Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Constitutive preconditioning: the anoxia tolerant freshwater turtle as a model organism of the preconditioned phenotype
Time:2019-05-02
Number:9597
Author Affiliations

Conditioning Medicine, 2019. 2(2):90-105.
Abstract
While mammalian brains are highly susceptible to the oxygen deprivation of ischemia or hypoxia, some vertebrates are considered facultative anaerobes that can survive extended periods of anoxia. The freshwater turtles Chrysemys picta and Trachemys scripta are able to tolerate days of anoxia at room temperature to months without oxygen at 3°C. Extended anoxic survival depends on (1) preventing anoxic depolarization of the brain, (2) entrance into a state of deep hypometabolism, and (3) preventing cellular injury via the suppression of apoptotic pathways and an upregulation of protective mechanisms. Many of the cellular and genomic mechanisms employed by anoxia tolerant organisms also appear in studies of mammalian preconditioning, but are expressed to a greater extent in the turtle. These include the opening of KATP channels, increased adenosine release, activation of mitogen activated protein kinases and the PI3K/Akt pathway, and an upregulation of heat shock proteins. More recent studies are focusing on changes in gene expression and epigenetics including the downregulation of cell death pathways and increases in anti-oxidant gene expression, with microRNA regulation of gene expression currently of interest in studies of both preconditioning and anoxia tolerant animals.
Investigating mechanisms of anoxia tolerance utilized by facultative anaerobes may suggest new therapeutic targets for diseases of ischemia and hypoxia.
Keywords: apoptosis; Chrysemys picta; hypometabolism; ischemia; microRNA; Trachemys scripta
Abstract
While mammalian brains are highly susceptible to the oxygen deprivation of ischemia or hypoxia, some vertebrates are considered facultative anaerobes that can survive extended periods of anoxia. The freshwater turtles Chrysemys picta and Trachemys scripta are able to tolerate days of anoxia at room temperature to months without oxygen at 3°C. Extended anoxic survival depends on (1) preventing anoxic depolarization of the brain, (2) entrance into a state of deep hypometabolism, and (3) preventing cellular injury via the suppression of apoptotic pathways and an upregulation of protective mechanisms. Many of the cellular and genomic mechanisms employed by anoxia tolerant organisms also appear in studies of mammalian preconditioning, but are expressed to a greater extent in the turtle. These include the opening of KATP channels, increased adenosine release, activation of mitogen activated protein kinases and the PI3K/Akt pathway, and an upregulation of heat shock proteins. More recent studies are focusing on changes in gene expression and epigenetics including the downregulation of cell death pathways and increases in anti-oxidant gene expression, with microRNA regulation of gene expression currently of interest in studies of both preconditioning and anoxia tolerant animals.
Investigating mechanisms of anoxia tolerance utilized by facultative anaerobes may suggest new therapeutic targets for diseases of ischemia and hypoxia.
Keywords: apoptosis; Chrysemys picta; hypometabolism; ischemia; microRNA; Trachemys scripta
Introduction
The mammalian brain is well known to be sensitive to oxygen deprivation, under hypoxic conditions, for example, or during an ischemic stroke. While making up only 2% of the body mass, the human brain may use up to 20% of systemic oxygen; because of this high metabolic rate, any interruption in oxygen supply results in a well-described pathological cellular cascade that results in brain damage and death (Lutz et al., 2003). Upon disruption of oxidative phosphorylation, ATP levels fall, thus ATP-dependent neuronal processes, including ion transport and neurotransmitter re-uptake decline as well (Kopp et al., 1984; Santos et al., 1996). Without pumping, ion gradients fail, neurons depolarize, and excitatory neurotransmitters, including dopamine (DA) and glutamate rise due to both decreased reuptake and an increase in vesicular and non-vesicular release (Dawson et al., 2000; Rossi et al., 2000). Overstimulation of glutamate N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid (AMPA) receptors increases intracellular calcium levels (Brooks and Kauppinen, 1993; Bickler and Hansen, 1994). Excess free intracellular calcium in turn is taken up by the mitochondria until opposing pumps are overwhelmed and calcium is released back into the cytoplasm (Snowdowne et al., 1985; Villalba et al., 1994). This triggers multiple internal cascades that result in cell damage and death, including activation of lipases, endonucleases, and proteases (Lipton, 1999). Mitochondrial-dependent apoptosis follows formation of the mitochondrial permeability transition pore (MPTP) and cytochrome c release, with a shift in the balance of pro- and anti-apoptotic members of the B cell lymphoma 2 (Bcl-2) protein family (Lee et al., 2001; Kotipatruni et al., 2011). Hypoxia and reoxygenation (such as reperfusion following ischemia) can also induce oxidative stress, with the mitochondria as both the primary source of reactive oxygen species (ROS) and a significant target of ROS damage (Sanderson et al., 2012). The brain is especially susceptible to oxidative stress due to its concentration of unsaturated fatty acids, high iron content, and low antioxidant capacity (Solberg et al., 2012), and oxidative damage can continue to develop over a period of days. Ischemic stroke is thus a leading cause of death and morbidity in the developed world, and few effective treatments are available (Benjamin et al., 2017).
In the face of this pathological cascade, however, the brain responds with cellular and molecular mechanisms that work to counteract these processes and preserve neuronal integrity and function. These mechanisms include, to list just a few areas of investigation, the release of adenosine that is thought to reduce neuronal excitability and neurotransmitter release (Hoehn and White, 1990a, 1990b), the activation of mitogen activated protein kinases (MAPKs), the phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) pathway, and the activation of heat shock proteins. More recent studies focus on changes in gene expression and epigenetics (as expertly reviewed in e.g. Yang et al., 2017 and Yu et al., 2018), including the downregulation of cell death pathways and increases in anti-oxidant gene expression. Triggering these protective mechanisms via a sub-lethal stressor, that enable the cells or organism to survive a later, more extensive stress that would otherwise be lethal, is the basis of preconditioning (PC), as first describe by Murry et al. (1986) in the myocardium and later shown to occur in the brain (Liu et al., 1992). While a brief ischemic period is often used in laboratory studies to induce ischemic preconditioning (IPC), other stressors can also increase brain ischemic tolerance, including hypothermia, hypoxia, cortical spreading depression, or oxidative stress (Yang et al., 2017). With a more practical aspect in mind, recent studies are examining other forms of PC, including exercise and remote PC that can be more readily utilized therapeutically.
Constitutive adaptations that promote anoxic survival
Not all organisms share the mammalian vulnerability to neuronal oxygen deprivation, however; neonatal rats may survive periods of anoxia as much as 25 times longer than adults (Bickler et al., 2003), while hibernating mammals such as the Arctic ground squirrel (Drew et al., 2004), diving animals (Folkow et al., 2008), and the fossorial naked mole rat (Nathaniel et al., 2009; Chung et al., 2016; Park et al., 2017) tolerate extended periods of hypoxia without brain damage. The most impressive facultative anaerobes, however, can survive weeks to months in the complete absence of oxygen, and include the crucian carp Carassius carassius (Nilsson et al., 1993; Hylland et al., 1997), and several species of North American pond turtles, with the best studied being the painted turtle Chrysemys picta and the red eared pond slider Trachemys scripta (Lutz et al., 2003; Milton and Prentice, 2007). These animals have undergone millions of years of selective pressure to ensure survival; in the case of both the carp and turtles, winter hibernation occurs over extended periods under ice in ponds that become anoxic (Ultsch, 2006). There appear to be two critical mechanisms that enable anoxic survival: (1) the rapid entrance into a state of deep hypometabolism, coupled to enhanced glycogen stores that can fuel extended periods of glycolysis (Lutz et al., 2003) and (2) the abrogation of cellular injury during both anoxia and recovery via the suppression of pathological pathways and the upregulation of protective mechanisms. By entering a deep hypometabolic state, anoxia tolerant organisms are able to reduce energy demand to meet the reduced supply provided by anaerobic glycolysis, and thus prevent anoxic depolarization and the subsequent activation of apoptotic cascades (Lutz et al., 2003; Milton and Dawson-Scully, 2013). In addition to withstanding extended periods of complete anoxia, turtles also tolerate ischemic conditions for up to 24 hrs. They tolerate not just a lack of oxygen but also glucose deprivation and the ionic and pH perturbations associated with ischemic tissues (Pamenter et al., 2012). As explored below, many of the mechanisms that are employed to induce neuronal anoxia tolerance overlap with those currently under investigation in PC studies (Fig. 1), making C. picta and T. scripta the ultimate adaptive model organisms of preconditioned cell survival.
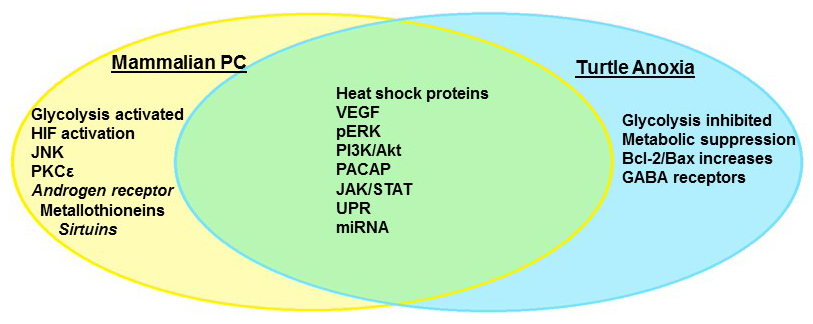
In a new window | Download PPT
Figure 1: Anoxia tolerance in the freshwater turtle and mammalian preconditioning share many molecular pathways that comprise the “survival phenotype.” Some mechanisms are unique to facultative anaerobes such as the turtle, which repress overall metabolism to survive extended anoxia.
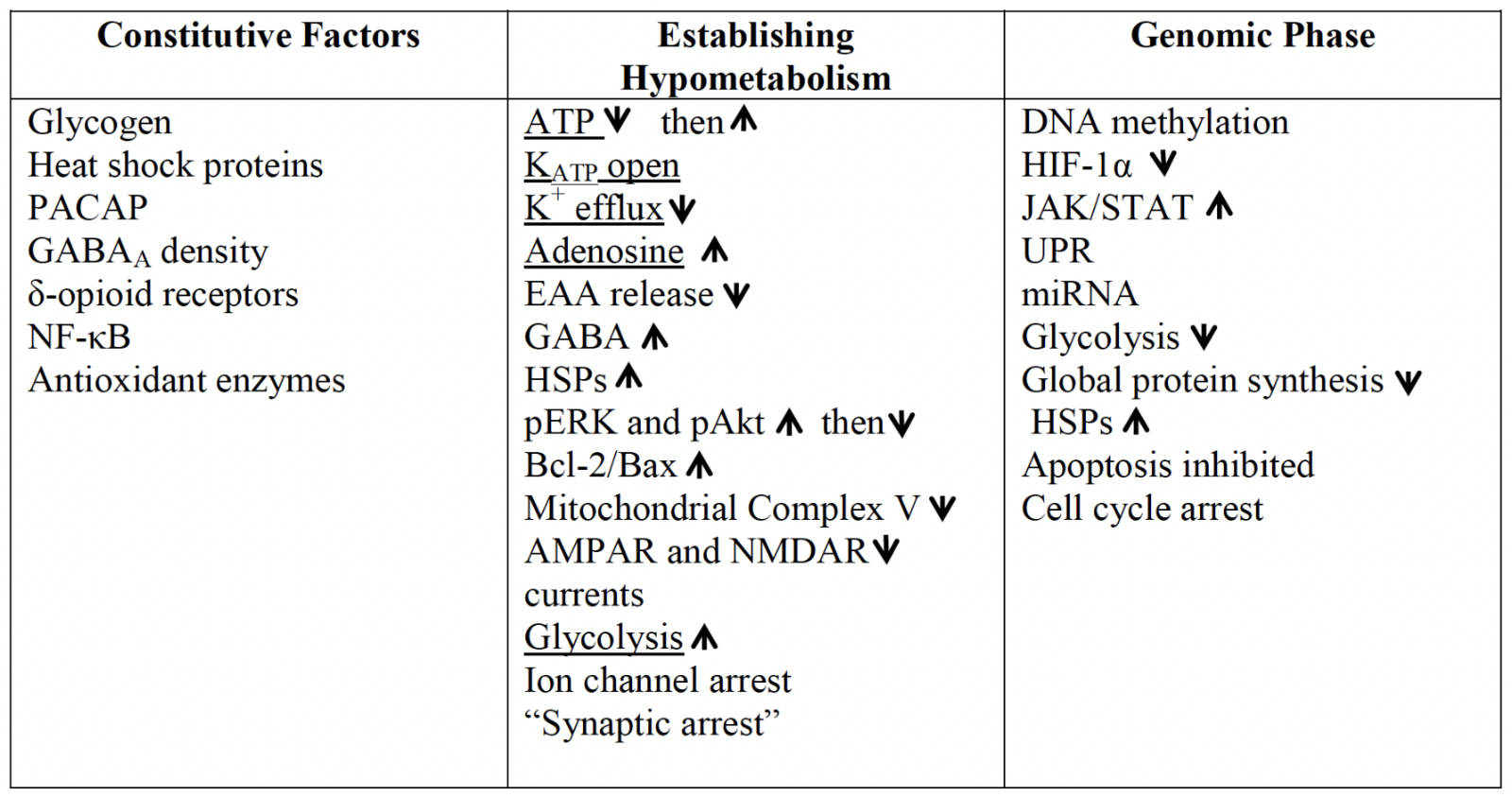
Many factors that enable anoxic survival in T. scripta are, in fact, constitutively present in normoxia. While some may be the result of inherent differences in basal metabolic rates, including much lower ion channel densities and lower aerobic enzyme activity (Lutz et al., 2003), others appear to be specific adaptations that extend anoxic survival (Table I). These include high intrinsic inhibitory potential, with the same density of γ-aminobutyric acid type A (GABAA) receptors as is seen in the rat brain (Lutz and Leone-Kabler, 1995), and delta-opioid receptor levels in the turtle cortex more than four times higher than in mammals, with a higher binding affinity (Xia and Haddad, 2001). While the subject of controversial findings in the past, evidence appears to be in favor of delta-opioid receptor activation as a protective measure against ischemia-reperfusion in the mammalian brain (Sheng et al., 2018). Other inherently protective mechanisms include much higher levels of adenylate cyclase activating polypeptide (PACAP38) than in the rat or human brain (Reglödi et al., 2001), elevated brain glycogen stores that permit extended glycolysis (Lutz et al., 2003), and high constitutive levels of the transcription regulator nuclear factor κB (NF-κB; Lutz and Prentice, 2002). NF-κB is a transcription factor central to the expression of anti-apoptotic and anti-inflammatory stress responsive genes that may be protective in brain ischemia (Simmons et al., 2016), though other studies report that under inflammatory conditions increases in NF-κB promote damage (Abraham, 2000). Anoxia tolerant turtles also exhibit detectable basal levels of inducible heat shock protein 70 (Hsp72), the cognate protein Hsp73 (Prentice et al., 2004), Hsp90 (Ramaglia and Buck, 2004; Stecyk et al., 2012), and other HSPs including heme oxygenase (HO-1; Hsp32), Grp94, Hsp60, and Hsp27 (Kesaraju et al., 2009a). Recovery from the anoxic state is likely to be aided as well by high levels of antioxidant enzymes, including high constitutive activities of catalase, superoxide dismutase (SOD), and alkyl hydroperoxide (Willmore and Storey, 1997), a high affinity glutathione reductase that is relatively insensitive to pH (Willmore and Storey, 2007), and cortex ascorbate levels 2- to 3- times higher than in the mammalian brain (Rice et al., 1994).
Table I: An overview of some of the factors coordinating anoxic survival in the turtle brain. Constitutive factors present even in the normoxic turtle, and genomic changes that occur to establish the survival phenotype, are shared with many elements of mammalian preconditioning but may occur to a greater extent in the turtle. While some of the mechanisms which enable the brain to avoid depolarization are common to PC (underlined), the coordinated downregulation of energetically expensive processes to ~20% of normoxic levels in the first 1–2 hrs without oxygen is reflective of true facultative anaerobes.
Numerous studies have shown PACAP to be protective in the ischemic heart (Alston et al., 2011; Danyadi et al., 2014; Banki et al., 2015) and retina (Atlasz et al., 2008, 2009, 2010; D’Alessandro et al., 2014), as well as brain (Banks et al., 1996; Uchida et al., 1996; Reglodi et al., 2000; Somogyvári-Vigh et al., 2000). The mechanisms of neuroprotection by PACAP38 are not fully understood, but appear to be through both a reduction of inflammation (Ohtaki et al., 2006; Dejda et al., 2011) and via pathways that promote cell viability by reducing cytochrome c/caspase-3 induced apoptosis, increasing BDNF and Bcl-2 protein expression, and modulating the expression of NMDA receptor subunits that would thus reduce glutamate induced toxicity (Ohtaki et al., 2008; Kaneko et al., 2018). In the turtle, high PACAP38 levels may protect against anoxia induced retinal damage (Rábl et al., 2002) and preliminary evidence from my laboratory shows that a PACAP38 agonist promotes cell survival in primary turtle neuronal cell cultures under conditions of either H2O2 exposure or 4 hr anoxia and anoxia/reoxygenation, while the antagonist PACAP6-38 increases cell death (Nayak and Milton, unpublished obs.).
Many studies, of course, have shown a link between the ischemic tolerance of PC (Nishi et al., 1993) and elevated HSPs, including Hsp70, Hsp60, HO-1, Hsp27, and the glucose- regulated proteins Grp94 and Grp78 (Kato et al., 1994; Geddes et al., 1996; Chen et al., 1996; Massa et al., 1996; Kirino, 2002; Valentim et al., 2001; Badin et al., 2006; Hwang et al., 2007; Brea et al., 2015). Both Hsc73 and Hsp72 increase over time following transient middle cerebral artery occlusion (tMCAO) in the rat (Li et al., 2016) (as also occurs in the anoxic turtle brain (Prentice et al., 2004; Kesaraju et al., 2009a)) and are thought to be key to survival in the penumbra. HO-1 overexpression also reduces infarct size in the rat focal ischemic model (Panahian et al., 1999), and neurons overexpressing HO-1 are more resistant to glutamate excitotoxicity (Chen et al., 2000). Of the HSPs examined in the turtle brain, only the endoplasmic reticulum (ER)-specific HSP Grp78 did not increase upon anoxic exposure or recovery (Kesaraju et al., 2009b), though changes have been reported in the anoxic heart and liver (Krivoruchko and Storey, 2013). In the turtle, anoxic increases in Hsp72 surprisingly do not appear to block apoptosis; knocking down Hsp72 in cell culture increased cell death (Fig. 2) but not cytochrome c release or caspase activation (Kesaraju et al., 2014). The most immediate effect of blocking Hsp72 expression was a near 6-fold increase in reactive oxygen species (ROS) production upon reoxygenation following anoxia, which significantly increased cell death (Kesaraju et al., 2014). ROS production is otherwise suppressed (Milton et al., 2007; Pamenter et al., 2007), which is likely a key factor promoting survival upon restoration of oxygen in these organisms.
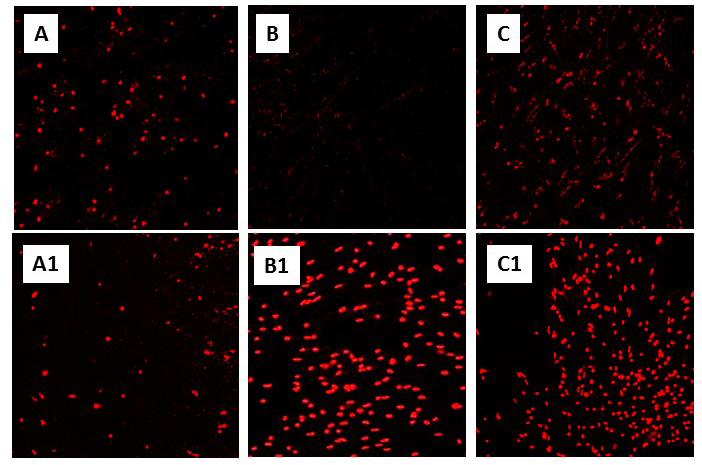
In a new window | Download PPT
Figure 2: Knockdown of Hsp72 significantly increases cell death in primary neuronal cultures of T. scripta (see “Constitutive adaptations that promote anoxic survival” in text, and Kesaraju et al., 2014). Representative propidium iodide (PI) staining of (A) and (A1): Normoxic control and siRNA treated cultures, respectively, (B) and (B1): 4 hr anoxic controls and siRNA treated, (C) and (C1): Anoxia/reoxygenation controls and siRNA treated cultures. Neuronal cultures were prepared from T. scripta cortex as described in Milton et al., (2007) and exposed to normoxia (air/5% CO2), 4 hr anoxia (90% N2/5% He/5% CO2), or 4 hr anoxia followed by 2 hr reoxygenation. Specific siRNA sequences against turtle Hsp72 and a scrambled siRNA control were transfected into cells with Lipofectamine-2000. A greater concentration of siRNA was required to achieve an equivalent knockdown (40-60%) in anoxic and anoxic/reoxygenation cultures (250 pM) than in normoxic ones (100 pM) due to the greater upregulation of HSPs in anoxia.
Establishing hypometabolism
As in mammalian models of PC, which result in both immediate effects and a delayed window of tolerance, turtles endure complete anoxia with a similarly bi-phasic response, with immediate adaptations that permit survival of a temporary decline in energy availability and the establishment of the hypometabolic state, and longer term changes that allow for extended survival without oxygen. These result from both physiological and molecular changes, with more recent studies focusing not only on alterations in protein level and function, but genomic and epigenetic controls.
The initial exposure to anoxia results in a temporary, partial decline in ATP levels that return to basal levels within the first hour without oxygen (Lutz et al., 1984). That decline in ATP supply, as energy demand outstrips glycolytic production, in turn produces a 12-fold increase in adenosine (AD) levels (Nilsson and Lutz, 1992) and increased adenosine receptor affinity (Lutz and Manuel, 1999), as well as the opening of ATP-dependent potassium channels (KATP), which in turn mediate a down-regulation of K+ efflux (Pék-Scott and Lutz, 1998). Such a downregulation of K+ current, as well as whole cell conductance, occurs in cardiac IPC models as well (Irie et al., 2003; Das and Sarkar, 2012). It has long been known that both KATP and AD are involved in PC in the mammalian brain (Pérez-Pinzón et al., 1996, 1997; Pérez-Pinzón and Born, 1999), with AD in the turtle having a similar suite of effects that induce neuroprotection in anoxia, including increased cerebral blood flow (Hylland et al., 1994), reduced ion flux (Pek and Lutz, 1997), effects on excitotoxin release, and modulation of protective molecular pathways. In the turtle brain, AD promotes NMDA receptor suppression (Buck and Bickler, 1995, 1998) and internalization from the cell membrane (Bickler et al., 2000). Blockade of AD A1 receptors (A1R) induces depolarization of the anoxic turtle cerebellum (Lutz and Nilsson, 1997). As in the mammalian brain (Sciotti et al., 1992), AD receptor blockade increases the release of excitatory neurotransmitters, including glutamate (Milton et al., 2002) and DA (Milton and Lutz, 2005). Decreased release of both glutamate and DA is also modulated initially by the opening of KATP, though this effect is transient and does not alter the release of either compound after the first 1-2 hrs after anoxia (Milton and Lutz, 2005; Thompson et al., 2007). While the focus in mammalian PC has been primarily on mitochondrial KATP, other KATP channels may also play a role, including sarcolemmal channels in cardiac cells (Gao et al., 2016), also influenced by adenosine (Yang et al., 2016), and plasmalemma channels (Bantel et al., 2009). Both mitochondrial and plasma membrane KATP are likely to play a role in turtle anoxia tolerance.
Later in anoxia, AD and GABAA receptors maintain the reduced release of glutamate; reuptake of the neurotransmitter continues, however, such that synaptic concentrations don’t change despite both reduced release and reuptake (Thompson et al., 2007). The reduction in glutamate release is accompanied by a ≥50% decrease in both AMPA and NMDA receptor currents (Pamenter et al., 2008a, 2008b, 2008c; Zivkovic and Buck, 2010). As with glutamate, the decrease in DA release in the initial 1-2 hrs after anoxia is followed later in anoxia (4 hrs) by continued release and reuptake despite the energy cost (Milton and Lutz, 2005), together suggesting that therapeutic approaches for stroke that totally block glutamate function may have failed due to an underlying need to maintain synaptic connectivity or allow for full operational recovery (Milton et al., 2002). The decrease in excitatory activity occurs simultaneously with massive increases in the level of the inhibitory neurotransmitter GABA of ~90-fold over basal level, along with the increased AD levels. GABA suppresses spontaneous electrical activity through (1) increased chloride conductance at the post-synaptic GABAA receptor, and (2) GABAB receptor-mediated decreases in presynaptic glutamate release (Pamenter et al., 2011). Together, the suppression of excitatory activity and the increased inhibition result in “synaptic arrest” (Buck and Pamenter, 2018) and a comatose like state (Fernandes et al., 1997).
AD is considered important in the ‘triggering” phase of mammalian PC via its role in decreasing glutamate release and inhibiting calcium influx, but is also thought to act in delayed protection through second messengers and signal transduction (Yang et al., 2017). In the turtle brain, AD modulates activation of both the PI3K/Akt pathway and several MAPKs. The MAPKs control intracellular events in response to various external stressors, including oxidative stress and anoxia, altering gene expression and post-translational modifications of proteins that ultimately change cell function (Martindale and Holbrook, 2002). Stimulation of A1R has been shown to decrease the activation of both c-Jun-N-terminal kinase (JNK) and p38, to increase extracellular signal regulated kinase (ERK) activation and Akt, and to decrease Bad activation while increasing Bcl-X in mammals (Ciccarelli et al., 2007; Miyawaki et al., 2008).
The PI3K/Akt pathway is generally considered to be anti-apoptotic, in part via phosphorylation of the Bcl-2 family member Bad and suppressing its pro-apoptotic activity (Dudek et al., 1997; Miyawaki et al., 2008). It has recently been demonstrated that a novel Akt activator decreases infarct volume and improves neurological function in MCAO rats, accompanied by increases in Bcl-2 and decreased Bax expression (Luan et al., 2018), and inhibition of Akt in mammalian stroke models increases cell death (Li et al., 2017; Ye et al., 2019). Similarly, the pharmacological blockade of either Akt or p-ERK increases cell death of turtle neurons in vitro (Nayak et al., 2016) and increases ROS production (Milton et al., 2008). Activated ERK increases more than 6-fold in the first hour of anoxia in the turtle brain, then returns to basal levels in 4 hrs (Milton et al., 2008). Both in vitro and in vivo, blockade of the A1R abrogates the initial anoxic increases in p-ERK and p-Akt but increases p-p38 and p-JNK levels (Milton et al., 2008; Nayak et al., 2011). A1R blockade also prevents anoxia-induced increases in the Bcl- 2/Bax ratio in the turtle; whereas in mammals ischemia or hypoxia increase Bax levels and promote cell death (Hara et al., 1996; Niwa et al., 1997; Antonawich et al., 1998). In the turtle, Bcl-2 levels in anoxia increase more than Bax (Nayak et al., 2011).
One downstream effect of these pro-survival mechanism is the inhibition of apoptosis, which in anoxia or ischemia occurs largely through formation of the MPTP, release of cytochrome c, and caspase activation (McClintock et al., 2002). The increases in Bcl-2 and suppression of Bax abrogate this result in turtles, but the effects of anoxia on turtle mitochondria also differ from mammals in other ways. In mammalian cells, oxygen deprivation leads to depolarization of the inner mitochondrial membrane, and F1F0 ATP synthase (complex V of the electron transport chain) runs in reverse such that mitochondria become a major site of ATP consumption in the cell (Rouslin et al., 1990; St-Pierre et al., 2000; Boutilier and St-Pierre, 2002). Galli et al. (2013) demonstrated that this is prevented by a significant downregulation of Complex V in the anoxic turtle heart over 2 weeks (Galli et al., 2013), with a more that 80% inhibition recently confirmed in the turtle brain (Pamenter et al., 2016a) and liver as well (Gomez and Richards, 2018a). In addition, activation of mitochondrial KATP in the turtle results in increased cytoplasmic calcium concentrations and thus reduced NMDAR activity, as the uncoupled mitochondria reduce calcium uptake via the uniporter (Pamenter et al., 2008d). While activation of mKATP has been associated with PKCε in mammalian brain (Raval et al., 2007) and heart (Waza et al., 2014), this is apparently not the case in turtles (Hawrysh et al., 2016). PKCε has also been shown to play a role in AD mediated preconditioning via the Akt pathway in mammals, acting in turn through ERK (Lange-Asschenfeldt et al., 2004).
Genomic changes permit long term anoxic survival
While cellular adaptations in the turtle allow for immediate survival in the face of oxygen deprivation and ATP decline, as in mammalian PC models, survival over the long term requires changes at the genomic and epigenetic level, though such changes are highly selective as transcription and translation are energetically demanding. In a transcriptomic study of anoxia responses in C. picta, very few genes in fact were significantly affected by 24 hr anoxia. Those that were altered (only 19 genes of 13,236 had a change ≥2x) were primarily immediate early genes such as Fos, Jun, and JunB. (Keenan et al., 2015). Surprisingly, no genes related to ion channels or synaptic transmission changed; those that did change were related to transcriptional, translational, and metabolic arrest. A similar study in T. scripta also showed relatively few changes to mRNA levels in response to anoxia at either high or low temperatures (Couturier et al., 2019), implying that alterations in neuronal gene expression in response to anoxia occur primarily by post-transcriptional, translational, or post-translational mechanisms, particularly as protein synthesis largely ceases (Brooks et al., 1993; Land et al., 1993; Fraser et al., 2001), with the known exceptions of some stress responsive proteins (Kesaraju et al., 2009b; Nayak et al., 2011, 2016) and some increased protein synthesis in the liver (Szereszewski and Storey, 2018).
Much research in both anoxia-tolerant organisms and in mammalian PC has lately focused on the genomic and epigenetic changes that occur. In PC models these changes are part of the ‘genomic phase’ required for long-term protection (Della-Morte et al., 2012), but the regulatory mechanisms are poorly understood (Yang et al., 2017). The genomic phase of IPC involves changes to hundreds of genes, initiated by the nuclear translocation of transcription factors in response to intracellular signal transduction, resulting in “genomic reprogramming” within the cell (Yang et al., 2017). While different forms of PC induce significantly different suites of genes (Stenzel-Poore et al., 2007; Cuomo et al., 2018), PC confers a ‘pro-survival’ phenotype, with various studies showing changes associated with heat shock proteins, metallothioneins, ion channels, MAPKs, the Janus kinase/signal transducer and activator of transcription (Jak/STAT) pathway, vascular endothelial growth factor (VEGF), a decrease in cell death pathways, and increases in anti-oxidant genes (reviewed in Yang et al., 2017). These changes in protein expression in IPC are thought to rely largely on three proteins: the androgen receptor (which has not been explored in anoxia tolerant organisms), hypoxia inducible factor (HIF)-1, and NFκB (Scornavacca et al., 2012). While NFκB does indeed increase as an immediate early gene in the turtle brain (Lutz and Prentice, 2002), interestingly, HIF-1α does not appear to play a significant role in anoxia tolerance in the turtle, with only a few anoxia-responsive genes under the control of HIF (Storey, 2006). In the similarly anoxia-tolerant crucian carp, HIF binding is more strongly affected by cold temperatures than hypoxia, with hypoxia responses varying by tissue and over time (Rissanen et al., 2006). Work in my lab examined whole brain total HIF-1α protein levels and that in the nuclear fraction in T. scripta. While HIF-1α was readily detected in the normoxic brain, no further increase occurred during anoxia; by contrast nuclear levels of HIF-1α protein decreased significantly in anoxia, reflected in decreased binding activity in anoxia as evidenced by eletrophoretic mobility shift assay. Furthermore, induction of HIF-1 through a small molecule activator, Tilorone, significantly increased cell death during anoxia, suggesting that in the turtle, anoxic suppression, rather than enhancement, of the nuclear translocation of HIF-1α may be a factor allowing metabolic downregulation and anoxic survival (Milton et al., 2010). By contrast, high basal levels of VEGF transcripts and protein increased further in the anoxic brain both in vivo (Fig. 3) and in cultured turtle neurons, despite the lack of HIF activation. In the turtle, the lack of HIF activation should perhaps not be surprising, as HIF binding activates numerous genes in response to hypoxia (Yu et al., 2018). HIF-responsive genes may be protective in anoxia-sensitive systems (e.g. erythropoietin, angiopoietins, pyruvate dehydrogenase kinase [PDK]-1) (Abu El-Asrar et al., 2007; Adluri et al., 2011; Abe et al., 2012) and reduce ischemia/reperfusion injury, but many pathways activated by HIF in mammals are actively suppressed in anoxia-tolerant organisms to induce hypometabolism (Smith et al., 2015; Wijenayake et al., 2018). Activation of PDK-1, for example, reduces pyruvate entry into the mitochondria and thus increases glycolytic rates (Alexander-Shani et al., 2017), whereas glycolysis is actively suppressed in T. scripta (Brooks and Storey, 1989, 1993) through post-translational modifications (Mattice et al., 2018) and overall DNA methylation (Wijenayake and Storey, 2016). Some studies in mammalian systems have also noted that suppression of HIF may be neuroprotective (Chen et al., 2008, 2009).
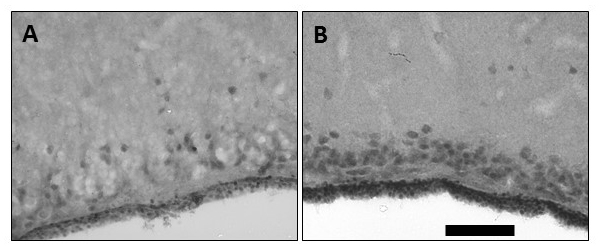
In a new window | Download PPT
Figure 3: Imunohistochemical labeling of vascular endothelial growth factor (VEGF). VEGF labeling in frontal sections of the perfusion-fixed turtle brain showing (A) basal expression in some cortical neurons and ependymal region of a control animal, and (B) enhanced expression in cortical neurons and ependymal region at the end of 24 hr anoxia. The calibration bar is equivalent to 100 μm.
The JAK/STAT pathway is also upregulated in the anoxic turtle, though this appears to be tissue specific as it is activated in turtle liver but not muscle (Bansal et al., 2016). JAK/STAT is involved in transmitting extracellular signals to the nucleus, targeting genes that promote cell survival and growth, acting in part through the PI3K/Akt and ERK pathways, which are both protective in the anoxic turtle brain (Milton et al., 2008; Nayak et al., 2011; Nayak et al., 2016). Similarly, the Unfolded Protein Response (UPR) acts as a signaling pathway, triggering downstream responses that promote cell survival. Cellular stress that results in misfolded secretory proteins in the ER lumen triggers the UPR, which works to re-establish homeostasis and thus may contribute to the ischemia-resistant phenotype of preconditioned cells (Bickler et al., 2015). However, the UPR can initiate apoptosis if the degree of protein misfolding is overwhelming, (Nakka et al., 2010). Downstream actions of the UPR may involve three different pathways: activating transcription factor 6 (ATF6), pERK, and serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1 α (IRE1α), which are all upregulated at different times following hypoxic PC in the rat hippocampal slice (Bickler et al., 2015). While these pathways are generally considered protective (i.e. ATF6 translocation to the nucleus activates the ER-targeted chaperone, GRP78 (Doroudgar et al., 2009)), taurine protection of rat neuronal cell cultures during hypoxia/hypoxia-reoxygenation occurred through a reduction in cleaved ATF6 and decreased IRE1 levels (Prentice et al., 2017). Two characteristic chaperones of the UPR, GRP78 and GRP94, increase at both the transcriptional and protein level in the anoxic turtle heart, kidney, and liver (Krivoruchko and Storey, 2013), though only GRP94, and not GRP78, change significantly in the anoxic turtle brain (Kesaraju et al., 2009c). The anoxic turtle heart, kidney, and liver also show increased levels of eukaryotic initiation factor 2 (eIF2α), activation of the PERK-regulated transcription factor, activating transcription factor 4 (ATF4), and significantly increasing the downstream targets of ATF4, ATF3 and GADD34 (Krivoruchko and Storey, 2013).
Epigenetics
While a number of genomic responses such as those above have been well described in IPC, the study of epigenetic responses is still in its infancy, though stroke is associated with increases in both DNA methylation and histone acetylation (Krupinski et al., 2018). Generally studied epigenetic mechanisms include non-coding RNAs (often microRNAs), global SUMOylation, methylation, and histone modification (Yang et al., 2017). Epigenetics is also of increasing interest in the study of anoxia tolerant organisms, as numerous studies, for example, suggest a conserved microRNA (miRNA) response to environmental stress that reprioritizes the synthesis of different protein types (Biggar and Storey, 2018). Different miRNAs may be up- or down-regulated, however, with differing effects on survival (Peng et al., 2013; Chi et al., 2014). Individual miRNAs may target the transcripts for hundreds of genes, while conversely an individual mRNA can be targeted by multiple miRNAs. Thus, there is the potential for significant regulatory potential and crosstalk (Luo and Zhang, 2009; Maziere and Enright, 2007), with computational methods increasingly applied to infer miRNA functions (Liu et al., 2014).
In the anoxic turtle, overall protein synthesis is suppressed as an energy saving mechanism, with decreased turnover in the brain (Fraser et al., 2001; Smith et al., 2015), liver (Hochachka et al., 1996), and even in a number of proteins in the turtle heart mitochondria (Gomez and Richards, 2018b), which may explain part of the anoxic decrease in complex V activity (Gomez and Richards, 2018b). As suggested by Biggar and Storey (2017), decreases in protein synthesis could be through decreased availability of mRNA substrates, via regulation of ribosomes, or by the regulation of mRNA availability to ribosomes, as through miRNA. In the turtle, though, there is no apparent change in total mRNA (Riggs et al., 2018) or in the mRNA levels of the genes that most consistently decrease in anoxia (Douglas et al., 1994; Rouble et al., 2014).
Increases in miRNA, though, can regulate a number of cell processes, including metabolism (Rottiers and Näär, 2012 and Vienberg et al., 2017), gene translation and expression, and general cell proliferation, development, and differentiation (Zhang et al., 2019a). MiRNAs are reported to play a role in the pathogenesis of stroke, including excitotoxicity, oxidative stress, inflammation, apoptosis, angiogenesis, and neurogenesis (Peng et al., 2013; Chi et al., 2014; Wang et al., 2015). Therefore therapies targeted to specific miRNAs may decrease secondary brain damage or increase regeneration after stroke (Lopez et al., 2017). MicroRNA MiR-21, for example, represses translation of proteins that lead to apoptosis and inflammation (Xu et al., 2014), and increases significantly in the penumbra 2-7 days post-reperfusion in the rat MCAO model (Buller et al., 2010), while mimetics of miR-19b decrease ROS and cell death in PC12 cells undergoing hypoxia/reoxygenation. Interestingly, the miR-19b mimetic improves both pAkt/Akt and the Bcl-2/Bax ratios (Liu et al., 2019), both shown to be important parts of anoxic survival in the turtle brain (Nayak et al.,2011, 2016). Similarly, one target of MiR-21 is phosphatase and tensin homolog (PTEN), which when not inhibited dephosphorylates phosphatidylinositol (3,4,5)-triphosphate (PIP3), preventing it from activating Akt and increasing caspase cleavage (Lopez et al., 2017). Other microRNAs affect heat shock proteins (Chi et al., 2014; Wang et al., 2015).
The small non-coding RNA (snRNA) catalog of the brain of several vertebrates tolerant to varying degrees of anoxia was recently determined by Riggs et al. (2018), including the less anoxia tolerant epaulet shark (Hemiscyllium ocellatum) and leopard frog (Rana pipiens), and the highly anoxia tolerant carp and turtle (C. carassius and C. picta). MiRNAs were the most abundant form of snRNA in each case, and the majority of those miRNAs were known stress responsive miRNAs (Riggs et al., 2018), as was also recently reported in the embryo of the anoxia tolerant annual killifish (Riggs and Podrabsky, 2017). The majority of the miRNAs (67% of the C. picta catalog) did not annotate to any previously described RNA, and while the profiles of the snRNAs were broadly conserved in normoxia, the changes in anoxia were unique to each species, suggesting that multiple strategies are available to achieve the same endpoint. All four species shared the normoxic expression of a number of non-coding RNAs (ncRNAs) identified as stress responsive, including miRNAs miR-26, -30, -92, -99, -100, -125, -126, 128, -143, -181, -204, and miR-222 (Riggs et al., 2018). Two anoxia-induced responses were unique to the turtle: miR-182 increased in anoxia but declined dramatically upon reoxygenation, and miR-6497 increased both in anoxia and further by 2 weeks of recovery. MiR-182 also increases in abundance in response to IPC in the mouse brain (Lee et al., 2010). Interestingly, in killifish, mitochondrial miRNAs are highly differentially expressed in anoxia, but by contrast only a few cataloged mitoRNA sequences responded to anoxia in the turtle (Riggs et al., 2018) despite significant alterations to mitochondrial function in anoxia, including a significant decline in aerobic capacity and a mild uncoupling of the mitochondrial H+ gradient (Pamenter et al., 2016b).
Tissues other than the brain also show alterations in miRNA levels in response to anoxia and/or cold temperatures in turtles, with miRNA and tissue specific increases and decreases. Biggar and Storey (2015) report an increase in miR-16 in the liver and heart of turtles frozen to - 3°C, while miR-21 increased only in the heart (Biggar and Storey, 2015a). Tissue specific responses were also seen in the liver, kidney, white muscle, and spleen of turtles submerged for 5 or 20 hrs in anoxic cold (5°C) water (Biggar and Storey, 2017). Interestingly, the authors predicted significantly greater targets for the miRNAs at cold temperatures than at 37°C, with evidence again of the highly complex regulatory nature of miRNAs: at 1°C, they suggest that miR-16 would be predicted to target 2153 sites across 820 genes, with miR-21 targeting 1479 sites within 756 genes (Biggar and Storey, 2015a, 2017). The largest fraction of the predicted targets was associated with metabolic or cellular processes, such as cell cycle regulation. This is unsurprising, as metabolic processes are greatly inhibited as a key facet of the hypometabolic state, and continuing the cell cycle is also energetically expensive. Thus Biggar and Storey (2012) reported that cyclin D1, regarded as a key factor in the initiation of cell proliferation, declines significantly in the liver and kidney during anoxic submergence in the turtle. As there were no changes in mRNA transcript levels, the change was likely post-transcriptional. The cyclin D1 mRNA has conserved binding sites for both miR-16-1 and miR-15a, and levels of both miRNAs increase in anoxia (Biggar and Storey, 2012), suggesting a role for these ncRNAs in cell cycle suppression. The increases reported in miR-21, on the other hand, may promote cell survival by repressing apoptosis (Bao et al., 2019). In mammalian studies, targeted transcripts of miR-21include caspase-3, APAF-1, and programmed cell death protein-4 (PDCD-4) (Chan et al., 2005; Frankel et al., 2008; Wang and Lee, 2008; Carletti et al., 2010; Liu et al., 2016). Increases in miR-21 also promote cell survival through the PTEN/Akt pathway and ERK (Mao et al., 2017; Chai et al., 2018).
While miRNAs generally inhibit the translation or degradation of proteins by binding to their complementary mRNA targets, some interactions have been documented that increase mRNA-specific translation (Biggar and Storey, 2015b), or decreases in miRNA may release previously repressed targets. For example in mammals, an overexpression of miR-34a decreases Bcl-2 and increases apoptosis in neurons of the auditory cortex (Huang et al., 2017), though in other systems the induction of p53 increases miR-34a and is cytoprotective (Bhatt et al., 2010). In turtles, Biggar and Storey (2015) report a decrease in miR-34a in the liver and heart at -3°C, but this decrease could in turn result in an increase in the expression of deacetylase sirtuin 1 (Sirt1) (Yamakuchi et al., 2008).
The sirtuins collectively coordinate the regulation of various metabolic pathways and processes in response to a change in energy status within the cell, including glycolysis, gluconeogenesis, the cell cycle, and cell survival (Chang and Guarente, 2014). Thus, the sirtuins have been shown to play important roles in IPC, with evidence for roles in DNA repair, mitochondrial function, blood flow and neuroinflammation, synaptic function, nicotinamide adenine dinucleotide (NAD)+ metabolism, and in antioxidant activation (Koronowski and Perez-Pinzon, 2015). Resveratrol induced neuroprotection, for instance, is lost in sirt1 knockout mice subjected to MCAO, with metabolomics showing changes in glucose metabolism (e.g. glycolysis, the pentose phosphate pathway, and increased expression of the glucose transporter GLUT1), while glycolytic ATP production is impaired in the knockout slice. While the sirtuins have not been investigated in the anoxic turtle, a potential role for them has been suggested in hibernating ground squirrels (Rouble and Storey, 2015), and hibernation is similarly defined by a hypometabolic state. The potential role of sirtuins in anoxia tolerance is likely to vary by SIRT and by tissue, as their roles are complex and varied (Koronowski et al., 2017). Future research should examine Sirt1 in the turtle, for example, as it is associated with the nucleus and acts through activation of transcriptional coactivator peroxisome proliferator-activated receptor γ co-activator 1-α (PGC1α) (Higashida et al., 2013). PGC1 is associated with increases in superoxide dismutase (SOD) activity and mitochondrial uncoupling protein 2 (UCP2), and anoxic turtles show both a mild mitochondrial uncoupling (Pamenter et al., 2016a) and an increase in SOD activity upon recovery from anoxia (Willmore and Storey, 1997). Glycolytic pathways, however, are suppressed rather than activated, which more closely resembles the effects of SIRT suppression. A Sirt5 deficiency, for example, suppresses mitochondrial ATP production (Zhang et al., 2019b), while glycolytic ATP production is impaired in sirt1 deficient mice (Koronowski et al., 2017).
Conclusion
Despite a large body of research, it is clear that by and large human clinical trials to induce acute neuroprotection have failed (Labiche and Grotta, 2004). A recent review notes that “acute neuroprotection and conditioning strategies face a common translation issue: a myriad of possibilities exist, but with no strategy to select optimal candidates” (Tauskela and Blondeau, 2018). The study of an animal, honed through millions of years of evolution and “constitutively preconditioned” to withstand both acute and long-term anoxia, and reoxygenation without pathology, may provide novel insights as to the underlying survival phenotype. Mammalian models of anoxia or ischemia activate both protective and pathological mechanisms simultaneously, with preconditioning triggering physiological and genomic changes that favor a “survival phenotype” when the animal is exposed to a later, potentially lethal period of ischemia or hypoxia. In animals adapted for extended anoxia, it is easier to distinguish pro-survival from pro-death signaling, as pathological mechanisms are largely suppressed, while protective pathways are enhanced. As described in this review, many of the mechanisms employed by anoxia tolerant turtles to survive extended periods without oxygen overlap with those identified as critical factors in PC, though the turtle responses are often more robust. Identification of additional mechanisms, and further exploration of novel areas of study such as the miRNAs, may provide new therapeutic targets for diseases of hypoxia and ischemia.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Acknowledgments
The author acknowledges Dr. Gauri Nayak and Dr. Shailaja Kesaraju for contributions to the previously unpublished research described in this manuscript. Funding for the work was provided by an AHA grant and NIH grant 1R15AG033374-01 to SLM, and the FAU Foundation.
References
Sarah L. Milton1
1Department of Biological Sciences, Florida Atlantic University, 777 Glades Rd, Boca Raton, FL.
Corresponding author:
Sarah L. Milton
Email: smilton@fau.edu
In a new window | Download PPT
Figure 1: Anoxia tolerance in the freshwater turtle and mammalian preconditioning share many molecular pathways that comprise the “survival phenotype.” Some mechanisms are unique to facultative anaerobes such as the turtle, which repress overall metabolism to survive extended anoxia.
In a new window | Download PPT
Figure 2: Knockdown of Hsp72 significantly increases cell death in primary neuronal cultures of T. scripta (see “Constitutive adaptations that promote anoxic survival” in text, and Kesaraju et al., 2014). Representative propidium iodide (PI) staining of (A) and (A1): Normoxic control and siRNA treated cultures, respectively, (B) and (B1): 4 hr anoxic controls and siRNA treated, (C) and (C1): Anoxia/reoxygenation controls and siRNA treated cultures. Neuronal cultures were prepared from T. scripta cortex as described in Milton et al., (2007) and exposed to normoxia (air/5% CO2), 4 hr anoxia (90% N2/5% He/5% CO2), or 4 hr anoxia followed by 2 hr reoxygenation. Specific siRNA sequences against turtle Hsp72 and a scrambled siRNA control were transfected into cells with Lipofectamine-2000. A greater concentration of siRNA was required to achieve an equivalent knockdown (40-60%) in anoxic and anoxic/reoxygenation cultures (250 pM) than in normoxic ones (100 pM) due to the greater upregulation of HSPs in anoxia.
In a new window | Download PPT
Figure 3: Imunohistochemical labeling of vascular endothelial growth factor (VEGF). VEGF labeling in frontal sections of the perfusion-fixed turtle brain showing (A) basal expression in some cortical neurons and ependymal region of a control animal, and (B) enhanced expression in cortical neurons and ependymal region at the end of 24 hr anoxia. The calibration bar is equivalent to 100 μm.
Table I: An overview of some of the factors coordinating anoxic survival in the turtle brain. Constitutive factors present even in the normoxic turtle, and genomic changes that occur to establish the survival phenotype, are shared with many elements of mammalian preconditioning but may occur to a greater extent in the turtle. While some of the mechanisms which enable the brain to avoid depolarization are common to PC (underlined), the coordinated downregulation of energetically expensive processes to ~20% of normoxic levels in the first 1–2 hrs without oxygen is reflective of true facultative anaerobes.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 9597 | 16 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA