Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
A Dual Role for Hyperbaric Oxygen in Stroke Neuroprotection: Preconditioning of the Brain and Stem Cells
Time:2018-06-30
Number:11970
Author Affiliations

Conditioning Medicine, 2018. 1(4):171-186.
Abstract
Stroke continues to be an extremely prevalent disease and poses a great challenge in developing safe and effective therapeutic options. Hyperbaric oxygen therapy (HBOT) has demonstrated significant pre-clinical effectiveness for the treatment of acute ischemic stroke, and limited potential in treating chronic neurological deficits. Reported benefits include reductions in oxidative stress, inflammation, neural apoptosis, and improved physiological metrics such as edema and oxygen perfusion, all of which contribute to improved functional recovery. This pre-clinical evidence has failed to translate into an effective evidence-based therapy, however, due in large part to significant inconsistencies in treatment protocols and design of clinical studies. While the medical community works to standardize clinical protocols in an effort to advance HBOT for acute stroke, pre-clinical investigations continue to probe novel applications of HBOT in an effort to optimize stroke neuroprotection. One such promising strategy is HBOT preconditioning. Based upon the premise of mild oxidative stress priming the brain for tolerating the full-blown oxidative stress inherent in stroke, HBOT preconditioning has displayed extensive efficacy. Here, we first review the pre-clinical and clinical evidence supporting HBOT delivery following ischemic stroke and then discuss the scientific basis for HBOT preconditioning as a neuroprotective strategy. Finally, we propose the innovative concept of stem cell preconditioning, in tandem with brain preconditioning, as a promising regenerative pathway for maximizing the application of HBOT for ischemic stroke treatment.
Keywords: preconditioning, ischemia, neuroprotection, neurodegeneration, cell therapy, regenerative medicine
Abstract
Stroke continues to be an extremely prevalent disease and poses a great challenge in developing safe and effective therapeutic options. Hyperbaric oxygen therapy (HBOT) has demonstrated significant pre-clinical effectiveness for the treatment of acute ischemic stroke, and limited potential in treating chronic neurological deficits. Reported benefits include reductions in oxidative stress, inflammation, neural apoptosis, and improved physiological metrics such as edema and oxygen perfusion, all of which contribute to improved functional recovery. This pre-clinical evidence has failed to translate into an effective evidence-based therapy, however, due in large part to significant inconsistencies in treatment protocols and design of clinical studies. While the medical community works to standardize clinical protocols in an effort to advance HBOT for acute stroke, pre-clinical investigations continue to probe novel applications of HBOT in an effort to optimize stroke neuroprotection. One such promising strategy is HBOT preconditioning. Based upon the premise of mild oxidative stress priming the brain for tolerating the full-blown oxidative stress inherent in stroke, HBOT preconditioning has displayed extensive efficacy. Here, we first review the pre-clinical and clinical evidence supporting HBOT delivery following ischemic stroke and then discuss the scientific basis for HBOT preconditioning as a neuroprotective strategy. Finally, we propose the innovative concept of stem cell preconditioning, in tandem with brain preconditioning, as a promising regenerative pathway for maximizing the application of HBOT for ischemic stroke treatment.
Keywords: preconditioning, ischemia, neuroprotection, neurodegeneration, cell therapy, regenerative medicine
1.0 Introduction
Stroke is defined as a sudden loss of blood supply to brain tissue resulting from either hemorrhagic or ischemic pathology causing severe neurological deficit (Jickling et al., 2014, George et al., 2017). In the United States, stroke is the fifth leading cause of death, with an occurrence rate of roughly 800,000 per year (George et al., 2017). Stroke can be broadly classified into hemorrhagic and ischemic stroke, with the latter accounting for 87% of all strokes (Go et al., 2014). Hemorrhagic stroke is overwhelmingly fatal, and thus provides a smaller therapeutic opportunity. Thrombolytic tissue plasminogen activator (TPA) is currently the only Food and Drug Administration (FDA)-approved intervention available for ischemic stroke, and is approved for use only within the first 4.5 hours following onset; delivery beyond this window is associated with a stark increased occurrence of severe hemorrhagic transformation (Knecht et al., 2017). Someone in the U.S suffers from stroke every 4 seconds, and with limited therapeutic options, new and effective stroke treatments are an urgent necessity.
Ischemic stroke pathology is characterized by abrupt blood vessel occlusion, causing ischemic damage to the area of the brain supplied by the occluded artery. During the acute phase, this primary anoxic environment induces a cascade of excitotoxicity, oxidative stress, and microglial activation throughout the infarcted region, resulting in extensive neural death (Stonesifer et al., 2017). Reactive oxygen species (ROS) weaken vasculature and create ionic imbalances, which lead to an abnormal increase of water movement into the intracellular space, resulting in edema (Stokum et al., 2016). In the subacute phase, cytokines, chemokines, and matrix metalloproteases (MMPs) are released, contributing to neuroinflammation (Lakhan et al., 2009). Elevated expression of MMPs increases blood-brain barrier (BBB) permeability, allowing migratory waves of leukocytes into the infarct area and exacerbating inflammatory activity (Lakhan et al., 2009). Multiple cell phenotypes which comprise the neurovascular unit (Lo et al., 2009) within the penumbra – the area of brain tissue surrounding the infarcted core – are susceptible to the abovementioned pathological mechanisms as well. These cells, however, are salvageable, and their protection is of great interest in rescuing the motor and cognitive functional deficits which follow stroke (Wetterling et al., 2016).
Hyperbaric oxygen treatment (HBOT) is a non-invasive therapy performed in a pressurized chamber where pure oxygen is administered at above-normal atmospheric pressure. This condition improves oxygenation from lungs to systemic organs, enhancing biomolecular processes specifically in ischemic conditions (Thom, 2011). Under increased oxygenation in the infarct core and penumbra, HBOT can reduce secondary brain injury effects such as apoptotic pathway initiation, oxidative stress, and rampant inflammation (Ostrowski et al., 2017). HBOT has shown promising restorative effects in a wide range of pathological contexts, including within TBI, SCI, stroke and other non-neurological maladies such as carbon monoxide poisoning, gangrene, and arterial gas embolism (Baratz-Goldstein et al., 2017). Among the neurological pathologies, in general, few effective treatment options are offered and severe debilitation and/or death are common outcomes. Current HBOT experimental studies have demonstrated improvement in facilitation of cerebral oxygenation, metabolism, angiogenesis, and reduction in inflammation in stroke and non-stroke disease states (Zhai et al., 2016). HBOT studies aim to effectively attenuate ischemic-related damage, as well as utilize various other mechanisms, all in an effort to improve the quality of life of affected patients. Here, we review the current status of HBOT for ischemic stroke, present scientific support and rationale for HBOT preconditioning in the stroke brain, and finally propose HBOT priming of stem cell transplantation as a promising and novel paradigm for stroke therapy.
2.0 HBOT in Ischemic Stroke: Potential Benefits and Limitations
Early investigations began 50 years ago on the results of hyperoxia following stroke; however, HBOT was viewed as more dangerous than beneficial, and the application of its therapeutic value was abandoned. In the last 20 years, this tapered view of HBOT has shifted towards a favorable assessment of its potential application in stroke as further research has revealed the ability of HBOT to reduce the severity of infarct volume if administered during the reperfusion window (Hu et al., 2017). This, however, creates a very limited opportunity for effective treatment as determination of reperfusion will require imaging modalities to inform the appropriate course of action, and by the time such assessment is completed the narrow effective timeframe for HBOT has likely passed (Chang et al., 2000). Other studies showed that delayed HBOT past the effective window of treatment resulted in worse outcomes versus the normobaric groups, potentially due to the role of ROS in glutamate-induced excitotoxic cell death (Singhal et al., 2002). Thus, indiscriminate use of HBOT is inadvisable, as treatment with excess oxygen can cause additional harm, such as obstructive pulmonary disease (Singhal et al., 2002).
Normobaric oxygen has also been critically evaluated as a potential treatment for stroke, with therapeutic efficacy being both reported and refuted (Padma et al., 2010; Singhal et al., 2005; Shi et al., 2016). While normobaric oxygen therapy does increase dissolved O2 available within the blood, it has not been demonstrated to exert other neuroprotective effects which are central to HBOT effectiveness both post- and pre-stroke, and is thus less intriguing when exploring preconditioning paradigms (Padma et al., 2010; Singhal et al., 2005; Shi et al., 2016).
To date, two primary methods of utilizing HBOT have been employed -- post-stroke and preconditioning. Following stroke, the goal of HBOT is to induce hyperoxia during the ischemic and reperfusion periods or expose the subject to repeated treatments once past the initial early treatment window (Zhai et al., 2016). Current preconditioning treatments focus on exposing the individual (albeit endogenous cells) to a mild stressor, therefore increasing tolerance to future stressors (Godman et al., 2010).
2.1 Preclinical functional outcomes of post-stroke HBOT
A typical HBOT treatment is conducted at 2.5 atmospheres (ATM) for a period of 60-90 minutes (Ostrowski et al., 2016), although protocols vary outside of this framework. If the pressure exceeds 2.5 ATM during HBOT, oxygen toxicity as well as increased oxidative stress can present throughout the body. Moreover, as the pressure increases above the recommended level, the risk of seizure activity increases drastically (Zhai et al., 2016). The primary goal of acute HBOT treatment is to increase oxygen levels in the ischemic region during stroke occlusion, in pursuit of minimizing hypoxic damage. Once past the initial treatment window, repeated HBOT treatments over several days have been shown to promote stimulation of endogenous repair processes (Wang et al., 2008).
The potency of HBOT appears to wane significantly as treatment initiation is delayed. Following an ischemic event, several studies have demonstrated effectiveness of HBOT when administered within 30 to 60 minutes of stroke, as evidenced by reduced infarct volume and improved behavioral scores (Chang et al., 2000; Hu et al., 2017). While useful as a proof-of-concept, this highly acute treatment timeframe provides little translational value. Importantly, though, investigations have found effectiveness with HBOT at less restrictive time points, such as HBOT (2.5 ATA for 2 hours) at 6 hours after reperfusion (Yin et al., 2003), and HBOT (3 ATA, 1 hour) at 3 and 6, but not 12, hours after reperfusion (Lou et al., 2004). While single-session HBOT has been reported as effective when initiated up to 18 hours (Xue et al., 2008) and 48 hours (Mu et al., 2013) after stroke, therapeutic effects with HBOT at more extended timepoints (i.e. 12+ hours after reperfusion) will generally necessitate more extensive and tailored treatment protocols. Delayed repeat treatment, with HBOT (2.5 ATA, 2 hours) initiated at 6 or 24 hours following stroke and delivered for 6 consecutive days, resulted in amelioration of infarct size and neurological deficits (Yin et al., 2005). Another report found significant chronic improvement in behavioral and histological outcomes following MCAO when exposed to an aggressive regimen of 15 HBOT sessions (2.5 ATA, 90 min) 5 times per week over 3 weeks, initiated 24 hours after stroke (Lee et al., 2013). Scarce evidence exists for the effectiveness of HBOT when initiated more than 48 hours post-stroke; a single study using a severe model of focal ischemia found that one session of HBOT (3 ATA, 1 hour) exhibited significant neuroprotective effects when delivered up to 72 hours post-stroke (Veltkamp et al., 2005).
2.2 Clinical results of HBOT in stroke
There have been only a handful of randomized controlled clinical trials involving HBOT to date, resulting in inconclusive results (Bennett et al., 2014). Many different factors likely contribute to the varying clinical results, including the unstable clinical status of acute stroke patients, preventing them from receiving HBOT during the early effective treatment window of 3-5 hours in humans (Sanchez, 2013). Moreover, wide variations in treatment protocols and stage of patient enrollment (i.e. more acute, sub-acute and chronic) likely inhibit the ability to draw reliable conclusions and compare outcomes across trials. Nonetheless, clinical success for HBOT has been achieved; for example, the use of HBOT has been shown to reduce levels of cerebral and myocardial biomarkers and reduce the length of intensive care unit stays (Li et al., 2011). Although clinical trials for HBOT have not been deemed overwhelmingly successful, the potential clinical significance of HBOT cannot be ignored, and further trials to elucidate the best methods should be pursued (Hu et al., 2016). With an increasing number of clinical trials being conducted, cerebral plasticity has been identified as a benefit of HBOT; however, these trials lack the necessary results to unequivocally conclude efficacy (Zhai et al., 2016). In a recent clinical study, the efficacy of HBOT in restoring memory function for chronic stroke patients was examined, revealing significant memory improvement following HBOT treatment, which was accompanied by an increased brain metabolic rate (Ploughman et al., 2015). Another clinical study utilized HBOT 5 days per week for patients with chronic stroke, and resulted in significant improvements in memory and attention testing scores (Hadanny et al., 2015). The timing of HBOT application has been examined through several clinical studies. Consistent with the preclinical studies discussed above, these studies found that the earlier HBOT is initiated in relation to the initial ischemic event, the greater its therapeutic effectiveness (Chang et al., 2000; Ding et al., 2014). Once past 12 hours post-ischemia, the benefits of single treatment HBOT are drastically reduced, although the application of repetitive HBOT use in the sub-acute stroke brain has documented neurogenic effects (Hu et al., 2014). Apparent from the completed clinical trials of HBOT for stroke is the consistent safety of this therapeutic strategy, despite inconsistent efficacy. These studies elucidate the clinical potential of using HBOT for chronic stroke patients, but the data are far more varied in acute stroke patients.
3.0 Unpacking the Mechanisms of Action of HBOT in Stroke
3.1 Physiological and metabolic effects
Neurons are particularly susceptible to oxygen deprivation, and thus the primary mechanism of acute HBOT delivery is to increase perfusion and oxygenation of at-risk tissue. Studies have consistently demonstrated that HBOT therapy can enhance arterial oxygen saturation and increase tissue oxygen content through enhanced cerebral microcirculation (Matchett et al., 2009; Zhai et al., 2016). Moreover, HBOT enhances BBB stability through various molecular mechanisms including MMP regulation (Ding et al., 2014), decreases intracranial pressure, and relieves cerebral edema (Zhai et al., 2016). Secondary effects of HBOT in the stroke brain may result from metabolic regulation such as reducing extracellular glutamate levels, which contribute to excitotoxic death and neural dysfunction (Gao-Yu et al., 2011).
3.2 Antioxidant effects
As will be discussed later, introducing high O2 levels can actually induce oxidative stress; however, in an apparent paradox, HBOT has been consistently shown to confer oxidative protection against stroke-induced ROS and nitrosative species (Li et al., 2008). HBOT treatment following stroke has been shown to reduce the levels of pro-oxidative enzymes such as malondialdehyde and to increase the antioxidant activity of CAT and SOD (Li et al., 2008). Other studies have found reduced stroke-generated ROS, such as hydroxyl free radicals in the striatum following HBOT therapy (Yang et al., 2010). Complex effects of HBOT on nitric oxide synthase have also been implicated in its antioxidant protective properties (Zhou et al., 2012). The counterintuitive effects of HBOT on reducing oxidative damage may result both from our incomplete understanding of HBOT mechanisms and from variations in experimental pressure and duration of treatment sessions. Moderate treatment protocols may simultaneously induce a degree of oxidative stress yet compensate for this through other antioxidant mechanisms. Regardless, additional studies into the intricacies of HBOT’s effects on oxidative pathways are necessary.
3.3 Anti-inflammatory effects
Runaway inflammation is recognized as a cornerstone of stroke secondary cell-death pathology, and its sequestration is known to improve functional outcomes (Borlongan et al., 2012). General markers of inflammation, such as tumor necrosis factor alpha, have been observed to decrease in HBOT-treated animals following stroke (Yu et al., 2015), while specific subpopulations of immune cells, such as CD40+ microglia, have also been demonstrated to decline following HBOT (Lavrnja et al., 2015). Inhibition of leukocyte accumulation within the ischemic area was found in HBOT-treated animals and was attributed to a reduction in the levels of inflammatory chemokines (Rink et al., 2010). Similarly, a study found that HBOT reduced myeloperoxidase activity – an indirect measure of inflammatory response – and inhibited neutrophil infiltration (Miljkovic-Lolic et al., 2003). The COX-2 signaling pathway has been proposed as a possible underlying mechanism of the HBOT-mediated reduction in inflammation (Cheng et al., 2011).
3.4 Additional neuroprotective mechanisms
A plethora of molecular pathways have been shown to be modulated by HBOT, which contribute to reduction in apoptosis and preservation of neural tissue. Many of these are intimately connected with the mechanisms described in this section, as neuronal apoptosis is an endpoint outcome commonly resulting from ROS, metabolic restriction, and inflammatory response; however, discrete pathways may also contribute to the protective effects of HBOT. Among those reported are a reduction in HIF-1α (Sun et al., 2008), decreased cortical and hippocampal caspase-3 (Calvert et al., 2003), increased growth factors such as GDNF and NGF (Zhang et al., 2012), and mitochondrial regulation (Lou et al., 2006). Finally, direct effects on glial cells have been suggested to assist in preserving susceptible neurons (Gunther et al., 2005).
4.0 Implications of HBOT in Other Neurological and Non-neurological Conditions
4.1 HBOT in acute and chronic TBI
A large number of studies have demonstrated the safety and effectiveness of HBOT in diverse models of traumatic brain injury (TBI), especially within the acute phase (Zhang et al., 2012; Lim et al., 2013; Wee et al., 2015; Lim et al., 2017). One such study evaluated HBOT (2.8 ATA, 45 min, twice a day for three consecutive days) initiated 3 hours after a dynamic cortical deformation model of TBI, and revealed significant histopathological alterations indicating HBOT effectiveness (Vlodavsky et al., 2006). In another study, mice with moderate closed head weight drop traumatic brain injury (mTBI) treated with HBOT (2 ATA, 60 min, four consecutive days) initiated at either 3 hours or 7 days post-injury exhibited significant recovery of learning and cognitive abilities compared to non-treated controls, displaying performance comparable to sham mice (Baratz-Goldstein et al., 2017). Despite wide-ranging variations in pre-clinical and clinical HBOT protocol, a meta-analysis of its clinical application in acute TBI revealed legitimate and consistent effectiveness in conferring neuroprotection (Wang et al., 2016). Clinical evaluations have revealed the potential of HBOT to confer neurorestorative effects in the chronic TBI brain as well; a recent study which initiated HBOT at 6 months to 27 years post-injury in human patients found upregulated angiogenesis and cerebral perfusion associated with improvement in memory, executive functions, information processing speed, and global cognitive scores as measured by an objective computerized exam (Tal et al., 2017). Additional studies of delayed HBOT for chronic TBI have supported these findings (Harch et al., 2012; Boussi-Gross et al., 2013; Tal et al., 2015; Harch et al., 2017), while other studies have provided discrepant results, such as HBOT (1.5 ATA, 90 min) given 60 days post-TBI for 15 consecutive days resulting in no beneficial effects (Yang et al., 2014).
4.2 HBOT in spinal cord injury
Several studies have demonstrated that spinal cord injuries (SCIs) are exacerbated more by secondary injury response mechanisms than by the primary insult (Geng et al., 2015). Secondary cell death mechanisms initiate a cascade of biomolecular events inducing reactive oxidative damage, astrocytic glial scarring, and infiltration of glia, lymphocytes, activated monocytes, and phagocytic macrophages, which may be amenable to HBOT (Oyinbo, 2011). Interestingly, HBOT experimental studies have shown positive therapeutic effects in SCI by providing a neuroprotective microenvironment that decreases anoxic conditions and enhances neuronal regeneration (Wang et al., 2014). For example, a recent report found significant positive alterations in oxidative pathway enzymes, apoptotic markers, and inflammatory mediators (Shams et al., 2017). Other studies have found preservation of BBB integrity following SCI and reversal of motor deficits (Sun et al., 2017). Patients with SCI treated with HBOT exhibited significant improvement of neurological function compared to control group patients, as well as preservation of various neuron physiological functions, such as evoked potential amplitude and conduction velocity (Tan et al., 2018). Postulated mechanisms of action of HBOT in SCI mirror those to be discussed later, but include increased expression of vascular endothelial growth factor (VEGF), axonal regeneration, and decreased apoptosis (Fu et al., 2017).
4.3 HBOT in other pathological contexts
Various disease pathologies that involve molecular cascades accompanied by oxidative stress, inflammation, and ischemia have found therapeutic effects from HBOT (Daruwalla et al., 2006; McDonagh et al., 2007; Danesh-Sani et al., 2012; Borab et al., 2017). Patients with maladies involving tissue hypoxia, including diabetic ulcers, have been examined as HBOT candidates (Londahl, 2012; Londahl, 2013), and acute coronary syndrome benefits from HBOT (Shuvy et al., 2013; Bennett et al., 2015). In addition, disease-specific mechanisms can underlie HBOT effectiveness, such as the antimicrobial properties of HBOT for necrotizing soft tissue infections (Bhutani et al., 2012) or increased gas-dissolution for air embolisms (Perez et al., 2017). Psychological symptoms, such as post-traumatic stress disorder (PTSD) and post-concussive syndrome, which may develop secondary to brain injury, have also displayed favorable responses to HBOT (Boussi-Gross et al., 2013). One study found a reduction in PTSD symptoms following 40 sessions of HBOT (1.5 ATA, 60 min) over the course of 30 days (Harch et al., 2009; Harch et al., 2012). Treated subjects experienced improvements in cognitive function, decreased anxiety/depression, and improved cerebral vascular blood flow in the white matter region (Harch et al., 2017). Finally, spontaneous physiological abnormalities including autism spectrum disorders have been reported as amenable to HBOT (Rossignol et al., 2006), although the evidence for these claims is controversial (Xiong et al., 2016). In summary, apparent from the extensive research into HBOT for non-stroke disorders is its consistent safety profile when delivered within normal protocols for appropriate disease indications.
5.0 Pre-clinical Findings with HBOT Preconditioning for Stroke
A growing body of evidence indicates the therapeutic potential of HBOT delivered prior to ischemic onset (Hu et al., 2016). The introduction of hyperoxia to a healthy brain induces mild oxidative stress, enabling endogenous cells to develop a greater tolerance to a future insult. Concerns regarding the efficacy of HBOT following stroke have been raised due to oxygen’s role in pathways associated with worse outcomes following stroke, such as glutamate-induced excitotoxic cell death (Singhal et al., 2005), as well as general lack of consistent and effective clinical HBOT treatment protocols. However, HBOT preconditioning could circumvent these concerns as mild oxidative stress is not coupled with more significant cerebrovascular events that initiate various apoptotic pathways; rather, HBOT conditions the cells to withstand future cerebrovascular events and their accompanying oxidative damage (Ostrowski et al., 2016). Patient populations at a particularly high risk for stroke (such as those with combinations of key factors such as morbid obesity, hypertension, low physical activity, diabetes mellitus, smoking, history of stroke, etc.) may benefit from preemptive HBOT. Similarly, patients who are diagnosed with carotid artery plaque are considered high-risk patients and could be candidates for preconditioning strategies to minimize damage from potential future strokes (Zhang et al., 2017). Indeed, as the ability of clinicians to accurately predict probable stroke cases advances via imaging techniques, such as magnetic resonance T2 mapping (Chai et al., 2017), preconditioning therapies, in particular HBOT, will undoubtedly increase in value.
The first published pre-clinical study of HBOT preconditioning in ischemic stroke reported that this strategy conferred ischemic tolerance and prevented neuronal death within the hippocampus of the gerbil brain following forebrain ischemia inflicted 48 hours after the final session (Wada et al., 1996). Subsequent studies showed that HBOT was protective against transient, but not permanent, stroke in a dose-dependent manner, and that a regimen of 5 treatments (2.5 ATA, 1 hour) over consecutive days was more effective than 3 treatments over consecutive days in rescuing functional deficits when initiated 24 hours before transient middle cerebral artery occlusion (MCAO) (Xiong et al., 2000). Independent laboratories have supported these findings, with one study, for example, demonstrating that HBOT (2.5 ATA. 1 hour, twice daily) conferred neurological and histopathological protection from MCAO inflicted 24 hours after the final session (Li et al., 2008). Other investigators have opted for more aggressive treatment protocols – such as 3.5 ATA for 1 hour, five consecutive days – finding significant histopathological signals of neuroprotection following forebrain ischemic stroke inflicted 12 hours after the final HBOT session (Yamashita et al., 2009). The preemptive “therapeutic window” of HBOT has also been explored, with HBOT delivered 24 hours before injury bestowing neuroprotection, but not at 72 hours (Hirata et al., 2007). Importantly, the length of this window may be a function of the intensity and number of HBOT sessions delivered prior to injury.
The mechanisms underlying the effects of HBOT undoubtedly involve several pathways that work in parallel, or independently, to induce preconditioning in the brain (Francis et al., 2017). While the mechanisms of post-injury HBOT were mentioned previously, in the following sections we present a detailed overview of the preclinical studies describing the mechanisms of action proposed to confer therapeutic protection for HBOT delivered before injury onset.
5.1 Preparation for oxidative stress
The chief mechanism of action for HBOT implicated to confer neuroprotection appears to be its action as an oxidative preconditioning agent (Gao et al., 2017). Prolonged hypoxia contributes to oxidative stress, antioxidant system imbalance, and eventual tissue injury. Preconditioning with HBOT can exert a protective role by priming brain tissue for oxidant stress, making it less susceptible to stroke-induced injury mechanisms. It is widely accepted that increased production of ROS and reactive nitrogen species (RNS), such as peroxynitrite or NO2, makes a major contribution to the development of CNS oxygen toxicity. Cells have evolved several defense strategies against ROS involving antioxidant enzymes, including superoxide dismutase (SOD) to scavenge superoxide, catalases (CAT) and peroxidases to break down hydrogen peroxide, and glutathione S-transferase to neutralize lipid peroxides, as well as their auxiliary enzymes, glutathione reductase (GRX) and glucose-6-phosphate dehydrogenase (G6PD). Low levels of ROS stimulate adaptive responses by increasing the cellular activity of these enzymatic antioxidants, while pathological levels of ROS, such as those produced under conditions of hyperoxia, can overwhelm the antioxidative capacity of the cells and cause oxidative injury. This oxidative stress manifests as protein oxidation, DNA damage with increased mutational rates, and lipid peroxidation, resulting in membrane damage, metabolic perturbation and death (Pisoschi et al., 2015). HBOT produces an elevated O2 partial pressure and increased mitochondrial generation of H2O2, elevating ROS production (Hu et al., 2016). However, several lines of investigation have shown that HBOT induces an initial oxidative stress that acts as a trigger mechanism prompting anti-oxidative responses (Gao et al., 2017).
In a model of focal cerebral ischemia, HBOT preconditioning induced an increase in the activity of SOD and CAT in the brain tissue associated with decreased mortality rate, improved neurological recovery, and lessened neuronal injury (Li et al., 2008). In addition, malondialdehyde (MDA) content, a marker of lipid peroxidation and oxidative stress, decreased in the ischemic penumbra and hippocampus (Li et al., 2008). Since HBOT preconditioning stimulates ROS production, one possible explanation is that modest levels of ROS stimulate compensatory increases of CAT and SOD, which scavenge excessive ROS and attenuate the lipid peroxidation following stroke (Li et al., 2008). Moreover, in a spinal cord ischemia animal model, HBOT preconditioning similarly increased CAT and SOD activities, with the administration of a CAT inhibitor (3-amino-1,2,4-triazole) before ischemia, attenuating the spinal cord ischemic tolerance induced by HBOT preconditioning (Nie et al., 2006). In addition, administration of a free radical scavenger, dimethylthiourea, before preconditioning reversed the increased activities of both enzymes in spinal cord tissue (Nie et al., 2006). The results indicate that an initial oxidative stress upregulates the antioxidant enzyme activities, playing an important role in the formation of the tolerance against ischemic injury by HBOT preconditioning.
Repeated exposure to non-convulsive HBOT provides protection against central nervous system (CNS) oxygen toxicity by decreasing levels of GRX and G6PD, and conferring a significant increase in glutathione peroxidase (GSH-Px) activity, as well as by increasing glutathione S-transferase (GST) activity (Arieli et al., 2014). G6PD catalyzes the oxidation of glucose to generate NADPH from NADP+ while NADPH-oxidase catalyzes the production of superoxide by oxidation of NADPH (Arieli et al., 2014). Hence, by downregulating G6PD activity, HBOT may indirectly reduce oxidative stress. In addition, the decrease of G6PD activity is associated with a downregulation in the activity of GRX and an increase in GSH-Px, indicating that HBOT may upregulate antioxidants and downregulate pro-oxidant enzymes.
In the healthy brain, HBOT induces an increase of heat shock proteins (HSPs), which play important roles in cellular repair and protective mechanisms. HBOT preconditioning has been shown to upregulate HSP70 specifically (Ni et al., 2013), which exerts protective effects including prevention of protein aggregation, refolding of partially denatured proteins, reduction of inflammatory responses, and inhibition of apoptosis (Brown, 2007). Moreover, in vitro studies have shown that HBOT preconditioning protects neurons against oxidative injury and oxygen-glucose deprivation (OGD) by upregulating HSP32 expression (Li et al., 2008; Huang et al., 2014). HSP32, also named heme oxygenase-1, degrades heme into three products: carbon monoxide (CO), ferrous iron, and biliverdin. Free heme is produced mainly through the oxidation of hemoproteins, including hemoglobin, myoglobin, and neuroglobin. In the center of heme is a Fe atom that can react with H2O2 and gives rise to toxic hydroxyl radicals. Catalysis of heme by HSP32 produces ferritin release, and its accumulation provokes iron sequestration and thus may provide protection against oxidative damage (Li et al., 2008; Huang et al., 2014). In addition, ROS and NO are two well-established inducers of HSP32; of note, HBOT-induced HSP32 expression is mediated via the ROS/p38 MAPK/Nrf2 pathway and by MEK1/2/Bach1-mediated negative regulation (Huang et al., 2016). Oxidative stress resulting in free radical generation should encourage HSP expression, as these studies confirm. However, a study showed no induction of HSP72 expression within peripheral blood mononuclear cells (PBMC) following a single HBOT exposure in healthy males, indicating the importance of cell-specific response to HBOT (Vince et al., 2010).
Another protective effect of HBOT preconditioning against oxidative stress may involve expression of many Nrf2-regulated antioxidant genes. The Nrf2 signaling pathway has the potential to activate over 200 antioxidant and cytoprotective genes (Srivastava et al., 2013). HBOT preconditioning was shown to increase the levels of Nrf2 and enhance some of its target genes such as key proteins for intracellular GSH synthesis and transit (GST, GCL, cGT and MRP1), molecular chaperones (HSP32 and HSPA1A), and anti-oxidative enzymes (SOD1, GST) (Xu et al., 2014; Huang et al., 2016; Perdrizet, 2016; Xue et al., 2016; Zhai et al., 2016). The neuroprotective mechanism of HBOT preconditioning is also mediated by upregulating SirT1 expression in at least three different ways: (1) upstream regulation for fasting-induced activation of the Nrf2 pathway by affecting the activity of the PPAR-ƴ/PGC1-1α complex that binds to Nrf2 promoter and activates its expression; (2) inhibition of apoptosis by increasing the protein expression of anti-apoptotic Bcl-2, decreased pro-apoptotic cleaved caspase-3, deacetylating p53; (3) upregulation of FoxO, promoting the expression of SOD and CAT in response to oxidative stress (Zeng et al., 2012; Yan et al., 2013; Bian et al., 2015; Xue et al., 2016; Ding et al., 2017).
Exposure to HBOT is associated with increased levels of nitric oxide (NO) (Goldstein et al., 2006; Liu et al., 2008; Arieli et al., 2014). NO acts as an important neurotransmitter and plays a dual role in both neuroprotection and neurotoxicity depending on the NO synthase (NOS) isoform, the cell type by which it is produced, as well as the temporal stage after ischemic onset (Chen et al., 2017). Immediately after brain ischemia, NO release from endothelial NOS (eNOS) is protective mainly by promoting vasodilation; however, after ischemia develops, NO produced by overactivation of neuronal NOS (nNOS) and expression of iNOS both contribute to brain damage. While nNOS-derived NO decreases neurogenesis, NO produced by eNOS and iNOS seems to stimulate it (Sawada et al., 2009). NO, as a vasodilator of cerebral vessels, can increase tissue oxygenation, yet may also increase the delivery of ROS to tissue. In addition, NO can combine with oxygen radicals to form the potent oxidant peroxynitrite and induce nitrosative stress. However, it was shown that HBOT preconditioning has a protective effect involving alterations in the enzymatic activity of the antioxidant system and lower levels of peroxynitrite mainly in the hippocampus (Arieli et al., 2014).
The increase of eNOS and nNOS mRNA/proteins in hypothalamus and hippocampus, as well as the elevated NO, may enhance the sensitivity to convulsions after repeated HBOT exposures, potentially leading to seizures during the subsequent oxygen exposures (Liu et al., 2008). Interestingly, the increase of Mn-SOD, CAT and Bcl-2 and the reduction of apoptosis seem to be mediated by NO, because the neuroprotective effect of HBOT preconditioning is abolished by a nonspecific NOS inhibitor, L-NAME (Wang et al., 2009). These results suggest that NO after HBOT preconditioning exerts both neuroprotection and neurotoxicity, indicating that further studies are needed to better understand the role of NO in HBOT preconditioning.
5.2 Reduction of apoptosis, activation of autophagy, and promotion of cell survival
ROS can react with macromolecular components and induce the cells to undergo necrosis or apoptosis. Inhibition of thioredoxin reductases (TrxR), one of the major redox systems in cells involved in the control of cellular redox balance, has been shown to result in (Li et al., 2009) generation of ROS and induced cell apoptosis in neuronal cell lines (Seyfried et al., 2007). In a post-traumatic stress disorder (PTSD)-induced rat model, HBOT preconditioning upregulated the expression of TrxR-1 and TrxR-2 mRNA in the hippocampus concurrent with a reduction in neuronal apoptosis and preserved viable neurons (Peng et al., 2010). Further evidence demonstrated that HBOT preconditioning lessens apoptosis via mitochondrial pathway modulation. In particular, a decrease in the activity of capase-3 and -9, and reduced cytoplasm cytochrome c levels were shown to upregulate the ratio of Bcl-2 and Bax proteins associated with reduced brain edema, decreased infarction volume, and improved neurological recovery (Li et al., 2008; Li et al., 2009; Wang et al., 2010; Lu et al., 2012; Lu et al., 2013). Moreover, the reduction in apoptosis was associated with Mn-SOD and CAT increase and elevated NO after HBOT preconditioning (Wang et al., 2009). Additionally, HBOT preconditioning reduces early apoptosis and apoptosis progression through induction of BDNF and suppression of p38/MAPK phosphorylation (Ostrowski et al., 2008; Yamashita et al., 2009). As mentioned above, HBOT preconditioning can limit apoptosis through the upregulation of SirT1 expression, which increases expression of anti-apoptotic Bcl-2, decreases pro-apoptotic cleaved caspase-3, and deacetylates p53 (Yan et al., 2013).
Interestingly, ROS also regulate starvation-induced autophagy, which is clearly a survival mechanism, partly through the class III phosphoinositide 3-kinase pathway (Wang et al., 2010). It was shown that HBOT preconditioning significantly increases the level of protein expression of LC3-II and Beclin 1 and induces autophagosome formation in the ischemic penumbra following ischemia in rat brain (Yan et al., 2011). Therefore, autophagy can be activated by ROS and may confer neuroprotection. Another mechanism by which HBOT preconditioning can promote cell survival is by reducing matrix MMP-9 activity/tissue expression, which plays a deleterious role after global cerebral ischemia. HBOT preconditioning suppressed post-ischemic MMP-9 activity, CA1 cell damage, and improved functional performance (Ostrowski et al., 2010).
Finally, HBOT preconditioning can promote proliferation and counter cell loss through different mechanisms including activation of the Wnt signaling pathway, secretion of vascular endothelial growth factor (VEGF) and upregulation of HIF-1 (Wang et al., 2007). It was shown that the levels of VEGF, VEGFR2, Raf-1, MEK1/2, and phospho-extracellular signal-regulated kinase (ERK) 1/2 protein were boosted by HBOT, which was associated with NSC proliferation and migration to the lesion area, and improved neurological function (Yang et al., 2017).
5.3 Immunosuppression and immunopreparation
Reducing and preventing aberrant inflammation is another prominent mechanism of HBOT preconditioning (Hu et al., 2016). HBOT preconditioning was shown to reduce the expression of neurotoxicity microglia alongside a decrease in TNF-α and neuronal degeneration, all of which is connected with reduced cerebral edema and amelioration of motor dysfunction after intracerebral hemorrhage (Yang et al., 2015). Moreover, HBOT preconditioning reduced cyclooxygenase-2 (COX-2) expression (which is involved in post-ischemic neuroinflammation), increased the level of surviving neurons in the CA1, attenuated post-operative brain edema, and improved neurological outcomes after global ischemia and surgical brain injury (Jadhav et al., 2009; Cheng et al., 2011). Preconditioning with HBOT also regulates the expression of Osteopontin (OPN), which was shown to reduce the expression of interleukin (IL)-1β/nuclear factor-κ-gene binding (NFκB) and to augment protein kinase B (Akt) (Hu et al., 2015). HBOT preconditioning could significantly mitigate cognitive impairment and preserve other physiological functions via reduction of systemic and hippocampal pro-inflammatory cytokines and caspase-3 activity (Gomez et al., 2012; Sun et al., 2014).
5.4 Preservation of blood-brain barrier, edema minimization, and angiogenesis
HBOT preconditioning has been shown to attenuate brain edema and improve neurological outcomes following surgical brain injury (SBI), ischemic and hemorrhagic stroke, TBI and high altitude exposure (Qin et al., 2007; Hu et al., 2008; Jadhav et al., 2010; Lin et al., 2012; Soejima et al., 2012; Soejima et al., 2013, Fang et al., 2015; Hu et al., 2015; Guo et al., 2016). HBOT preconditioning may reduce edema and protect the BBB by suppressing the inflammatory response. It was shown that HBOT preconditioning decreased both infarction and hemorrhage volumes, and improved neurobehavioral function through reduced expression of the NLRP3 inflammasome and its downstream targets after hemorrhagic transformation (Guo et al., 2016). Moreover, HBOT preconditioning reduces cerebral vasospasm and stabilizes the blood–brain barrier (BBB) by increasing OPN expression in the brain, inhibiting IL-1β/NFκB, and suppressing MMP-9 (Hu et al., 2015). In support of this, some studies showed that HBOT preconditioning improved neurological deficits and reduced hemorrhagic volume via decreasing HIF-1α and its downstream MMP-2 and MMP-9 (Hu et al., 2008; Ostrowski et al., 2008; Soejima et al., 2013). In addition, it was shown that HBOT preconditioning may depend on the induction of MMP-9 in the pre-ischemic phase and may be in part mediated by exhaustion of MMP-9 stores in cerebral tissues (Ostrowski et al., 2008).
HBOT preconditioning-induced HSP-70 overexpression in the hippocampus can significantly attenuate brain edema, cognitive deficits, and hippocampal oxidative stress (Lin et al., 2012). It was indicated that HBOT preconditioning can induce cytoprotective effects on human microvascular endothelial cells via upregulation of Nrf2 and HSP32 or heme oxigenase-1 (Godman et al., 2010). However, more recently it was shown that HBOT improved neurological deficits, infarction volume, BBB disruption, and hemorrhagic transformation without including the activation of these proteins in a focal cerebral ischemia model (Soejima et al., 2012).
Reduced intracerebral edema may also be achieved with HBOT preconditioning by downregulating the expression of aquaporin 4 (AQP-4), a key factor that effects the water and electrolyte balance in the CNS, thus impeding intracerebral hemorrhage and protecting neural tissue (Fang et al., 2015). On the other hand, it was shown that levels of both AQP-4 and VEGF were significantly increased in cultured astrocytes after HBOT preconditioning (Wang et al., 2016). These findings suggest that HBOT preconditioning is also able to promote transient and regulated BBB opening, which may contribute to the induction of cerebral ischemic tolerance as well as representing a possible strategy for promoting drug transport into the CNS (Wang et al., 2016). In addition, it was shown that preconditioning with HBOT protects against brain edema formation following intracerebral hemorrhage by activation of the p44/42 MAPK pathway, whose activation has been linked to ischemic tolerance (Qin et al., 2007). Changes in tight junction protein (TJP) expression can lead to the loss of BBB integrity and BBB breakdown. Interestingly, it was shown that HBOT preconditioning protected the integrity of the BBB in an in vitro model through modulation of occludin and ZO-1 expression under hypoxic conditions (Hao et al., 2016).
Finally, HBOT preconditioning may exert protective effects on energy metabolism and tissue perfusion by 1) stabilizing the glucose level; 2) decreasing the lactate/pyruvate ratios and glycerol in the peri-infarct area; 3) inhibiting the increase of the glutamate level; and 4) upregulating Ang-2, which is associated with increased microvessel density in the penumbra, reduced brain injury, and improved neurological function after focal cerebral ischemia (Gao-Yu et al., 2011; Duan et al., 2015).
5.5 Considerations for HBOT preconditioning protocols
The constituent components of HBOT (hyperoxia and hyperbaricity) are critical for the induction of tolerance against ischemic injury since simple hyperbaricity (2.5 ATA, 21% O2) did not induce ischemic tolerance (Dong et al., 2002). Interestingly, there is a close link between hyperoxia and hypoxia (Mik, 2011). HBOT and hypoxic preconditioning elicit similar preconditioning efficacy in neonatal brain, but invoke different defenses against oxidative stress (Freiberger et al., 2006). Usually, the HBOT preconditioning is carried out at 2-3 ATA and the duration of each exposure ranges from 60 to 90 minutes (Theodoraki et al., 2011; Liu et al., 2012; Losada et al., 2014). The interval of 24 hours is the most commonly applied in HBOT preconditioning (Theodoraki et al., 2011; Liu et al., 2012). Serial exposition for 3 or 5 days at HBOT preconditioning can induce ischemic tolerance in a dose-dependent manner (Xiong et al., 2000; Dong et al., 2002). Interestingly, HBOT-induced neuroprotection against ischemic injury appears to have a time window, showing protection at 6 h, 12 h and 24 h pre-stroke, but not at 72 h (Hirata et al., 2007). However, neuroprotection induced by preconditioning consists of biphasic time windows defined by immediate and delayed preconditioning effects (Yokobori et al., 2013; Hu et al., 2016). Immediate preconditioning is observed within 1 h after the preconditioning stimulus and is characterized by transient changes in activity of ion channels, secondary messengers, and enzyme activity (Yokobori et al., 2013). Delayed preconditioning consists of intracellular changes that develop more slowly and manifest as long-lasting alterations in gene expression and protein expression profiles (Yokobori et al., 2013).
6.0 HBOT-primed Stem Cells as a Promising Therapy
Present within discrete niches of the adult brain, stem cells exert neuroprotective and neurorestorative effects following stroke by migrating to infarcted and damaged regions whereby they utilize multi-pronged therapeutic tactics to confer their benefits (Sullivan et al., 2015). Among the mechanisms employed by these undifferentiated cells are bystander effects, such as secretion of trophic factors, anti-inflammatory molecules, apoptotic pathway regulators, and angiogenic factors (Napoli et al., 2018) and, to a much lesser extent, direct cell replacement of damaged neural tissue (Stonesifer et al., 2017). Unfortunately, the ability of unaided endogenous stem cells to initiate robust recovery following stroke is extremely limited. In this light, therapeutic modalities that enhance the brain’s intrinsic reparative capacity are highly attractive. A growing body of literature supports enhancing the effects of HBOT on endogenous stem cells as a prominent mechanism of action in the post-stroke brain (Thom et al., 2006). This knowledge – in addition to supporting the previously discussed concepts of HBOT as a standalone therapy for stroke or prophylactic treatment – indicates a possible role for HBOT in conditioning of stem cells prior to transplantation. The following sections will discuss the effects of HBOT on stem cells, and how these effects may be lent favorably to a stem cell preconditioning paradigm.
6.1 HBOT effects on endogenous and transplant stem cell populations
HBOT exerts dynamic effects on various populations of stem cells in vivo. Perhaps most prominently, HBOT enhances the total number of available endogenous stem cells by increasing the quantity of circulating stem cells in a pressure-sensitive manner (Thom et al., 2006; Dhar et al., 2012; Heyboer et al., 2014) and upregulating neural stem cell proliferation within the neurogenic niches of the adult brain (Yang et al., 2008). This phenomenon has been demonstrated in multiple injury models including heightened sub-ventricular zone (SVZ) and sub-granular zone (SGZ) proliferation following ischemic brain damage (Yang et al., 2008; Wei et al., 2015), increased neural stem cell proliferation in the piriform cortex in the vascular dementia brain (Zhang et al., 2010), and upregualtion of both neural stem cell proliferation (Yang et al., 2017) and circulating stem cells (Shandley et al., 2017) following TBI. Moreover, this effect has been observed in non-oxy-injured animals (Heyboer et al., 2014), insinuating the potential for therapeutic effectiveness independent of pathological idiosyncrasies, as well as for pre-injury applications. In addition to encouraging proliferation of neural stem cells, HBOT promotes their migration to areas of injury, as demonstrated in a rat model of TBI (Yang et al., 2017). Furthermore, of the various stem cells which are released into circulation following HBOT, particular subsets – such as vasogenic endothelial progenitor cells (EPCs) – may enact particularly potent effects in the stroke brain (Thom et al., 2011). Paired with the mechanisms described in Sections 3 and 5, the presence of heightened stem cell numbers in the periphery and brain likely contribute to the therapeutic benefits of HBOT observed in the stroke brain.
Of great interest are the mechanisms by which HBOT increases available stem cells within the body. The circulating stem cells released following HBOT originate from the bone marrow, and their departure into circulation is largely attributed to heightened nitric oxide synthesis by eNOS (Heyboer et al., 2014). Experiments using eNOS-knockout mice and nitric oxide synthase inhibitors support this concept (Thom et al., 2006). The mechanisms underlying HBOT-induced upregulation of cerebral neurogenesis, however, are more elusive and likely multi-factorial. A number of key signaling molecules and growth factors modulated by HBOT have been identified and may be responsible for the increase (Mu et al., 2011). Among these, HBOT is known to exert a stabilizing effect on HIF-1α, a transcription factor specifically activated by hypoxia, slowing its degradation by the prolyl hydroxylase pathway (Milosevic et al., 2009) and allowing for increased activation of the Wnt/β-catenin signaling pathway, which has been directly linked to neural stem cell proliferation (Qi et al., 2017). Supporting this, Wnt-3 expression was observed to increase substantially in HBOT-treated ischemic-hypoxia neonate rats in parallel with elevated SVZ neurogenesis (Wang et al., 2007). Particularly interesting, studies have reported that high levels of ROS within proliferative NSCs may serve secondary messenger functions which contribute to their self-renewal, and that pharmacological inhibition of ROS reduces their proliferative capacity (La Belle et al., 2011). Thus, an HBOT-induced increase in ROS levels may increase NSC levels by enhancing signaling pathways which are intrinsically linked to NSC survival and proliferation.
HBOT may also promote neural stem cell proliferation via upregulation of key regulatory molecules such as vascular endothelial growth factor (VEGF) (a downstream target of HIF-1α), its receptor (VEGFR2) (Yang et al., 2017), ERK (Jiang et al., 2015), and CREB (Zhu et al., 2004; Mendoza-Paredes et al., 2008) – all of which play roles in neurogenic pathways (Lu et al., 2011). HIF-1α induces the expression of hundreds of gene products in response to hypoxia or ischemia involved in processes such as angiogenesis, glycolysis, inflammation, proliferation (Aljitawi et al., 2016) and growth, which collectively promote cell survival and neuroprotection after brain injury (Milosevic et al., 2009; Hu et al., 2016). It was shown that erythropoietin (EPO), another target gene of HIF-1α, was upregulated in the cerebral cortex and hippocampus and prevented changes to BBB permeability, decreased brain edema, reduced infarction volumes, and improved neurobehavioral outcome after HBOT (Gu et al., 2008; Peng et al., 2008). Conversely, it has also been shown that a systemic reduction in EPO levels by HBOT promotes bone marrow homing and engraftment after allogeneic umbilical cord blood (UCB)-derived hematopoietic stem/progenitor cell (HSPC) transplantation (Aljitawi et al., 2016). In addition, the increased nuclear expression of HIF-1α following HBOT is associated with increased expression of CXCR4 (Chen et al., 2017). A recent study showed that the HIF-1α-CXCR4 pathway can promote proliferation of neural crest stem/progenitor cells (NCSCs) by decreasing nuclear expression of the cyclin-dependent kinase inhibitor CDKN1A (p21CIP1/WAF1) and by SDF-1/CXCL12 signaling mediated by receptor CXCR4 under hypoxia (Chen et al., 2017). Moreover, after HBOT, increased cytoplasmic expression of TPM1 and decreased nuclear expression of TP53 and CDKN1A resulted in decreased apoptosis and increased proliferation of NCSCs (Chen et al., 2017). Finally, HBOT-induced vascularization may increase neurogenesis by providing metabolic support to under-perfused neurogenic areas (Tal et al., 2017).
6.2 HBOT and exogenous stem cells
A careful analysis of combining HBOT with stem cell transplantation offers insight into potential underlying mechanisms and interplay between the two treatments. This combinatorial approach has been studied in a number of neurological and non-neurological contexts including TBI (Zhou et al., 2016), SCI (Geng et al., 2015) and diabetes mellitus (Estrada et al., 2008). One study demonstrated that HBOT improves graft survival within the bone marrow, peripheral blood, and spleen following umbilical cord blood stem cell transplantation in a rodent model of whole-body irradiation injury (Aljitawi et al., 2014). Similar results were found in a model of SCI, with greater MSC graft survival found in HBOT+cell therapy animals than in cell therapy alone (Geng et al., 2015). Animals receiving the combination therapy were also observed to have an assuaged inflammatory response, with lower levels of pro-inflammatory mediators TNF-α, IL-6, and IFN-α at various time points when compared to animals receiving cell therapy alone (Geng et al., 2015). In a traumatic nerve crush model, transplantation of human amniotic fluid mesenchymal stem cell (AFS) delivered with adjunctive HBOT displayed synergistic effects (Pan et al., 2009). Among the improvements reported in this study were a suppressed inflammatory response in combination therapy animals, upregulated nerve regeneration metrics such as neurofilament production and s-100 expression, and, interestingly, a dramatic decrease in expression of the apoptosis marker TUNEL in the transplanted AFS (Pan et al., 2009).
6.3 Effects of HBOT in vitro: potential for stem cell priming
In light of the broad effects of HBOT on endogenous and transplanted stem cells, further investigations into the effects of HBOT on stem cells in vitro are warranted. Importantly, many of the molecular signaling pathways which are discussed above (i.e. Wnt/β-catenin, VEGF/VEGFR2, and CREB) are likely mediated by non-stem cell host tissue secretions. Thus, while the knowledge obtained from studies of HBOT on stem cells in vivo guides our scientific inquiries, the mechanisms, both stem cell- and non-stem cell-mediated, must be considered. Moreover, potential adverse effects of HBOT on stem cells should also be examined. Indeed, in vitro study of HBOT on stem cells has found unique effects on stem cell cultures. In contrast to the paradigm described previously of HBOT-mediated increase in stem cell proliferation, HBOT was shown to decrease cell survival in mesenchymal stem cell (MSC) cultures (Schulze et al., 2017). The enhanced oxygen tension delivered by HBOT increases the formation of ROS and exerts oxidative stress on cells (Thom, 2009; Cheung et al., 2018). Similar results have been reported with other stem cell populations, including umbilical cord blood stem cells (Cheung et al., 2018).
The therapeutic potential of HBOT priming stem cells for subsequent transplantation will also require careful evaluation, as exposure to oxidative stress in vitro may improve stem cell resiliency to oxidative stress upon transplantation (employing a similar conceptual framework as described for HBOT preconditioning in Section 5). With this in consideration, we propose that in vitro HBOT priming may have genetic, molecular, or transcriptomic effects on stem cells which increase their therapeutic potential, and further, that preliminary HBOT-induced oxidative stress may improve the resiliency of these stem cells to the harmful microenvironment of the post-stroke brain upon transplantation.
This strategy of HBOT-primed stem cells has been employed in a limited fashion in other pathological contexts; human adipose-derived stem cells were demonstrated to have improved extracellular matrix-secreting capabilities following transplant into a rabbit cartilage defect model when primed with 2.5 ATA HBOT for 1 hour (Dai et al., 2015). In another study, increased O2 exposure in vitro prompted the maturation and differentiation of human embryonic stem cell-derived pancreatic progenitor cells into the desired β-cell lineage (Cechin et al., 2014).
Precedent evidence on priming exists for this therapeutic approach in stroke as well. Hypoxic preconditioning of stem cells (5% O2 for 24 hours) has been shown to increase graft survival post-transplantation in a hemorrhagic stroke mouse model (Wakai et al., 2016) and an ischemic stroke model (23031629), as well as to enhance stem cell migratory/homing ability (Hu et al., 2011; Lee et al., 2013; Wei et al., 2013). By preparing stem cells for the metabolic challenges posed by the microenvironment of damaged tissue, post-transplantation cell survival and function are bolstered (Wei et al., 2012). Hypoxia is a major contributor to low graft survival following post-stroke transplantation and serves as the premise for hypoxic preconditioning, yet it is not the only prevalent challenge posed to cells within the post-stroke brain. Rampant oxidative stress contributes significantly to both endogenous cell and graft cell death (Chen et al., 2011; Xing et al., 2012). As such, exposing stem cells to mild/moderate oxidative stress before transplantation may extend their survival time in a similar manner to hypoxic precondition by permitting their genetic and phenotypic acclimation to a state of oxidative stress.
HBOT may also alter and enhance the stem cell secretome, boosting its effectiveness within the stroke brain. In vitro experiments have revealed that HBOT exerts a notable effect on the secretion profile of stem cells, including a number of proteins implicated in: 1) the oxidative stress response such as vasorin, thrombospondin-4, thioredoxin, heat shock protein (HSP) 90, HSP70, and gamma-glutamylcyclotransferase; and 2) various neuroprotective pathways such as peroxiredoxin, cystatin C, laminin, syndecan and thymosin-beta (Schulze et al., 2017). The increased cellular nitric oxide recorded following HBOT consequently upregulates expression of certain growth factors including VEGF and transforming growth factor-beta 1 (TFGb1) (Venetsanou et al., 2012). Moreover, using MSC cultures, HBOT was shown to increase expression of placental growth factor (PlGF) in a dose-dependent manner, which was associated with increased MSC tubule formation and enhanced migratory ability (Shyu et al., 2008). Interestingly, HBOT has been shown to prevent differentiation of stem cells in culture (Cheung et al., 2018). Depending on the treatment strategy of transplantation (i.e. direct cell replacement paradigms such as with oligodendrocyte progenitor cell transplantation, or immunomodulation paradigms such as with MSC transplantation), the ability to promote or maintain naïve stem cell states may be an additional benefit of HBOT priming.
Interestingly, HBOT-preconditioned stem cells may be an effective adjunctive to HBOT-preconditioning of the host. Given the extensive evidence supporting the neuroprotective capacity of HBOT-preconditioning on the healthy brain (see Section 5), stem cell preconditioning could also be executed in tandem to offer a readily available cell therapy option following stroke. This could present a potent dual therapy approach for patients at an exceptionally high risk for stroke, utilizing both a preemptive neuroprotective modality as well as a post hoc neurorestorative biologic.
In summary, the ability of HBOT to prolong graft survival via oxidative stress conditioning, prevent premature differentiation, enhance migratory capacity, promote injury homing, upregulate trophic factors within the secretome, and encourage anti-inflammatory mediation all indicate the great potential of HBOT to advance the therapeutic efficacy of stem cells transplanted in the stroke brain alone, or in synchrony with host preconditioning therapies.
7.0 Future Directions and Conclusion
Preclinical studies have demonstrated the efficacy of HBOT in preserving vulnerable neural tissue and improving functional outcomes in various stroke models. A wide range of HBOT regimens have been reported as effective, with 1) pressures ranging from 2.0 ATA-3.0ATA, 2) durations lasting from 45 min-2 hrs, and 3) time of initiation ranging from immediately after stroke to 48+ hours. Unfortunately, the wide range of effective parameters has prevented a consensus regimen from being employed in clinical trials, slowing the progress of HBOT translation. Compounding this issue, human effectiveness has not been well proven due to a lack of high-quality multicenter randomized controlled trials. The successful translation of clinical HBOT will largely rest on the field’s ability to organize and implement standardization and rigorous methodology in its clinical trials. Nonetheless, the basic science field has continued to advance our understanding of the mechanisms of HBOT in the injured and healthy brain, and this has paved the way for the emergence of HBOT preconditioning paradigms. While preconditioning strategies are innovative and may hold real benefits for certain patient populations, obvious limitations exist regarding the ability to preemptively deliver stroke therapy; strokes are acute events which are unpredictable even in patients with extensive predispositions.
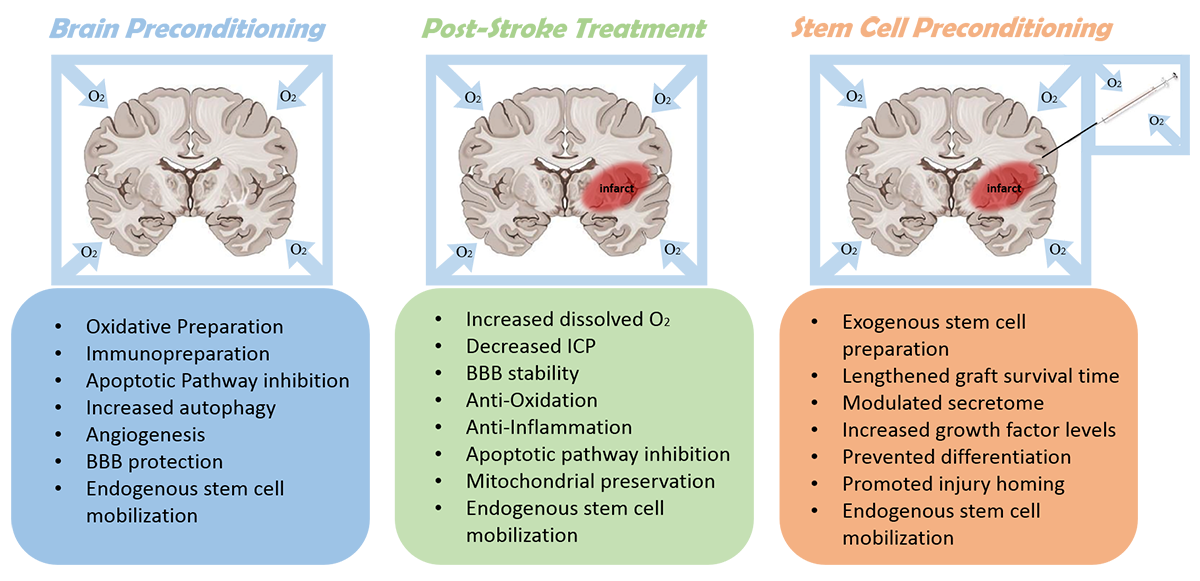
In a new window | Download PPT
Figure 1: A summary of prominent mechanisms of hyperbaric oxygen therapy for pre-stroke, post-stroke, and stem cell preconditioning applications. BBB - blood brain barrier; ICP - intracranial pressure.
In light of these challenges – namely, the difficulties in translating canonical HBOT therapy, and limited application of preconditioning – we propose here a novel application of HBOT as a preconditioning mechanism for stem cell transplantations (Figure 1). Based on the extensive literature, we speculate that oxidative preconditioning of stem cell grafts via HBOT may be an effective means of increasing graft survival and optimizing graft function within the post-ischemic environment. Advancing this concept will require due diligence in verifying the genetic, epigenetic, secretome, and functional influence that HBOT exerts on specific stem cell populations, and characterizing the in vivo response of HBOT-primed versus non-primed stem cells. This therapeutic strategy may be able to harness the promising laboratory findings that have been widely reported for both HBOT and stem cell transplantation following cerebrovascular accident, offering a hybrid approach of either standalone or combinatorial pre-conditioning strategies for conferring neuroprotection in the stroke brain.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Acknowledgments
CVB is funded by NIH R01NS071956, NIH R01 NS090962, NIH R21NS089851, NIH R21 NS094087, and VA Merit Review I01 BX001407.
References
Eric Eugene Paul Cosky 1
1Department of Neurological Surgery, Wayne State University School of Medicine, Detroit, MI.
Yuchuan Ding 1
1Department of Neurological Surgery, Wayne State University School of Medicine, Detroit, MI.
Corresponding author:
Eric Eugene Paul Cosky
Email: ecosky@med.wayne.edu
and
Yuchuan Ding
Email: yding@med.wayne.edu
In a new window | Download PPT
Figure 1: A summary of prominent mechanisms of hyperbaric oxygen therapy for pre-stroke, post-stroke, and stem cell preconditioning applications. BBB - blood brain barrier; ICP - intracranial pressure.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11970 | 60 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA