Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Differences and similarities in neuroprotective molecular pathways activated by distinct preconditioning inducers
Time:2018-06-30
Number:13016
Author Affiliations

Conditioning Medicine, 2018. 1(4):187-203.
Abstract
The identification of neuroprotective therapies is gaining great interest in the design of strategies to prevent neuronal death. However, up until now all tested approaches have been unsuccessful despite many years of clinical trials, mainly because of the numerous side effects observed in humans and absent in animals used at the preclinical level. The neuroprotective strategy known as brain preconditioning is becoming important with the increased understanding of the endogenous mechanisms able to induce tolerance. Over the years, several stimuli have been described as possible preconditioning inducers, and many molecular pathways have been proposed as plausible mechanisms to explain the neuroprotection. The nature of these mechanisms is strongly influenced by the temporal profile examined and the nature of the stimulus able to induce conditioning protection. The purpose of the present review is to summarize the most studied molecular mechanisms involved in the different kinds of conditioning, highlighting differences and similarities. Knowledge of these mechanisms may bring to light those molecular pathways that must be activated or inhibited in order to protect the brain. This review describes the possible differences and similarities among neuroprotective pathways involved in four different types of preconditioning stimuli: pharmacological, physical, surgical and thermoregulatory.
Abstract
The identification of neuroprotective therapies is gaining great interest in the design of strategies to prevent neuronal death. However, up until now all tested approaches have been unsuccessful despite many years of clinical trials, mainly because of the numerous side effects observed in humans and absent in animals used at the preclinical level. The neuroprotective strategy known as brain preconditioning is becoming important with the increased understanding of the endogenous mechanisms able to induce tolerance. Over the years, several stimuli have been described as possible preconditioning inducers, and many molecular pathways have been proposed as plausible mechanisms to explain the neuroprotection. The nature of these mechanisms is strongly influenced by the temporal profile examined and the nature of the stimulus able to induce conditioning protection. The purpose of the present review is to summarize the most studied molecular mechanisms involved in the different kinds of conditioning, highlighting differences and similarities. Knowledge of these mechanisms may bring to light those molecular pathways that must be activated or inhibited in order to protect the brain. This review describes the possible differences and similarities among neuroprotective pathways involved in four different types of preconditioning stimuli: pharmacological, physical, surgical and thermoregulatory.
1. Introduction
The ischemic tolerance (IT) paradigm has been described in several organs. It may be realized through different experimental strategies, and may represent a fundamental cell/organ response to certain types of injury (Barone et al., 1998). Ischemic tolerance in the brain was first described in 1990, when Kitagawa and collaborators demonstrated that two minutes of ischemic preconditioning in gerbils produced tolerance against global brain ischemia following a 24-hour interval, and that multiple brief preconditioning episodes were more protective than a single episode (Kitagawa et al., 1990). Over the years this strategy has been applied to several brain disorders, and several molecular mechanisms taking part in this protective process have been described. The mechanisms are usually classified in three distinct categories: triggers, mediators and effectors (Dirnagl et al., 2009). The nature of these mechanisms is strongly influenced by the temporal profile examined and the nature of the stimulus able to induce conditioning protection. The purpose of the present review is to summarize the most studied molecular mechanisms involved in the different kinds of conditioning, highlighting differences and similarities. Knowledge of these mechanisms may bring to light those molecular pathways that must be activated or inhibited in order to protect the brain.
Preconditioning triggers a radically different adaptive response and, in mammals, this response is characterized by at least two distinct time intervals of tolerance induced with respect to the preconditioned stimulus and subsequent ischemia. A short-lived protective phenotype can be induced within minutes of exposure to preconditioned stimuli, as a result of changes in ion channel permeability, protein phosphorylation and other post-translational modifications; this is known as rapid preconditioning. However, the phenomenon of ischemic tolerance is more effective if it occurs through gene activation and de novo protein synthesis; this "classic preconditioning" or delayed preconditioning takes many hours or even days to fully manifest (Gidday et al., 2006).
2. Physical Exercise
Among different conditioning stressors, physical exercise is capable of reinforcing the neurovascular unit, strengthening the blood-brain barrier, and improving angiogenesis, thus increasing the ability of the brain to enhance neuronal survival following vascular damage (Li et al., 2005; Cobianchi et al., 2017). It has been widely demonstrated that regular physical exercise improves abnormally elevated blood pressure, reduces obesity, improves blood glucose levels and ameliorates lipid metabolic disorders (Lee et al., 2003; Chrysohoou et al., 2005). Biological response and adaptations to stressors experienced with exercise provide protection against potential injury or cellular damage induced by a subsequent harmful stimulus. Generally, physical activity is an important modifiable risk factor, particularly for stroke and cardiovascular disease. Preconditioning exercise prior to ischemia can lead to protection from subsequent serious injury through the promotion of angiogenesis, mediation of inflammatory responses, inhibition of glutamate over-activation, protection of the blood-brain barrier, and inhibition of apoptosis (Zhang et al., 2001). The neuroprotective effects of exercise conditioning may be evoked by several mechanisms (Figure 1), all of which strengthen neuronal integrity and improve cell survival (Kochanski et al., 2014). Furthermore, animal studies have indicated that preconditioning exercise confers beneficial effects on cerebral ischemia, including enhanced survival rates, alleviation of oxidative damage, improvement of cerebral blood flow, and maintenance of neurovascular integrity (Ding et al., 2009, 2006; Zhang et al., 2014). The exercise-mediated increase in neurogenesis and in synaptic plasticity occurs through both increased release of neurotrophins, a class of growth factors able to prevent neurons from undergoing programmed cell death, and release of other growth factors. These processes result in a reinforcement of the blood-brain barrier, in an enhanced induction of protective astrocytosis, and in an amplification of angiogenesis at the level of the neurovascular unit (Neeper et al., 1996; Sakakima et al., 2012). A recent study showed that 1 or 2 weeks of pre-ischemic exercise did not reduce brain infarction after ischemic stroke compared with a non-exercise group, but that at least 3 weeks of pre-training was necessary for neuroprotection (Liebelt et al., 2010). Based on the above studies, it is evident that 2 or 3 weeks of pre-training is necessary to exert neuroprotection after ischemic stroke. Most animal studies have used two types of exercise manipulations: voluntary and forced (Hu et al., 2010; Zhang et al., 2014). Voluntary exercise permits the subject to exercise at will, mimicking human daily activity or manual labor (Cotman and Berchtold, 2002). Conversely, forced exercise demands that the subject exercise on a treadmill for 0.5 to 1 h for 5–7 days per week (Ding et al., 2006). Forced exercise could be regarded as a simulation of gym exercise as in the case of athletes. While these two types of exercise manipulations are different in volume and amount of time, they both have been shown to exert protection. In particular, forced exercise, as opposed to voluntary exercise, has been shown to confer a more substantial neuroprotection in animal models of cerebral ischemia, reducing ischemic volume and neurological deficits (Hayes et al., 2008).

In a new window | Download PPT
Figure 1: Neuroprotective molecular mechanisms and mediators activated by physical exercise-induced brain conditioning. The mechanisms inducing protection after exercise preconditioning are indicated with regards to pathways involved in inflammation, and in integrity of neurovascular unity, metabolic homeostasis and apoptotic death.
In addition, physical exercise extended over time (so-called “chronic exercise”) reduces the effects of inflammation following an ischemic insult through the downregulation of leukocyte invasion and fluid permeability in interstitial space and collectively by reducing neuron apoptosis. The model of chronic exercise was performed on female Sprague-Dawley rats subjected to 10 weeks of daily training on a treadmill where running speed, grade and duration was progressively increased to 25 m/min, 10% grade, for 2 h/day by the end of week 4, and this intensity was maintained during the rest of the training period (Lawler et al., 2016).
Exercise training benefit resides in the restoration of brain metabolism by preventing the metabolic dysfunction that occurs following an ischemic insult; this improvement is associated with an up-regulation of HIF-1α, whose neuroprotective effect has been demonstrated also during brain ischemic preconditioning (Valsecchi et al., 2011). Together, these mechanisms provide increased neuroprotection against cerebral ischemic insults, improving neuronal survival following a cerebrovascular event. Understanding the mechanisms that are triggered by this form of preconditioning is useful for therapeutic intervention to enhance brain neuroprotection. The sections below will show the main mechanisms involved in the protection mediated by physical exercise (Figure 1).
2.1 Maintenance of the integrity of the neurovascular unit
One of the better characterized mechanisms of chronic exercise preconditioning is the augmented integrity of the blood-brain barrier (BBB), which leads to decreased cerebral edema and reduced brain injury (Masada et al., 2001). The integrity of the neurovascular unit, composed of endothelial cells, glia and neurons, is essential for both structural stability and maintenance of appropriate permeability of the cerebral vasculature (Del Zoppo and Hallenbeck, 2000; Del Zoppo and Mabuchi, 2003).
Exercise preconditioning seems to improve BBB integrity by increasing the expression of the main basal laminar protein, collagen type IV, whose expression is usually strongly reduced in the ischemic core (Davis et al., 2007). Furthermore, cerebral microvasculature integrity depends on integrins that lose their affinity for lamina and collagen following ischemia. In response to exercise preconditioning, the disruption of integrin functionality is significantly attenuated (Ding et al., 2006), thus contributing to the stability of the BBB. The upregulation of integrins and collagen type IV represent one of the putative mechanisms of protection activated by exercise preconditioning. In a recent paper describing adult rats that exercised on a treadmill for 30 minutes each day for 3 weeks and were subjected to 2 hours of middle cerebral artery (MCA) occlusion, it was demonstrated that exercise pre-conditioning reduces brain injury by decreasing cerebral permeability and enhancing brain integrity after stroke (Ding et al., 2005).
2.2 Inflammatory response
Physical activity induces endogenous neuroprotection by reducing both the expression of inflammatory mediators and the accumulation of leukocytes during reperfusion. The beneficial effects of exercise on ischemic brain injury suggest multiple mechanisms underlying this induced neuroprotection (Zhang et al., 2016). Following acute ischemia, cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α) stimulate the expression of adhesion molecules such as P-selectin, E-selectin and intracellular adhesion molecule-1 (ICAM-1), thus triggering the increase in leukocyte infiltration into the brain parenchyma (Ding et al., 2005). In this phase, leukocytes may mediate reduced reperfusion following stroke and damage from free radicals, inflammatory cells, and endothelial dysfunctions. In 3-week-old rats exercised on a treadmill for 30 minutes each day for 3 weeks and then subjected to 2 hours of transient MCA occlusion, chronic preconditioning exercise prevented ICAM-1 over-expression, and, consequently, also leukocyte infiltration in the brain. Exercise preconditioning also limited the damage that occurred during reperfusion (Ding et al., 2005). Exercise preconditioning may attenuate the inflammatory response occurring after stroke damage and modulate TNF-α levels. Indeed, chronic small elevations of TNF-α levels that have been observed with ischemic exercise and preconditioning can cause neuronal tolerance to cytokine development and may promote angiogenesis. Such elevated levels of TNF-α lead to a decrease in the expression of its receptor, generating neuronal tolerance. In fact, TNF-α receptors are downregulated during exercise preconditioning in a model in which chronic stimulations of low TNF-α levels resulted in receptor desensitization. The decrease of TNF-α receptors renders less dangerous the presence of elevated levels of TNF-α occurring in the post-stroke period, leading to a better overall neuronal survival (Ding et al., 2005).
Similar to what occurs in the case of TNF-α receptors, a reduced expression of another class of receptors, toll-like receptors (TLR), has been observed after exercise preconditioning (McFarlin et al., 2006). Since TLRs take part in the immune response and probably in the cytokine cascade, leading to leukocyte infiltration, exercise preconditioning may reduce the post-stroke inflammatory cascade also through this mechanism (McFarlin et al., 2006).
We now have enough evidence to claim that exercise preconditioning reduces post-stroke systemic inflammation by influencing the expression of specific receptors rather than modulating the expression of inflammatory mediators.
2.3 Cell death and survival pathways
Neuronal death following ischemic reperfusion injury is mediated by a series of genes and regulatory proteins that trigger cascades leading to cell death or survival. Heat shock protein (Hsp70) and ERK-mediated signaling pathways have been shown to be involved in ischemia-induced apoptosis (Xu et al., 2006; Zhang et al., 2001). In addition to the role of TNF-α and Hsp70, anti-apoptosis genes such as BCL-2 and BCL-xL, and pro-apoptosis genes such as Bax, BAD, Bak, and HIF are responsible for the neuronal response under hypoxic conditions (Lazou et al., 2006). By inducing an overexpression of Hsp70, exercise preconditioning leads to increased levels of anti-apoptotic protein expression, also leading to increased neuroprotection (Chaudhry et al., 2010). In fact, Hsp70 mediates such neuroprotection by inducing a downregulation of proapoptotic proteins such as HIF and an upregulation of antiapoptotic molecules such as Bcl-2 (Matsumori et al., 2005). Besides this well-characterized role, Hsp70 may work in conjunction with other factors, rather than alone, to prevent apoptosis (Lee et al., 2001). Indeed, Hsp70 together with TNF-α works to correctly balance the relationship between pro- and anti-apoptotic genes (Goel et al., 2010). Exercise is therefore likely to generate high levels of Hsp70 and, working in tight relationship with the elevated levels of TNF-α, may attenuate the apoptotic cascade. Previous studies suggested that transient forebrain and global ischemia causes neuronal injury and loss in the CA1 region of the hippocampus, a brain region important for learning and memory (Olsson et al., 2003; Ouyang et al., 2007; Albasser et al., 2012). Exercise preconditioning rescued ischemia-induced hippocampal CA1 neuronal degeneration in a rat common carotid artery stroke model and thus prevented memory deficit (Shamsaei et al., 2015). Exercise prior to ischemic insult prevented hippocampal neuronal loss in CA1 and CA3 regions through BAX/BCL-2 ratio reduction and caspase-3 activation (Aboutaleb et al., 2015, 2016).
2.4 Metabolic alterations
Chronic exercise preconditioning has also been associated with changes in neuronal tissue metabolism (Lawler et al., 2016). Physical exercise preconditioning is known to ameliorate stroke-induced injury. In addition to several other mechanisms, the beneficial effect of pre-ischemic exercise following stroke is due to an upregulated capacity to maintain energy supplies. This has been demonstrated through increases in the regional use of oxygen and glucose. These changes are associated with the ability to generate ATP by utilizing less oxygen, thus creating a greater ability to produce energy during hypoxia (McCloskey et al., 2001; Querido and Sheel, 2007; Lawler et al., 2016). Another mediator belonging to the class of metabolic proteins produced after physical exercise is hypoxia-inducible factor-1α (HIF-1α). Physical exercise preconditioning signals a metabolic demand for higher ATP-generating capacity to cerebral tissues, indicated by a chronically elevated ADP/ATP ratio and upregulation of GLUT1, GLUT3, PFK, pAMPK, and HIF-1α after 3 weeks of exercise (Dornbos et al., 2013). In particular, HIF is strongly upregulated during different kinds of preconditioning, including exercise preconditioning, and induces both angiogenesis and glycolysis. Recently, a particularly relevant role played by HIF has emerged in several pathophysiological aspects of cerebral ischemia. In fact, neuron-specific knocking-out of HIF-1α induces an exacerbation of brain damage after transient cerebral ischemia and prevents preconditioning-induced neuroprotection, supporting its neuroprotective action (Valsecchi et al., 2011).
3. Hypothermia and Hyperthermia
Hypothermic preconditioning is performed by administering hypothermia of a specific depth and duration at a time point prior to the onset of ischemia, showing its maximum effects especially when cooling is initiated within a few hours of injury onset (Rzechorzek et al., 2015; Kim et al., 2008, 2015). Experimentally, whole-body cooling to temperatures of 31.5, 28.5, or 25,5 °C for 20 minutes showed a significant temperature-dependent reduction in brain infarction after focal cerebral ischemia in rats (Kochanski et al., 2013). Further studies demonstrated that hypothermic tolerance was not dependent on stimulus duration but rather on reaching a critical body temperature, as indicated by better neuroprotection obtained at 33°C compared to 34.5 °C (Yunoki et al., 2003).
On the other hand, hyperthermic stimulus is delivered through a controlled increase of the body temperature of the experimental model chosen, prior to the onset of ischemia. As mentioned, in a focal model of transient brain ischemia in rats, a 15-minute immersion of animals in a water bath set to 42°C determined a significant neuroprotective effect (Gidday et al., 2006; Yenari et al., 2012).
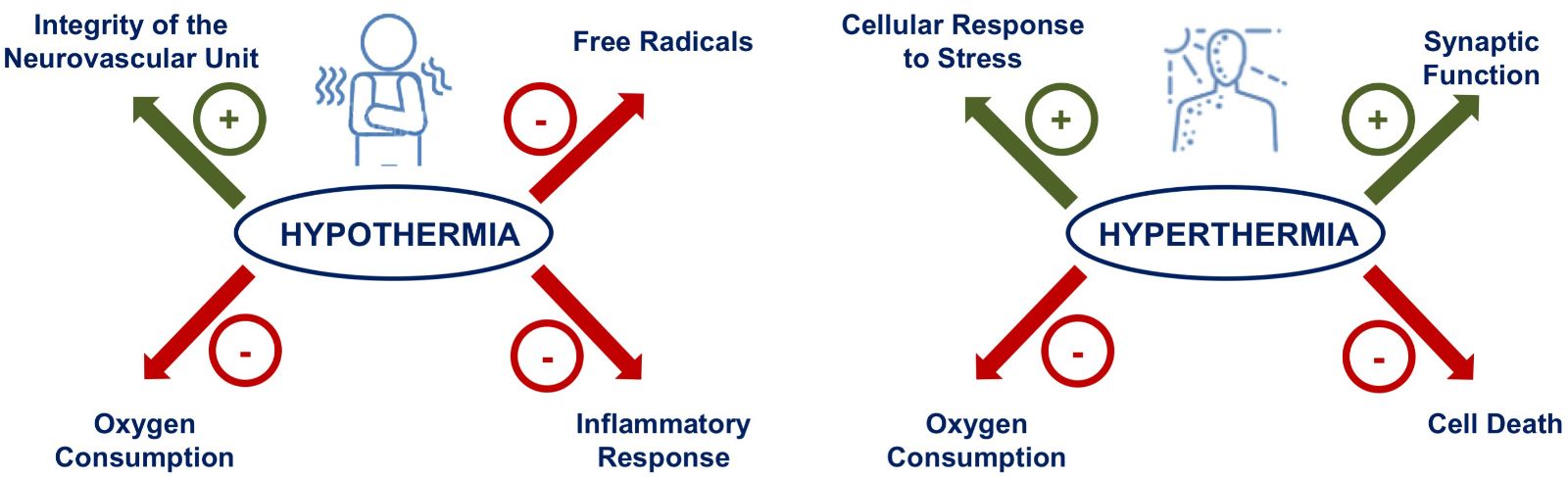
A number of explanations have been proposed for induced neuroprotection in response to preconditioning due to low (hypothermia) or high (hyperthermia) temperature, including several mechanisms that are usually related to the concept of “thermo-pharmacology“ (Muzzi et al., 2017; Yunoki et al., 2003; Nawashiro et al., 1997). These molecular mechanisms include reduction in cerebral metabolism (Polderman, 2009), decreased free radical formation (Karibe et al., 1994), preservation of blood-brain barrier integrity (Huang et al., 1999; Chi et al., 2001) and suppression of inflammation (Aibiki et al., 1999; Kimura et al., 2002; Luan et al., 2004) (Figure 2).
The best characterized of the preconditioning stimuli have been some heat shock proteins (HSP), hypoxia-inducible factor (HIF), and adenosine receptors. Mediators of these preconditioning stimuli will be discussed below.
In a new window | Download PPT
Figure 2: Neuroprotective molecular mechanisms and mediators activated by hypothermia and hyperthermia-induced brain conditioning. The mechanisms inducing protection after thermal conditioning are indicated with regards to pathways involved in inflammation, integrity of neurovascular unity, synaptic function, free radical production and oxygen consumption.
3.1 Heat shock proteins
Heat shock proteins (HSPs) are molecular chaperones that play a vital role in the cellular response to stress stimuli as they diminish the accumulation of denatured proteins and increase cell survival through a reduction of pro-apoptotic proteins (Kampinga and Bergink, 2016). Boosting the activity or expression of crucial HSPs has been proposed as a promising way to delay the onset of or to treat neurodegenerative diseases. As the name suggests, heat shock protein expression may be induced by hyperthermic stimulus prior to brain stress, although several fundamental HSPs are expressed at physiological temperature (37°). Indeed, only a subset of HSPs are induced by hyperthermia (Kampinga and Bergink, 2016).
In particular, preconditioning induced by both hypothermia and hyperthermia determines the over-expression of 72-kDa inducible heat shock protein (Hsp72) and 70-kDa inducible heat shock protein (Hsp70) in glia and endothelial cells as well as in neuroendocrine areas 24 hours after stimulation (Kelty et al., 2002; Kirino et al., 2002; Terao et al. 2009; Cullen et al., 1997; Yenari et al., 2005). The importance of the increase in Hsp72 has also been underlined in other studies in which hyperthermic preconditioning applied in rats subjected to 42°C for 15 min has been demonstrated to protect synaptic function (Kelty et al., 2002; Kirino et al., 2002; Yenari et al., 2005). When mouse brain slices were treated with exogenous Hsp72 in the absence of hyperthermic stimulation, similar synaptic protection was demonstrated, confirming the central protective role of Hsp72 (Kelty et al., 2002; Kirino et al., 2002).
3.2 MicroRNAs
MicroRNA (miRNA), a subset of non-coding RNA that plays a role in silencing mRNAs, have been involved in a recent report describing a model of head injury which showed that cooling alters the expression of different miRNAs (Bao et al., 2013). Some miRNAs, including miR-874 and miR-451, were the most affected: the cooling decreased the expression of both miRNAs at 7 hours, but miR-451 was increased from cooling to 24 hours compared to normothermia. Further research is needed to define their role in brain injury (Vemuganti et al., 2010; Truettner et al., 2011).
3.3 HIF-1α
HIF-1α plays an important role in the maintenance of oxygen homeostasis and is also involved in the tolerance conferred by hyperthermic preconditioning (Du et al., 2010). Current literature links HIF-1 to the development of hypothermia. A reasonable explanation for this relationship might be the energy depletion attenuating glycolysis and ATP production associated with hypothermia, which in turn leads to HIF-1 inhibition under stressful conditions like stroke (Umschweif et al., 2013). This effect may be linked to heat shock protein functions. In fact, it has been hypothesized that the overexpression of some HSPs induced by thermal preconditioning is controlled by the induction of HIF-1α, which confers resistance to harmful ischemia (Du et al., 2010; Huang et al., 1999).
In a new window | Download PPT
Figure 3: Neuroprotective molecular mechanisms and mediators activated by medical gas-induced brain conditioning. The mechanisms inducing protection after medical gas preconditioning are indicated with regards to pathways involved in inflammation, cell death, and ionic homeostasis.
3.4 Adenosine receptors
Adenosine is an endogenous neuroprotectant that can inhibit the release of excitatory amino acids. When ischemia occurs, adenosine can increase significantly. Adenosine inhibits synaptic transmission, decreases K+-stimulated glutamate release, and inhibits presynaptic calcium fluxes via adenosine A1 receptors (Liu et al., 2006). Notably, the protective response to brain conditioning induced by hyperthermia (Xu et al., 2002) is attenuated in animals receiving an adenosine receptor antagonist, thus suggesting the involvement of adenosine receptors in the protection elicited by thermal preconditioning (Yuan et al., 2004). This and other evidence suggest a crucial role for adenosine receptors in both hypothermic and hyperthermic tolerance (Yuan et al., 2004; Carlin et al., 2017). Indeed, administration of adenosine or adenosine 5'-monophosphate (AMP) can trigger a hypothermic, torpor-like state that mediates strong neuroprotection (Carlin et al., 2017). This mechanism of reduced metabolic activity could explain the effect of adenosine in mediating some of the protective actions elicited by thermal preconditioning.
4. Hypoxia and Ischemia
Preconditioning induced by hypoxia (HPC) or a subliminal ischemia can produce significant protective effects on neurons in experimental cells, animals and humans (Matsushima and Hakim,1995). Occlusion of the middle cerebral artery (MCAO) by intraluminal insertion of a nylon monofilament into the internal carotid artery is the most common model to induce focal cerebral ischemia in rats and mice (Takano et al., 1997; Durukan et al., 2008). This surgical procedure, if performed for a brief time, is an excellent method for inducing ischemic preconditioning (Pignataro et al., 2009, 2012). In the same way, intermittent hypoxia preconditioning can ameliorate nerve injury in the global cerebral ischemia-reperfusion model (Zhao et al., 2017).
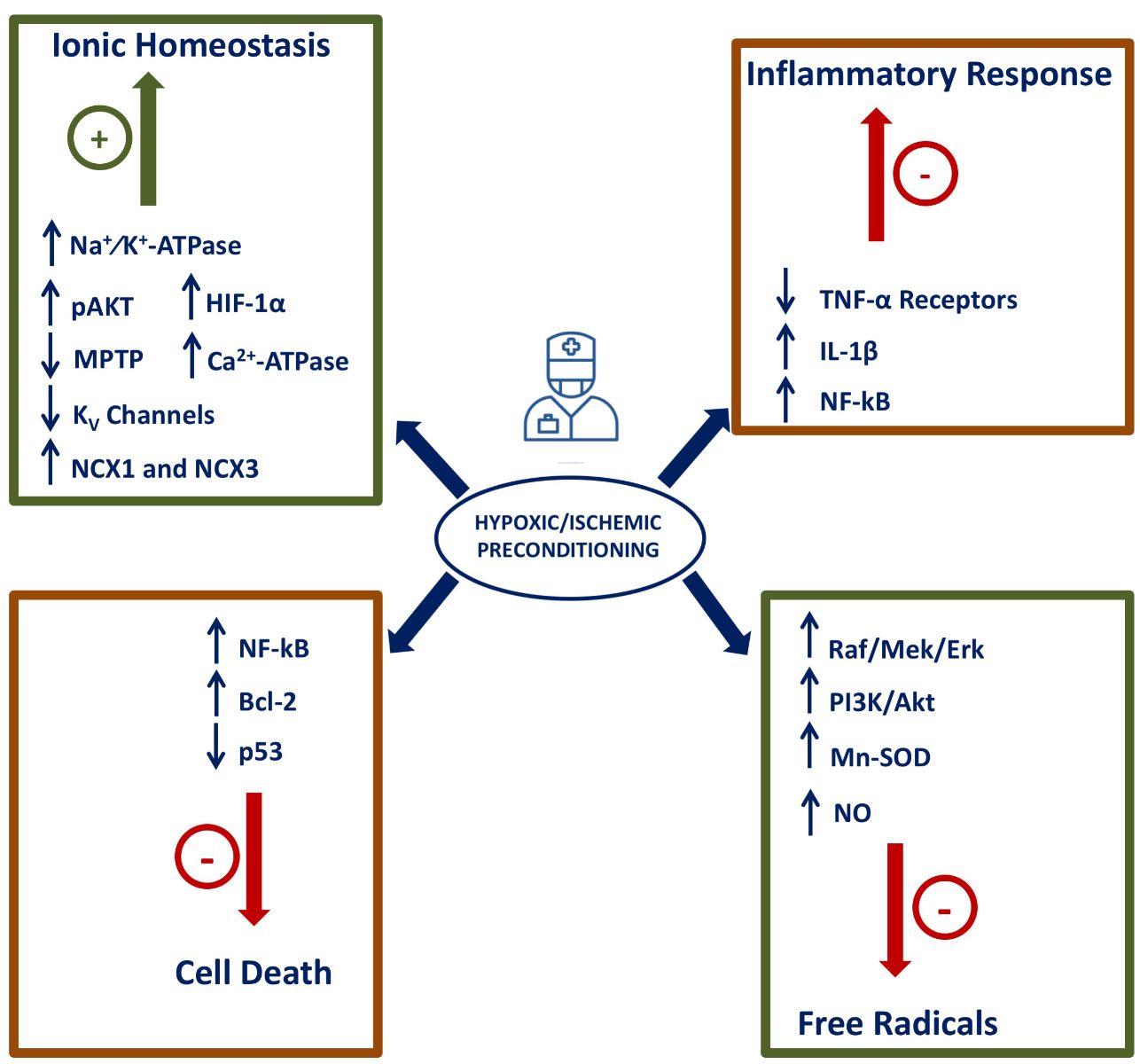
The mechanisms by which subliminal episodes of hypoxia and ischemia may confer neuroprotection have been partially elucidated, and they include: (1) the activation of hypoxia-inducible factor-1 alpha (HIF-1α) and of its target genes; (2) the activation of pro-survival pathways; (3) a general genomic reprogramming; (4) the activation of anti-inflammatory pathways; (5) modification of the metabolic processes; and (6) ionic homeostasis maintenance. In the present review, the main mechanisms involved in hypoxia/ischemia neuroprotection will be analyzed (Figure 4).
In a new window | Download PPT
Figure 4: Neuroprotective molecular mechanisms and mediators activated by hypoxia/ischemia-induced brain conditioning. The mechanisms inducing protection in preconditioning induced by ischemia or hypoxia are indicated in the figure, particularly pathways involved in inflammation, free radical production, metabolic homeostasis and pro-survival pathways.
4.1 Hypoxia-inducible factor-1 alpha
The involvement of hypoxia-inducible factor-1 alpha (HIF-1α) and the increased expression of its target genes in the neuroprotection induced by hypoxic preconditioning (HP) have been widely experimentally confirmed (Wacker et al., 2012). The transcriptional factor HIF-1α is a prominent player in hypoxia-inducible gene expression, which is stabilized by hypoxic stimuli and subsequently binds to HREs on many pro-survival genes across many cell types and species (Majmundar et al., 2010; Ratan et al., 2004; Semenza et al., 2009; Sharp and Bernaudin, 2004; Wenger, 2002). In particular, a dose-dependent increase in DNA binding and transcriptional activity of HIF-1α occurs during hypoxia and following reoxygenation (Tzeng et al., 2010). This mechanism is responsible for activating the transcription of over 200 genes including antiapoptotic factors, ion-gated channels, structural membrane proteins, and glycolysis-promoting enzymes (Majmundar et al., 2010; Semenza et al., 2009; Wenger, 2002). Interestingly, following ischemic insult, HIF-1α expression was widely evident in microvessels of preconditioned animals (Bergeron et al., 2000).
Among HIF target genes that are involved in stroke pathogenesis, the sodium–calcium exchanger-1 (NCX1), a ubiquitous plasma membrane protein regulating cellular calcium and sodium homeostasis in the brain, has aroused great interest. One of the mechanisms by which HIF-1 exerts its pro-survival role during ischemic preconditioning is the upregulation of NCX1 transcript and protein, whose neuroprotective action has been fully substantiated by recent studies (Valsecchi et al., 2011). Among other transcription factors involved in HPC, Nrf1, ATF2, JNK, MAPKs (ERK 1/2), c-fos, STAT3, mTOR, CREB and NFκB directly or indirectly target hypoxia-responsive gene expression, showing more extensive effects for HPC-mediated changes in gene expression compared to HIF-1α (Kenneth and Rocha, 2008; Majmundar et al., 2010; Ran et al., 2005; Rybnikova et al., 2009; Seta et al., 2002; Trachootham et al., 2008). Together these transcriptional factors might act by lowering the concentration of the excitatory amino acids aspartate and glutamate during HPC, which was proposed to contribute to the enhanced anoxia tolerance in this model (Xie et al., 1999). Moreover, it has been demonstrated that reduced activity of AMPA receptors was protective in a rat model of hypobaric hypoxia stimulus because it reduced the sensitivity of neurons to glutamate and/or downregulated glutamate receptors (Turovskaya et al., 2011; Zhang et al., 2006; Chang et al., 2006). In addition, adaptation to hypoxia of cells and tissues leads to the transcriptional induction of a series of genes that have important functions in iron metabolism. Transferrin (Tf) and transferrin receptor (TfR1) are two key proteins involved in iron uptake by mammalian cells. Hypoxia can increase iron uptake by cells as well as the expression of Tf and TfR1, both of which have been identified as hypoxia-inducible genes. It has also been reported that several other iron transport or regulation proteins, including ceruloplasmin (Cp), iron regulatory protein 1 (IRP1) and 2 (IRP2), and hepcidin, are regulated by HIF-1 in response to hypoxic conditions (Yang et al., 2010). Among other putative targets of HIF are the NO synthases iNOS and eNOS. Both NOS isoforms have been included among the possible targets of HIF in mediating neuroprotection induced by hypoxic preconditioning in neonate and adult brain (Gidday et al., 1999; Zhu et al., 2006; Vellimana et al., 2011).
4.2 Pro-survival pathways
Numerous reports support the idea that the activation of the pro-survival kinase Akt, through its phosphorylation, promoted by hypoxic brain conditioning, represents the trigger of other pro-survival targets, including the antiapoptotic protein BAD (Rybnikova et al., 2006; Wang et al., 2010), NFκB (Rybnikova et al., 2008), eNOS (Hashiguchi et al., 2004) and the anti-apoptotic survivin, a member of the “inhibitor of apoptosis” (IAP) gene family (Zhang et al., 2007), able to induce a rapid tolerance to an ischemic event. Neuronal NO synthase (nNOS) also seems to play a relevant role in neuroprotection induced by ischemic preconditioning in mature neurons (Gonzalez-Zulueta et al., 2000), involving N-methyl-d-aspartate (NMDA) receptor activation, Ca2+ influx, and new protein synthesis. In particular, an increase of NO levels is related to the modulation of the MPTP opening (Shiva et al., 2007) and the enhancement of activity and protein expression of mitochondrial Mn-SOD (Scorziello et al., 2007), thus affecting reactive oxygen species (ROS) production. These functions are mediated by two main pathways: the RAF/MEK/ERK cascade (Nandagopal et al., 2001) and the PI3K/Akt pathway (Brunet et al., 2001). On the role of survival kinases in ischemic preconditioning, different opinions are present in the scientific literature. Ischemia activates a process of protein phosphorylation (Shamloo and Wieloch, 1999) that persists for a few days, involving calcium/calmodulin-dependent protein kinase II (CaMKII) and mitogen-activated protein kinases (MAPK). Some authors report that this enhanced and excessive phosphorylation is blocked after preconditioning induction. On the other hand, the activation of Akt/protein kinase B occurring after a sublethal ischemia may contribute to the induction of tolerance (Yano et al., 2001, Pgnataro et al., 2012). Some studies suggest that the early phase of ischemic preconditioning is characterized by rapid post-translational modification of pre-existing proteins through signaling pathways that involve protein kinase C (PKC) (Speechly-Dick et al., 1994) and MAPK (Shampoo et al., 1999). The common idea is now that the over-phosphorylation of some kinases belonging to the so-called RISK (Reperfusion Injury Salvage Kinase) family takes part in the effects of ischemic preconditioning in promoting cell survival. Accumulating evidence suggest that other pharmacologic approaches for inducing tolerance may work by mechanisms quite similar to those induced by hypoxia (He et al., 2008).
4.3 Genomic reprogramming
Although based on the nature of hypoxic conditioning stimulus (i.e. its frequency, magnitude and duration), genes that regulate neuronal signal transduction, ionic homeostasis, metabolism, inflammation, apoptosis, and transcriptional activation are often upregulated by hypoxia (Bernaudin et al., 2002; Bickler and Fahlman, 2009; Tang et al., 2006; Gustavsson et al., 2005). Epigenetic transcriptional regulation by HPC can occur through interaction with hypoxia-response elements (HREs) on promoter sequences of genes targeted by different stressors. In a model of oligonucleotide preconditioning, cytosine-phosphate-guanine (CpG) administration reduced ischemic injury dramatically in both rodent and nonhuman primate models of experimental stroke, and this action was mediated by small RNA non-coding molecules, namely microRNAs (Bahjat et al, 2011; Stevens et al., 2008; Vartanian B et al., 2015). Several oxygen-dependent enzymes can serve as “receptors” for post-translational modifications and changes in gene expression induced by hypoxia. A crucial role of this mechanism of tolerance induction is mediated by mitochondria, wherein free radicals that originated from oxygen-nitrogen species formed upon hypoxia serve as subsequent signals for neuroprotection (Bailey et al., 2011). Another mediator is adenosine, which is formed during metabolic stress or oxygen demand, and is able to induce a genomic adaptative response that involves the whole neurovascular unit (Lin et al., 2008; Zhang and Lu, 1999). Delayed preconditioning is mediated by the synthesis of new protective proteins, regulated by the activation of transcriptional factors through PKC and tyrosine kinase signaling pathways. The regulation of gene expression leading to the apoptosis process seems to be related to ischemic tolerance. Indeed, during ischemic preconditioning, Bcl-2 protein expression is enhanced, thus preventing the delayed neuronal death that normally occurs in the penumbra region (Shimazaki et al., 1994), whereas p53 gene expression is markedly reduced (Tomasevic et al., 1999). Generally, protein synthesis is impaired by cerebral ischemia (Kleihues and Hossmann, 1971), but in the gerbil model it was established that preconditioning restores general protein levels (Nakagomi et al., 1993; Furuta et al., 1993): autoradiography analysis using isotope-labeled amino acids demonstrated that the pattern of amino acid incorporation in the CA1 neurons returned to a normal pattern within 24 hours. Several studies have shown that hypoxic preconditioning is able to increase manganese and copper-zinc superoxide dismutase (SOD1 and SOD2), heme oxygenase-1 (HO-1), glutathione peroxidase, and glutathione reductase, in proportion to the magnitude of the ischemic tolerance evoked (Alkan et al., 2008; Arthur et al., 2004; Duan et al., 1999; Garnier et al., 2001; Gorgias et al., 1996; Stroev et al., 2004; Zhu et al., 2007).
4.4 Inflammatory response
Following ischemia, cytokines such as IL1 and TNF-α stimulate the expression of adhesion molecules including intercellular adhesion molecule 1 (ICAM-1), and selectin P and E on endothelial cells, activating the inflammatory pathway within ischemic regions. As mentioned above, TNF-α represents the major pro-inflammatory cytokine with profound effects on the brain response to hypoxic damage. Chronic elevations of low TNF-α levels following exercise or ischemic preconditioning can cause neuronal tolerance to cytokine development and promote angiogenesis (Pradillo et al., 2005). Such elevated levels of TNF-α lead to a decrease in the expression of its receptor, generating neuronal tolerance. Similar to what occurs with TNF-α, recent studies have shown that ischemic preconditioning reduces other markers of systemic inflammation such as toll-like receptors that are involved in the immune response and probably in the cytokine cascade, leading to leukocyte infiltration (Stevens et al., 2008). In a model of transient global ischemia in which both common carotids were simultaneously occluded for either 2 or 3.5 min to produce transient global ischemia of the forebrain, daily injection of IL-1α over a 3-day period protected CA1 hippocampal neurons from 3.5 min of global ischemia in a dose-dependent manner as compared with IL-1α vehicle injections (Ohtsuki et al., 1996).
4.5 Metabolic contribution
Tissue and cellular metabolism usually runs lower in hypoxic environments, a manifestation of the well-known “Pasteur effect” which, in turn, leads to the reduction in protein synthesis and other adaptative responses (Buck and Pamenter, 2006). This mode of saving energy is mainly determined by glucose transporters (Ye et al., 2008), glycolytic enzymes (Jones and Bergeron, 2001), calcium influx (Bickler et al., 2009; Semenov et al., 2008), mitochondrial respiration (Buck and Pamenter, 2006), and the endoplasmic reticulum (Bickler et al., 2009). HPC then reprograms the metabolic response of cell/tissue to ischemia, as has been proposed for conditioning’s effect on the genome. A mechanism through which HPC induces tolerance is represented by preservation of postischemic Na+/K+-ATPase levels (Zhan et al., 2011). In neonatal rats it has been shown that HPC-induced neuroprotection was not due to tissue acidosis or depletion of high-energy phosphate reserves following ischemia, but was related to the restoration of high-energy phosphate reserves in the early hours of postischemic reperfusion (Vannucci et al., 1998). Accumulating evidence suggests that mitochondria are likely to be critical effectors of the ischemia-tolerant state (Correia et al., 2010; Dirnalg and Meisel, 2008; Zhan et al., 2002) given that with strong preservation of mitochondrial function, ATP production by aerobic respiration, the activation of ATP-sensitive potassium channel (KATP) and reduction of superoxide production secondary to fewer electrons from the electron transport chain are all involved in preconditioning signaling (Busija et al., 2008; Correia et al., 2010).
4.6 Restoration of ionic homeostasis
Recent evidence highlights that maintenance of ionic homeostasis plays a key role in propagating the preconditioning phenomenon (Cuomo et al., 2015). Several experiments performed both in vivo and in vitro showed that an improved capacity to preserve cellular ionic and pH homeostasis represents a determinant factor for ischemic tolerance (Pasuphathy and Miller, 2005). Indeed, in cortical neurons exposed to brief non-injurious oxygen and glucose deprivation (OGD), impairment in voltage-gated potassium channels (KV channels) has been observed. Moreover, in vivo experiments showed that ischemic preconditioning prevented the inhibition of Na+⁄K+-ATPase activity after brain ischemia in hippocampal and cortical neurons of rats subjected to ischemia (de Souza Wyse et al., 2000). In fact, neuronal stimulation induces changes in intracellular calcium concentration that in turn trigger several mechanisms mediating numerous nerve cell functions. In order to avoid the extended elevation of Ca2+ cell levels, which become toxic, and in order to allow cells to be able to respond to a new stimulus, several pathways work together to restore calcium levels. Among them there are Ca2+ binding proteins that avoid abnormal intracellular Ca2+ increase through sequestration into the endoplasmic reticulum and mitochondria, and through the extrusion across the plasma membrane (Zaidi, 2010). The latter process is operated mostly by the low affinity-high capacity Na+/Ca2+ exchanger (NCX) and by the high affinity-low capacity plasma membrane Ca2+-ATPase (PMCA). In certain conditions, Ca2+ flux across membranes is the predominant mechanism of Ca2+ removal from cytosol compared to refilling of stores, for example after activation of localized signals in the dendritic spines of neurons. In particular, in these cases, while PMCA transport proteins control the resting levels of this ion, NCX proteins play a role in calcium homeostasis following the increase of levels during a signal event. Regarding calcium homeostasis maintenance, preconditioning seems to induce an increase in NCX and PMCA activity (Ohta et al., 1996) and protein expression (Kato et al., 2005; Pignataro et al., 2008), and result in a reduction of intracellular [Ca2+] (Shimazaki et al., 1998). In fact, during cerebral ischemia experimentally induced in rats, NCX gene expression is reduced in the brain in a different manner depending on the exchanger isoforms and on the region involved in the insult (Pignataro et al., 2004; Boscia et al., 2006). In contrast, NCX1 and NCX3 isoforms increased after ischemic insult in preconditioned rats (Pignataro et al., 2012), and their silencing partially prevented preconditioning-mediated neuroprotection.
Moreover, it has been shown that p-Akt, by acting on NCX1 and NCX3 (Formisano et al., 2008), represents a fundamental transducer of the neuroprotection exerted by preconditioning (Pignataro et al., 2012). Recently it was demonstrated that NCX1 is a target gene for HIF-1α, and that after ischemic preconditioning induction HIF-1α expression is strongly augmented and exerts its prosurvival role through the upregulation of NCX1 transcript and protein (Valsecchi et al., 2011). In regular ischemic conditions, Ca2+ ions play an important deregulation role in mitochondria, enhancing the uncoupling of oxidative phosphorylation and causing the reduction of mitochondrial membrane potential with consequent mitochondrial permeability transitional pore (MPTP) opening (Dirnagl et al., 1999). The latter process represents a crucial event for cell death, but its inhibition is considered as an important step for cytoprotection observed after preconditioning stimulus. In this scenario, nitric oxide (NO) and protein kinases have been proposed as possible MPTP regulators (Shiva et al., 2007; Zhao et al., 2006). In 2015, an epigenetic regulation of sodium/calcium exchanger isoform 1 (NCX1), respectively, by two functional protein complexes was reported: REST/Sp3/HDAC1/HDAC2 and HIF-1/Sp1/p300. In particular, whereas the former downregulates NCX1 expression during brain ischemia, the latter upregulates it during preconditioning. Notably, the development of drugs that epigenetically regulate NCX1 by preventing its downregulation in stroke might be a new pharmacological avenue to ameliorate neuronal damage during brain ischemia (Formisano et al., 2015).
4.7 Inflammatory cytokines
Other defensive mechanisms activated during ischemic preconditioning are represented by the inflammatory cytokines. Among cytokines, tumor necrosis factor-alpha (TNF-α) or interleukin-1 beta (IL-1β) have been implicated in the mechanisms of ischemic tolerance (Ohtsuki et al., 1996; Wang et al., 2000). Furthermore, NF-kB, which is activated by various signals such as oxidative stress and intracellular Ca2+ elevation, is involved in the induction of neuroprotective gene products such as MnSOD and Bcl-2, and pretreatment with its inhibitor abolishes the neuroprotective effect of preconditioning (Blondeau et al., 2001).
4.8 Heat shock proteins
The stress response of the brain to a noxious environment through heat shock proteins is recognized to be an important process underlying ischemic tolerance induction. These proteins exhibit protective effects in the neurons by reducing protein misfoldings, preventing ER stress response, scavenging reactive oxygen species (ROS), and blocking caspase-mediated apoptosis. In fact, HSP gene expression is greatly enhanced, and the role of HSPs is known to be essential for cell survival because the stress response avoids the accumulation of denatured proteins that arise from various stresses. In post-ischemic hippocampal neurons, the processing of denatured proteins is disturbed (Ide et al., 1999), leading to their accumulation. However, when ischemic preconditioning is induced by preceding ischemia, Hsp70 increases in the hippocampal CA1 pyramidal cells of ischemic animals (Kirino et al., 1991), and so its experimental manipulation induces ischemic tolerance by reducing accumulation of misfolded proteins (Nakata et al., 1993).
4.9 Miscellany of other effector
It has been observed that hypoxic preconditioning reduces post-ischemic leukocyte adherence and diapedesis secondary to downregulation of adhesion molecule RNA expression in cortical venules, thus reducing inflammation (Stowe et al., 2011). Another mechanism of hypoxic preconditioning-induced tolerance seems to be mediated by the upregulation of Bcl-2 and Bcl-x levels in hippocampus and cortex of rats, accompanied by a reduction in the extent of Bax upregulation caused by subsequent severe hypoxic injury (Rybnikova et al., 2006). Like all conditioning stimuli, hypoxic preconditioning promotes adaptive changes in all components of the so-called neurovascular unit, neurons, endothelium and glial cells, starting with the increase in vessel diameters of leptomeningeal anastomoses (Woitzik et al., 2006). The reduction of infarct volume by hypoxic preconditioning is also achieved by a postischemic cerebrovascular angiogenic response able to increase cerebral vascular density (Gustavsson et al., 2005). Surprisingly, hypoxic conditioning is also able to improve basal lamina integrity as well as pericyte structure/function. Recently, the role of VEGF following a hypoxic preconditioning stimulus has been clarified. In adult mice, antagonism of one of two receptors for VEGF, VEGF-R2, blocked hypoxic preconditioning-induced tolerance to transient focal stroke in adult mice, thus indicating that this receptor mediates the majority of pro-angiogenic effects (Fan et al., 2011). Finally, in a study carried out on a global ischemia model, it was demonstrated that IPC protected hippocampal CA1 neurons from delayed death when the test insult was induced 1 to 3 days after IPC, and this effect was mediated by an increase in the expression of neuronal Ku70, a DNA repair protein (Sugawara et al., 2001).
5. Pharmacological Brain Conditioning
A large range of agents can induce pharmacological conditioning: gas anesthetics, thrombin, erythropoietin, deferoxamine, erythromycin, opioids, and lipopolysaccharide. They all act as upregulating defense mechanisms in the brain, with EPO and thrombin acting as endogenous compounds able to modify stroke and LPS and inducing upregulation of endogenous defense mechanisms. Accumulating evidence suggest that other pharmacologic approaches for inducing tolerance may work by mechanisms quite similar to those induced by hypoxia (He et al., 2008).
5.1 Medical gases
Among different stimuli that may induce preconditioning are medical gases commonly used as general anesthetics in clinical practice (Gidday et al., 2006) ( 3).
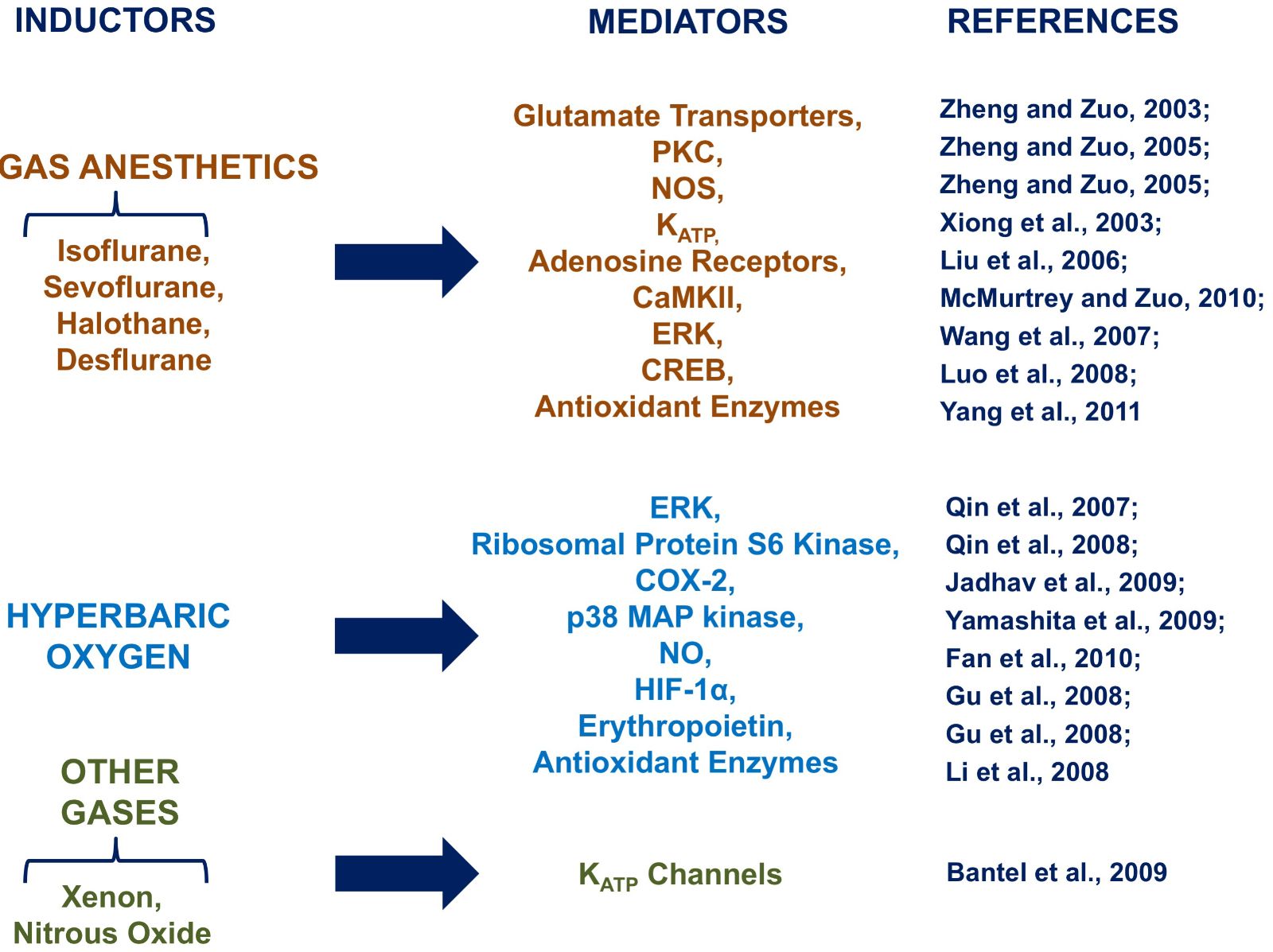
Multiple mechanisms have been proposed to contribute to isoflurane conditioning, including: signaling molecules, as free radicals (Sang et al., 2006); intracellular Ca2+ (Bickler et al., 2005); calcium/calmodulin-dependent protein kinase II (CAMKII) (McMurtrey and Zuo, 2010); and inducible NOS (Kapinya et al., 2002; Zhao and Zuo, 2004). The isoflurane preconditioning effect may also involve protein kinase C (PKC) and nitric oxide synthase (NOS) (Zheng and Zuo, 2005); however, ATP-sensitive potassium (KATP) channels may not play a role in this phenomenon (Zhang and Zuo, 2003), although conflicting results reveal its involvement in a delayed phase of preconditioning (Xiong et al., 2003; Kaneko et al., 2005). The role of adenosine in isoflurane conditioning has been recently demonstrated as A1 receptors have been shown to be implicated in an acute phase of protection in rats after focal brain ischemia (Liu et al., 2006). Inhibition of isoflurane-induced neuroprotection in rat cerebral slices by glutamate transporter inhibitors suggests a role of glutamate transporters in this effect (Zheng and Zuo, 2005; Wang et al., 2008). Similarly, glutamate transporters seem to be involved in the halothane-mediated preconditioning, at least in the acute phase of protection. Furthermore, sevoflurane-induced neuroprotection was inhibited by mitochondrial KATP channels, thus suggesting a role for these channels in this medical gas effect (Velly et al., 2009). It has recently been suggested in a study using rat hippocampal slices that ERK is also involved in sevoflurane conditioning (Wang et al., 2007). Molecules that are suggested to be involved in this protection include: free radicals (Yang et al., 2011), cAMP response element-binding (CREB) (Luo et al., 2008), and antioxidant enzymes (Yang et al., 2011). Interestingly, one published study suggests that inhibition of nuclear factor (NF)-kB, p38 MAP kinase, and subsequent neuroinflammation contributes to sevoflurane-induced neuroprotection (Wang et al., 2011). Moreover, since the protection is prevented by the use of an ERK inhibitor, this suggests a role for ERK in this protection (Ding et al., 2009).
With desflurane, the protection conferred was lost due to a glutamate transporter inhibitor (Wang et al., 2007), indicating that glutamate transporters may represent a common target in the preconditioning neuroprotection elicited by gas anesthetics.
Another mechanism of protection common to all gas anesthetics, including xenon and nitrous oxide, is the activation of mitochondrial KATP channels (Bantel et al. 2009; Weber et al. 2005).
Further, different molecular mechanisms have been suggested for hyperbaric oxygen (HBO)-mediated preconditioning. The role of kinases such as ERK and p38 MAPK (Qin et al., 2007; Yamashita et al., 2009) has been highlighted. Furthermore, HBO increases the expression of ribosomal protein S6 kinase, an enzyme that is involved in cerebral protein synthesis (Qin et al., 2008). Other identified mediators of HBO preconditioning are cyclooxygenase-2 (COX-2) (Jadhav et al., 2009, 2010) and nitric oxide (NO) (Fan et al., 2010). NO would induce autophagy activation and consequently increased expression of HIF-1α, (Gu et al., 2008), erythropoietin (Gu et al., 2008) and antioxidant enzymes (Li et al., 2008). In addition to the mechanisms discussed above, HBO has been shown to preserve regional cerebral blood flow and partial oxygen pressure in rat brain tissue with traumatic injury (Hu et al., 2010). It has been also supposed that free radicals can trigger HBO preconditioning effects, increasing expression of heme oxygenase 1 in primary spinal cord neuronal cultures (Li et al., 2007).
5.2 Thrombin
Thrombin is a protease that cleaves fibrinogen to fibrine in the coagulation cascade (Coughlin, 2000). Many of thrombin’s actions are mediated by protease-activated receptors (PARs), a kind of G-protein coupled receptor, activated by a proteolytic cleavage, expressed in neurons and astrocytes (Wang et al., 2002; Coughlin, 2000). Through these PARs, thrombin can affect cell shape, secretion, mobility, and metabolism (Coughlin, 2000). Other pathways that contribute to thrombin-induced brain injury are: the potentiation of NMDA receptors through activation of PAR1; the induction of endothelial hyperpermeability through Rho kinase and protein tyrosine kinase (van Nieuw Amerongen et al., 2000); the increase in substances as complement C9, TNF-α, matrix metalloproteinases; and upregulation of heat shock protein 27 (Hsp27). Among neuroprotective proteins activated by thrombin pathway is HIF-1α.
5.3 Erythropoietin
Erythropoietin is an endogenous cytokine able to mediate neuroprotection through its anti-apoptotic actions elicited by recruiting components of the Janus tyrosine kinase 2 pathway (JNK-2), increasing glutathione peroxidase, inhibiting recruitment of inflammatory cells and decreasing the production of various proinflammatory cytokines. Together these effects improve hemodynamics, stimulating angiogenesis and preventing compromise of the blood-brain barrier (Hasselbratt et al., 2006). Neuroprotective effects mediated by erythropoietin are seen within minutes and can last up to 3 days (Ruscher et al., 2002).
5.4 Deferoxamine
Deferoxamine is a potent iron chelator that has shown neuroprotective effects through a variety of mechanisms. Deferoxamine administered subcutaneously penetrated the blood-brain barrier and inhibited iron-mediated free radical formation, neurological deficits and levels of APE/Ref-1, a marker of oxidative DNA damage, in a rat model of intracerebral hemorrhage (Namura et al., 2003). Its administration has also been shown to significantly increase binding of HIF-α to DNA (Prass et al., 2003) thus reinforcing HIF-induced preconditioning neuroprotection.
5.5 Erythromycin
Erythromycin is an antibiotic of the macrolide family showing a beneficial effect when used as a preconditioning stimulus. The mechanism of action of this antibiotic as neuroprotectant is not completely clear; nevertheless, it is known to occur also through the reduction of cytokines, chemokines and iNOS, potent mediators of inflammatory neuronal damage (Koerner et al., 2007).
5.6 Opioids
Most commonly used for analgesia, opioids function by inhibiting nociceptive signal transmission (Lehmann, 1997). The mechanism of opioid preconditioning likely involves several pathways and has not yet been fully elucidated. It has been shown that preconditioning with morphine reduces lipopolysaccharide- and interferon-mediated injury to microglial cells in the brain () and improves blood flow to ischemic regions (Chi et al., 2010).
5.7 Lipopolysaccharide (LPS)
LPS is an integral component of the cell walls of gram-negative bacteria. One of the possible explanations for the neuroprotective role of this potent endotoxin is the induction of tumor necrosis factor-alpha and the stimulation of manganese superoxide dismutase. Both mechanisms may contribute to the reduction of free radical-mediated damage, finally inhibiting the apoptosis cascade (Mallard and Hagberg, 2007). LPS is also able to improve blood flow in the peri-infarct area minutes and days after occlusion (Furuya et al., 2005).
5.8 Anti-miRNA
MicroRNAs (miRNAs) are small (20-22 nucleotides) non-coding RNA able to regulate post-transcriptional gene expression by direct effects on messenger RNA (mRNA) translation. MicroRNAs have been shown to be regulated after non-harmful and harmful stimuli in the brain and to contribute to neuroprotective mechanisms. Several studies reveal that ischemic preconditioning regulates expression of miRNAs and their predicted targets in animals subjected to preconditioning, and further suggest that miRNAs could serve as effectors of ischemic preconditioning-induced tolerance (Saugstad, 2011). MicroRNAs are master regulators of gene-expression, and their regulation after a preconditioning stimulus could be causative of the general suppression of gene expression and make them a good candidate for future targeted therapies. Indeed, microRNA inhibitors, or anti-miRNA, represent a new class of compounds able to post-transcriptionally regulate targeted genes at the level of their RNA messengers. Among them, it has been recently shown that anti-miRNA-103 is capable of inducing a brain conditioning phenomenon in a rat model of transient brain ischemia (Vinciguerra et al., 2014). The mechanism of action of this locked nucleic acid (LNA) anti-miRNA blocked the detrimental increase of pathological miRNA-103-1 in the ischemic core responsible for the downregulation of neuroprotective plasma membrane sodium/calcium exchanger (NCX1) involved in the counteraction of sodium and calcium ion imbalance following stroke (Figure 4).
5.9 Beta-methylamino-L-alanine
Recently it has been shown that the cycad neurotoxin L-BMAA can be used as a preconditioning stimulus in SOD1 G93A mice and an in vivo model of amyotrophic lateral sclerosis (ALS). In fact, its administration can delay ALS progression by preventing the downregulation of sodium/calcium exchanger isoform 3 (NCX3), a membrane transporter able to handle the deregulation of ionic homeostasis occurring during ALS (Anzilotti et al., 2018).
5.10 TRAIL
TNF-related apoptosis-inducing ligand (TRAIL) is a member of the TNF superfamily released by microglia following focal brain ischemia. A recent study provided evidence that the neuroprotection elicited by ischemic preconditioning, consisting of 30 minutes of MCAO followed by 100 minutes of occlusion of the same brain artery (Pignataro et al., 2012), occurs either through upregulation of the TRAIL decoy receptor DcR2 or through downregulation of TRAIL and its death receptor DR5, indicating that PC prevents detrimental effects of TRAIL and sets into motion the cell survival machinery to rescue neurons from death (Cantarella et al., 2014).
5.11 Epigenetic modulators
The transcriptional repressor REST is a zinc finger protein that binds to a conserved 21-bp motif known as RE1 (repressor element 1, also called NRSE). In a recent paper it was shown for the first time that sodium/calcium exchanger isoform 1 (NCX1) is a new additional target gene for REST. In fact, it has been shown that this transcription factor selectively represses neuro beneficial NCX1 expression in the brain via the NCX1-RE1 sequence, causing a loss of neuroprotective effect obtained in a rat model of preconditioning performed by subjecting animals to 30 minutes of MCAO followed by 100 minutes of deleterious re-occlusion of the MCA (Formisano et al., 2013).
In this new promising field, recently it has been reported that resveratrol, a natural polyphenolic antioxidant with a well-documented epigenetic modifier activity, found in grape skin, grape products, and peanuts as well as in red wine, is able to influence stroke progression (Lanzilotta et al., 2013, 2015; Faggi et al., 2018) and, when used as a preconditioning stimulus, confers a long-lasting, 14-day neuroprotection (Khoury et al., 2016). Resveratrol’s long-lasting neuroprotective effect could be explained by the multitude of stroke-related pathways targeted by this drug. In particular, it has effects on NF-KB (Lanzilotta et al., 2013, 2015; Faggi et al., 2018) and on sirtuin 1, a NAD+-dependent histone deacetylase whose induction by resveratrol is able to reduce infarct volume as well as neurological deficits (Koronowski et al., 2017; Khoury et al., 2018).
6. Conclusions
The knowledge of molecular and cellular mechanisms underlying the induction and the maintenance of ischemic tolerance is still fragmentary; however, various studies have demonstrated that different events play key roles in the protection against ischemic stroke (Pignataro et al., 2009; Kirino, 2002).
Several mechanisms are involved in the protection mediated by brain conditioning, and some of them are strictly dependent upon the stimulus used to induce brain protection, while others are similar and among the most common preconditioning-stimuli (Figure 5).

In a new window | Download PPT
Figure 5: Shared mediators of neuroprotection elicited by different preconditioning stimuli.
Three proteins are important mediators of all types of preconditioning examined: TNF-α, HSP and HIF-α. Other proteins such as NO and ERK are considered mediators only of some types of preconditioning. Others, like ICAM-1 and survival factors, seem to be specific only for one typology of preconditioning. However, these observations need to be examined carefully as they could easily lead to incorrect conclusions. Indeed, it is possible that for some of these factors there are no studies to demonstrate their involvement in preconditioning. On the other hand, it is evident that the factors that appear to be consistently involved in protection represent excellent molecular targets to be further explored for possible future therapeutic applications.
The knowledge of such mechanisms may provide more direct opportunities for translational neuroprotection trials. Indeed, the present review, summarizing the specific pathways activated in different types of conditioning, would provide information about which mediators need to be activated or inhibited to protect the brain from ischemic injury.
7. Acknowledgments
This work was supported by the following grants: ConSLA from ARISLA to G.P.; POR Campania FESR 2007-2013 FARMABIONET (B25C1300023007) to G.P.; Programma Operativo Nazionale (PON_01602 and PON03PE_00146_1) from MIUR to L.A.; POR Campania FESR 2007-2013 MOVIE (B25C1300024007) to L.A.
8. Conflict of interest
The authors declare no competing financial interests.
References
Ornella Cuomo1#
1Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Via Pansini, 5, 80131, Naples, Italy.
Antonio Vinciguerra1#
1Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Via Pansini, 5, 80131, Naples, Italy
Pasquale Cepparulo1
1Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Via Pansini, 5, 80131, Naples, Italy
Serenella Anzilotti2
2IRCCS SDN Napoli, Naples Italy
Paola Brancaccio1
1Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Via Pansini, 5, 80131, Naples, Italy
Luigi Formisano1
1Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Via Pansini, 5, 80131, Naples, Italy
Lucio Annunziato2
2IRCCS SDN Napoli, Naples Italy
Giuseppe Pignataro1
1Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Via Pansini, 5, 80131, Naples, Italy
Corresponding author:
Giuseppe Pignataro
Email: giuseppe.pignataro@unina.it
#These authors contributed equally to this article.
In a new window | Download PPT
Figure 1: Neuroprotective molecular mechanisms and mediators activated by physical exercise-induced brain conditioning. The mechanisms inducing protection after exercise preconditioning are indicated with regards to pathways involved in inflammation, and in integrity of neurovascular unity, metabolic homeostasis and apoptotic death.
In a new window | Download PPT
Figure 2: Neuroprotective molecular mechanisms and mediators activated by hypothermia and hyperthermia-induced brain conditioning. The mechanisms inducing protection after thermal conditioning are indicated with regards to pathways involved in inflammation, integrity of neurovascular unity, synaptic function, free radical production and oxygen consumption.
In a new window | Download PPT
Figure 3: Neuroprotective molecular mechanisms and mediators activated by medical gas-induced brain conditioning. The mechanisms inducing protection after medical gas preconditioning are indicated with regards to pathways involved in inflammation, cell death, and ionic homeostasis.
In a new window | Download PPT
Figure 4: Neuroprotective molecular mechanisms and mediators activated by hypoxia/ischemia-induced brain conditioning. The mechanisms inducing protection in preconditioning induced by ischemia or hypoxia are indicated in the figure, particularly pathways involved in inflammation, free radical production, metabolic homeostasis and pro-survival pathways.
In a new window | Download PPT
Figure 5: Shared mediators of neuroprotection elicited by different preconditioning stimuli.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 13016 | 31 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA