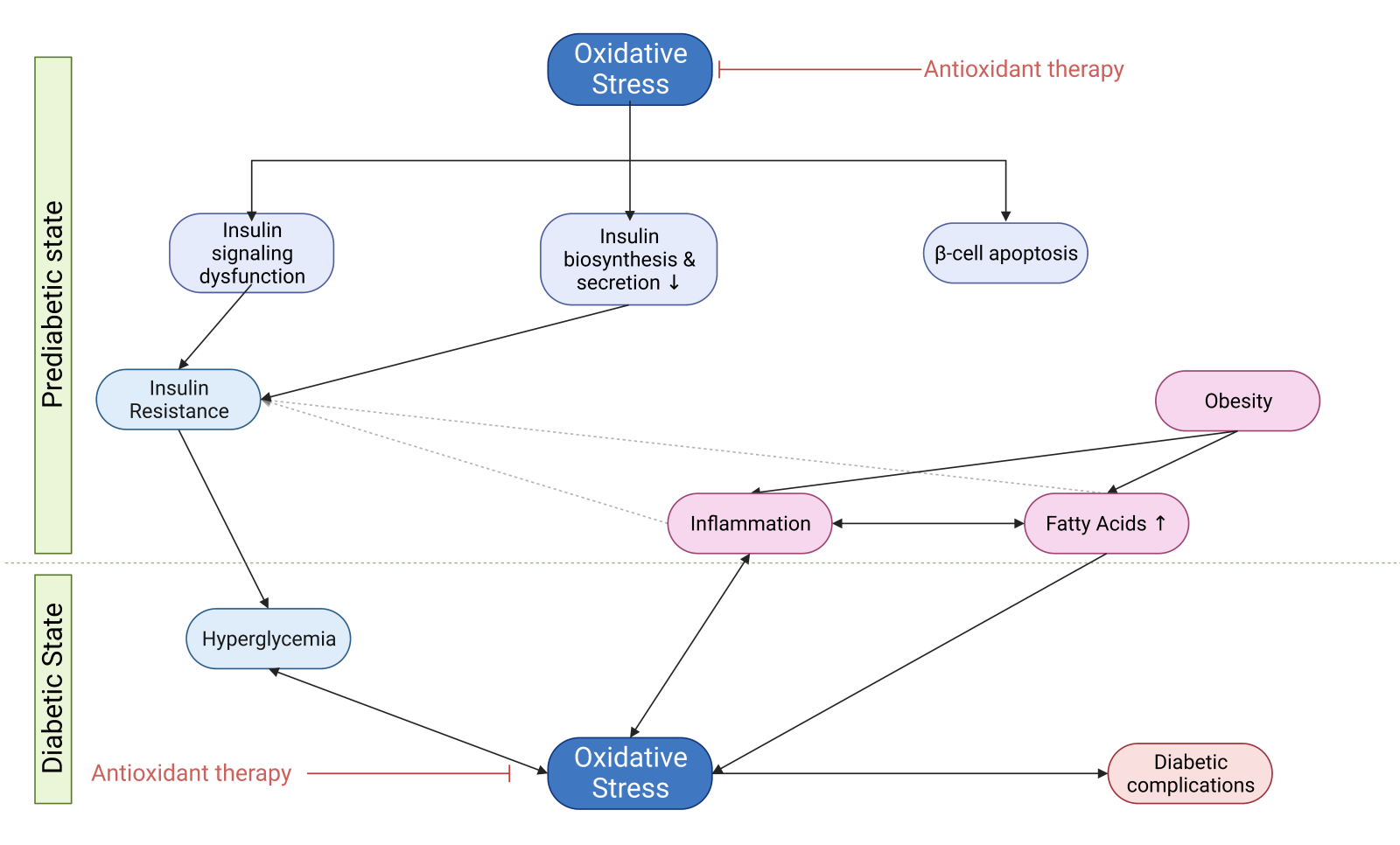
In a new window | Download PPT
Figure 3. Schematic representation of the reciprocal correlation between oxidative stress and DM. Oxidative stress is a major mechanism that leads to insulin resistance and β-cell apoptosis present in the prediabetic state. Obesity elevates levels of FFAs and promotes the inflammatory state that further exacerbates oxidative stress and insulin resistance. Consequently, hyperglycemia that characterizes the establishment of a diabetic state leads to more oxidative damage, which in turn triggers diabetic complications and feeds this reciprocal correlation, promoting a vicious circle. Therefore, antioxidant therapy may provide a reasonable antidiabetic approach since it ameliorates oxidative stress before and after the onset of DM. Abbreviations: FFAs, fee fatty acids (Created with BioRender.com.)
Based on these observations, it is reasonable to regard antioxidant therapy as a mechanism-based approach for diabetes complications, together with conventional anti-hyperglycemic therapeutic agents that control hyperglycemia. Our review evaluated various antioxidant agents currently under investigation that may play a central role in future DM treatment. Although a range of conventional antioxidants have shown moderate positive results against DM, novel mechanism-based approaches, such as Nrf-2 inhibitors and anti-AGE therapeutic agents, could provide a remarkably promising strategy against DM. Some of these mechanisms mediate the anti-hyperglycemic action of new therapeutic antidiabetic agents. Nevertheless, more research and clinical evidence are required to support this perspective, which remains controversial, and translate this perception to an actual clinical application with a favorable outcome for diabetic patients.
Conflicts of interest
The authors declare that they have no conflicts of interest. Ioanna Andreadou, who serves on the Editorial Board for Conditioning Medicine, did not participate at any level in the editorial review of this manuscript.
References
Nikolaos Bertos 1
1School of Pharmacy, National and Kapodistrian University of Athens.
Georgios Seiradakis1
1School of Pharmacy, National and Kapodistrian University of Athens.
Ioanna Andreadou1
1School of Pharmacy, National and Kapodistrian University of Athens.
Corresponding author:
Ioanna Andreadou
Email: jandread@pharm.uoa.gr
In a new window | Download PPT
Figure 1. The mechanisms through which oxidative stress leads to insulin expression and secretory deficiencies. (1) Increase of level and activity of c-Jun protein, leading to suppressed expression of MafA and subsequent regulation of insulin expression and secretion. (2) Activation of JNK kinase pathway, leading to translocation of FOXO-1 and subsequent opposite translocation of PDX-1. (3) Inactivation of PPARγ, resulting in PDX-1 and MafA degradation. Abbreviations: MafA, MAF bZIP transcription factor A; JNK, c-Jun N-terminal kinase; FOXO-1, Forkhead/winged helix box protein O1; PDX-1, pancreatic and duodenal homeobox -1; PPARγ, peroxisome proliferator activated receptor γ (Created with BioRender.com.)
In a new window | Download PPT
Figure 2. Schematic representation of the mechanisms through which hyperglycemia leads to oxidative stress and diabetic complications. Hyperglycemia induces oxidative stress through increased TCA cycle flux. Oxidative stress-mediated inactivation of GAPDH: a) leads to accumulation of glycolytic intermediates, which in turn trigger the five major pathogenetic pathways responsible for diabetic complications and further promotion of oxidative stress, and b) counteracts the accumulation of pyruvate to prevent excessive TCA cycle flux. Abbreviations: TCA, tricarboxylic Acid; PARP, poly-ADP ribose polymerase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; DAG, diacylglycerol; AGEs, advanced glycation end products; PKC, protein kinase C (Created with BioRender.com.)
In a new window | Download PPT
Figure 3. Schematic representation of the reciprocal correlation between oxidative stress and DM. Oxidative stress is a major mechanism that leads to insulin resistance and β-cell apoptosis present in the prediabetic state. Obesity elevates levels of FFAs and promotes the inflammatory state that further exacerbates oxidative stress and insulin resistance. Consequently, hyperglycemia that characterizes the establishment of a diabetic state leads to more oxidative damage, which in turn triggers diabetic complications and feeds this reciprocal correlation, promoting a vicious circle. Therefore, antioxidant therapy may provide a reasonable antidiabetic approach since it ameliorates oxidative stress before and after the onset of DM. Abbreviations: FFAs, fee fatty acids (Created with BioRender.com.)
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 7318 | 18 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA